

Abstract
In this study, we investigate whether chronic treatment with β-adrenoceptor (βAR) ligands with inverse agonist activity enhances myocardial β2AR-mediated atrial tension more than neutral antagonists in transgenic mice (TG35). These mice exhibit chronic adrenoceptor activation because they possess a greater number of constitutively active receptors than wild type mice due to cardiac-specific overexpression of human β2ARs. TG35 and wild type mice were chronically treated for 90 h with three inverse agonists, ICI-118,551, propranolol, and carvedilol, and one neutral antagonist, alprenolol. After 96 h, we compared the basal and isoprenaline-stimulated (10 μM) increase in atrial tension in treated or untreated TG35 mice and wild type mice. In parallel, to determine the effect of chronic βAR ligand treatment on the amounts of G protein receptor kinase-2 (GRK-2) and G proteins, we performed Western blotting on myocardial cytosolic and membrane proteins.
Atria from the TG35 mice treated with inverse agonists showed increases in the baseline tension compared to those from alprenolol/vehicle-treated mice. ICI-118,551 and propranolol treatment restored the elevated myocardial G-inhibitory protein (Giα) levels to that of wild type. Also, treatment with inverse agonists upregulated G-stimulatory protein (Gsα) levels and GRK2 above those levels in vehicle-treated TG35 or wild type mice. The increased baseline atrial tension was reversed by the addition of ICI-118,551.
Overall, our data suggests that inverse agonists enhance baseline atrial tension more than neutral antagonists. Based on this, we propose that upregulation of the active conformation of the β2ARs, Gsα protein and restoration of Giα as three possible mechanisms to explain this enhanced receptor activity.
Therefore, the favourable effects of some ligands used in pathological conditions involving chronic adrenoceptor activation may be due to the inverse agonist activity of the ligand.
Keywords: β-Adrenoceptors, left atria, G proteins, G protein coupled receptor kinase, inverse agonists, isoprenaline, transgenic mice
Introduction
In diseases such as congestive heart failure (CHF), chronic activation of cardiac β-adrenoceptors (βARs) by endogenous agonists like adrenaline and noradrenaline can result in diminished myocardial contractility. This chronic adrenoceptor activation is accompanied by reduction in β1AR number, uncoupling of β2AR from the Gs-protein, increase in Giα protein, and increase in GRK2 protein (G protein coupled receptor kinase or β-adrenergic receptor kinase, βARK1/2) (Castellano & Bohm, 1997). Similar changes are observed during chronic activation of βARs by infusing synthetic and exogenous βAR agonists (Muller et al., 1993; Zhou et al., 1995). Recent studies show that chronic treatment with some βAR antagonists increase survival in heart failure by improving myocardial contractility (Reviewed by Krum, 1997). However, other ligands that exhibit similar βAR subtype selectivity profiles are not equally effective in heart failure suggesting mechanisms independent of antagonism of agonist actions for these ligands.
Traditionally, inverse agonism has been determined by detecting changes in baseline (unstimulated) parameters. This requires enough spontaneously active receptors to reveal the inverse agonist properties of compounds. The two most extensively used techniques for increasing the number of spontaneously active receptors have been to overexpress the wild type receptor or to express a constitutively active mutant of the receptor (Milligan et al., 1995). In heart failure β-adrenoceptor numbers are decreased, and it is therefore unlikely that inverse agonists would have any effect on baseline parameters. However, recent data has suggested that even at low receptor densities, and with no detectable change in baseline, inverse agonists and neutral antagonists evoke different cellular responses (Berg et al., 1999, in press). In CHO cells transfected with relatively low levels of the 5-HT2C receptor (∼200 fm mg−1 protein), 24 h incubation with the inverse agonist for 5-HT2C receptors, SB 206553, produced different cellular effects than incubation with the neutral antagonist, 5-methoxygramine. The inverse agonist, SB206553 produced sensitization to 5-HT2C receptor agonists activating phospholipase C (PLC), and also sensitized the response to another agonist, ATP, activating purinergic receptors, which also mediates its response through PLC activation. This effect was observed even though SB 206553 had no effect on baseline IP accumulation; SB 206553 was determined to be an inverse agonist from studies in cells transfected with much higher densities (∼5–10 pm mg−1 protein) of the 5-HT2C receptor. These results suggest the cellular response of sensitization may be a more sensitive indicator of inverse agonist activity than inhibition of baseline. The neutral antagonist, 5-methoxygramine, did not produce sensitization and antagonized the sensitizing effect of SB 206553 (Berg et al., 1999, in press). Thus, cells recognize differences between antagonists and inverse agonists even when there is no change in baseline.
Many βAR ligands previously classified as antagonists are now known to possess inverse agonism which imparts these ligands with negative intrinsic activity because they reduce the number of constitutively active receptors (reviewed by Milligan et al., 1995; Milligan & Bond, 1997). Constitutively active receptors can couple to G proteins and elicit biological response even in the absence of an agonist. Inverse agonists, in contrast to neutral antagonists, prevent both agonist-dependent receptor activation (like neutral antagonists do) and inhibit constitutively active receptors. We have a transgenic mouse line (TG35) which display a 50 fold increase in their total βAR density than wild type mice due to cardiac-specific overexpression of human β2ARs. This receptor overexpression increases the number of constitutively active receptors that contribute to chronic activation of the adrenoceptors (Milano et al., 1994; Bond et al, 1995). Hence, the TG35 mice provide a model to study agonist-independent chronic βAR activation.
Our study is designed to assess in the TG35 hearts, whether βAR ligands exhibiting inverse agonism improve myocardial β2AR-mediated atrial tension under basal and agonist-stimulated conditions more effectively than ligands exhibiting pure antagonism. As an index of myocardial tension (contractility) in mice, we will measure basal and isoprenaline-stimulated increases in atrial tension after chronic treatment with four pharmacologically distinct βAR ligands. Also, we will examine the effect of this treatment on the amount of GRK2 and G proteins and correlate the results with the functional data.
Methods
We have designed experiments to address the question, whether βAR ligands exhibiting inverse agonism improve atrial contractile response more effectively than ligands exhibiting pure antagonism. TG and wild type (WT) mice were chronically treated with four pharmacologically distinct βAR ligands: Alprenolol, β1-, β2-, β3-AR neutral antagonist (1.2 mg kg−1 h−1), carvedilol, β1-, β2-AR, α1AR selective and partial inverse agonist at β2AR (0.4 mg kg−1 h−1), ICI-118,551, β2AR-selective, inverse agonist (0.7 mg kg−1 h−1), and propranolol, β1-, β2-AR preferential and partial inverse agonist at β2AR (0.4 mg kg−1 h−1). To achieve uniform delivery of drug solutions, all mice were implanted subcutaneously with osmotic mini-pumps for 90 h after which additional 6 h were allowed for the elimination of drugs. The mice were then sedated with pentobarbitone (Nembutal®), the heart excised and the left atrium isolated. The left atrium was then used for measuring the baseline and isoprenaline-stimulated increases in tension (functional studies). The remaining right atria and ventricles were homogenized and separated into membrane and cytosol fractions. These protein fractions were subjected to gel electrophoresis and immunoblotting to detect membrane proteins such as, Giα, Gsα, Gq/11α and the cytosolic protein, GRK2/3.
Left atrial tension measurement
Left atria were excised from the hearts of either untreated or βAR ligand treated WT or TG35 mice. Atria were suspended in modified Kreb's bicarbonate solution [(mM) NaHCO3 (pH 7.4) 25, NaCl 118, KCl 4.8, glucose 10, NaS2O5 0.1, EDTA 0.03] , CaCl2.2H2O supplemented with ascorbic acid (1.1×10−4M), cocaine (1×10−5M), corticosterone (4×10−5M), phentolamine (3×10−6M) and CGP-20712A (3×10−7M). Phentolamine, a nonselective α-adrenoceptor antagonist and CGP-20712A, a β1AR antagonist, were maintained in the buffer during our experiments to ensure that the responses observed are only due to β2AR-mediation only. The atria were paced at optimal frequency (3.2 Hz) for isometric tension development with a 3 ms pulse duration and voltage at threshold +20%. After equilibration period of 10–15 min, baseline tension was recorded. Response to 10 μM isoprenaline was recorded on the polygraph after tension (mg) reached a plateau (2–4 min).
Determination of inverse agonist activity
To determine the inverse agonist activity of the compounds we used the left atria of TG4 mice. These mice possess severe cardiac-specific overexpression of the human β2-adrenoceptor (approximately 200 fold the β-adrenoceptor densities of wild type mice; Bond et al., 1995). This overexpression produces maximal left atrial tension in the absence of agonist stimulation (Milano et al., 1994), and provides an ideal screen for inverse agonist properties of drugs. The inverse agonist activity of three compounds, ICI-118,551, propranolol, and alprenolol had previously been determined (Bond et al., 1995). For this study we performed experiments with carvedilol and repeated the studies with ICI-118,551 to determine the validity of the historical results.
Myocardial membrane and cytosolic protein preparation
The right atria and ventricles from the mice were dissected free of connective tissue and fat. The tissues were then chilled and minced in 1 ml of ice-cold homogenization buffer (50 mM HEPES, pH 7.2, 150 mM NaCl, 5 mM EDTA). The minced tissues were homogenized with a polytron and the volume was brought to 10 ml with the same buffer and the homogenate was spun at 800 ×g for 10 min at 4°C in a tabletop centrifuge. The supernatant was passed through two layers of cheesecloth and then spun at 18,000 r.p.m. for 10 min at 4°C in a Sorvall R5B. The supernatant was stored as cytosol and the membrane pellet was resuspended in buffer (5 ml) and homogenized with a glass-glass homogenizer (Dounce) followed by high speed centrifugation. The membranes obtained were stored as pellets at 70°C. For immunoblotting the technique described next, pellets were resuspended in 1 ml of suspension buffer (50 mM HEPES pH 7.2, 12 mM MgCl2).
Immunoblotting
Membrane and cytosolic proteins prepared from mice hearts were separated on 12% SDS-polyacrylamide gels and electroblotted onto polyvinyldifluoride (PVDF membrane). The PVDF membrane was then probed with specific antisera for GRK2 and G proteins. The immunoblotting procedure used is as follows: Equal protein loading of lanes before electrophoresis was confirmed by measuring total protein prior loading using BCA-protein assay reagent kit (Pierce) described earlier and coommassie blue staining of gels and membranes. Blotted PVDF membranes were blocked with 5% non-fat milk in PBS-T at room temperature for 1 h followed by incubation with primary antibody either for Giα/Gq/11α/Gsα or GRK2/3 in 2.5% blocking milk for 1 h RT. Blots were washed with PBS-T three times over 30 min and incubated with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG for 1 h followed by three PBS-T washes. Detection of protein bands was performed using a chemilumenescent method according to the manufacturer's instructions. The optical densities of the detected bands were quantified using a calibrated densitometer. Since Gq/11α protein was unchanged in WT and TG mice, PVDF membranes were stripped and reprobed with Gq/11α anti-sera to assure equal loading of lanes. The absorbance of the protein bands detected by the densitometer was proportional to the amount of the Gq/11α protein over a range of total protein concentration (0.5–40 μg). Only samples on the same blot were compared for analysis and protein sample from each mouse was subjected to Western blot analysis at least three times. The antisera were used at the following dilutions: Giα, Gsα, and Gq/11α (1 : 1000), and GRK2/3 (1 : 3500).
Drugs
Alprenolol, isoprenaline, phentolamine, propranolol, and PBS-Tween were obtained from Sigma®. ICI-118,551, CGP-20712A and pertussis toxin were purchased from Tocris Cookson®, Calbiochem® and RBI® respectively. Carvedilol (DMSO & water) was a gift from SmithKline Beecham. ECL kit was purchased from Amersham. Antisera for Gia and Gq/11α were obtained from Santa Cruz biotechnology®. Gsα antisera were a gift from Professor Graeme Milligan, University of Glasgow. GRK2/3 antisera and the founders for the colonies of transgenic mice were a gift from Professor Robert. J. Lefkowitz, Duke University.
Data analysis
For functional studies, baseline and isoprenaline-stimulated increase in atrial tension (measured in mg of developed isometric tension) in TG35 mice atria treated with βAR ligand are compared to that of untreated (NTX) TG35 mice. The densities of all protein bands are expressed as percentages of the density of protein bands obtained from untreated WT mice (100%). Data are shown as mean±s.e.mean and were analysed using one-way ANOVA followed by Bonferroni's correction as a post-hoc test. P<0.05 was considered statistically significant.
Results
Basal and isoprenaline-stimulated left atrial tension in TG35 mice
The basal atrial tension was greater in TG35 mice chronically treated with the inverse agonists, carvedilol, propranolol, and ICI-118,551 than in untreated and alprenolol-treated mice (Figure 1). We observed a 56, 35 and 46% increase in baseline atrial tension in TG35 mice treated with ICI-118,551, propranolol, and carvedilol respectively, compared to the atrial tension in untreated TG35 mice. However, the maximal atrial tension stimulated by isoprenaline in the treated mice was not significantly different from that of the untreated mice atria. The actual values for isometric tension (mg) are depicted in Table 1. In WT mice, none of the ligands tested had any effect on baseline or isoprenaline-induced maximal atrial tension (data not shown).
Figure 1.
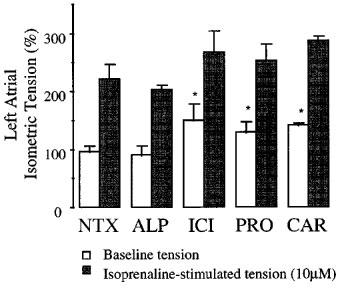
The effect of four βAR ligands on left atrial isometric tension in TG35 mice. Left atria were isolated from βAR ligand treated mice and mounted in the organ bath. Baseline and ISO (10 mM)-stimulated left atrial tension was recorded. The inverse agonists ICI-118,551, carvedilol, and propranolol treated atria showed an increase in the baseline tension, but no change in the ISO-stimulated maximal tension compared to the responses in atria from vehicle-treated TG35 mice. Shown mean values±s.e.mean. *P<0.05 vs NTX (baseline tension). NTX, untreated or vehicle-treated; ALP, Alprenolol; ICI, ICI-118,551; PRO, Propranolol; CAR, Carvedilol; ISO, Isoprenaline.
Table 1.
Left arterial isometric tension measurements in untreated and chronic βAR ligand-treated TG35 mice

Inverse agonist activity
Carvedilol produced a 42% maximal inhibition of baseline left atrial tension of TG4 mice (Figure 2). We also repeated experiments with ICI-118,551 to determine the validity of the historical controls published in Bond et al., 1995. The concentration-response curve to the inverse agonist effects of ICI-118,551 obtained was almost identical to the historical controls, with no significant differences in IC50 or maximal response (data not shown).
Figure 2.
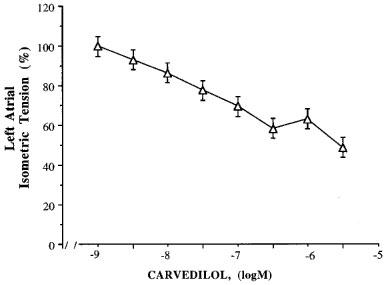
The effect of carvedilol on left atrial isometric tension of TG4 mice. Left atria were isolated from TG4 mice and mounted in the organ bath as previously described. A concentration response curve to carvedilol was performed. Shown are mean values± s.e.mean.
GRK2 and G protein Estimation
Chronic treatment of WT (Figure 3A) and TG35 mice (Figure 3B) with four different βAR ligands, alprenolol, ICI-118,551, propranolol, and carvedilol, showed distinct alterations in GRK2, Giα and Gsα proteins. TG35 mice possess a greater number of constitutively active receptors that contribute to the agonist-independent activation of the myocardial βARs. For TG35 mice, the myocardial proteins, Giα, Gsα and GRK2 were increased by 96, 38 and 82% respectively compared to WT mice in a fashion analogous to that observed with agonist-dependent overstimulation of adrenoceptors.
Figure 3.
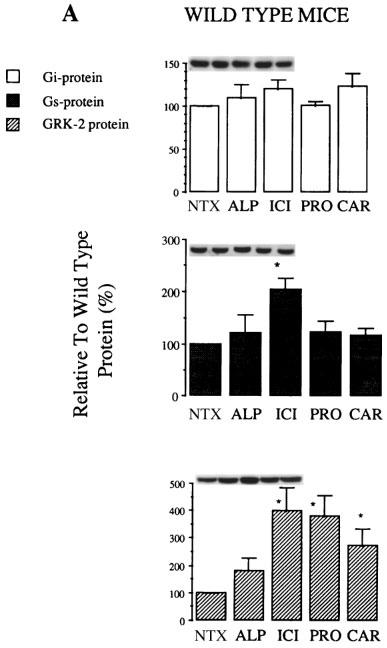
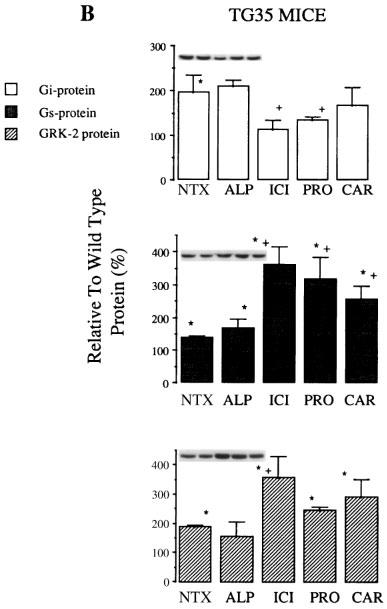
GRK2 and G-protein levels in WT (A) and TG35 (B) mice chronically treated with βAR ligands. WT and TG35 mice were implanted with 90 h mini-osmotic pumps to deliver either vehicle only (NTX) or ALP/ICI/PRO/CAR. After 96 h the mice were sacrificed and myocardial membranes and cytosol proteins were probed with Giα, Gsα or GRK2 anti-sera and specific bands were detected by ECL technique. Membranes were stripped reprobed with Gq/11α to assure uniform loading of membrane proteins. Only bands on the same blot were compared. All values shown are arbitrary densitometric units expressed as a percentage of WT (100%). ICI-118,551 and propranolol treatment restored Giα levels in TG35 to that of untreated WT. ICI-118,551, propranolol, and carvedilol upregulated Gsα levels in TG35 mice. GRK2 protein level was increased in all βAR ligand treated mice. *P<0.05 vs untreated WT and +P<0.05 treated TG35 vs untreated TG35 mice. NTX, untreated mice; ALP, Alprenolol; ICI, ICI-118,551; PRO, Propranolol; CAR, Carvedilol; ISO, Isoprenaline.
In TG35 mice, chronic treatment with ICI-118,551 and propranolol decreased the myocardial Giα levels, compared to untreated TG35 mice. While carvedilol and alprenolol treatment did not show similar reduction in Giα protein.
ICI-118,551 was different from the other inverse agonists in upregulating the Gsα in WT mice hearts by 259%. In TG35 mice treated with the partial inverse agonists, propranolol, and carvedilol, Gsα protein was increased by 216%, and 155% from the untreated WT hearts respectively. Alprenolol, a neutral antagonist, did not alter Gsα in the WT or TG35 mice.
GRK2 protein was increased in all WT and TG35 mice treated with inverse agonists compared to untreated WT mice. The order of increase in percentage of GRK2 protein in treated TG35 mice [343±69%(ICI)>280±58%(CAR)>236±11% (PRO)] are similar to the degree of inverse agonism exhibited by these ligands (ICI-118,551>carvedilol⩾propranolol>>alprenolol). In untreated TG35 mice hearts (NTX), GRK2 was increased by 82% compared to the WT mice and was not altered after treatment with alprenolol (Figure 3).
Reversal of the increase in baseline atrial tension by ICI-118,551
In TG35 mice treated with ICI-118,551, the enhanced baseline atrial tension was reduced by the addition of ICI-118,551 to the isolated atria (Figure 4). Thus, after chronic treatment with ICI-118,551, acute in vitro treatment with ICI-118,551 produced a 42% reduction in baseline. We have previously shown that in untreated TG35 mice, the maximal effect of ICI-118551 was a 20% reduction in baseline (Bond et al., 1995).
Figure 4.

The effect of ICI-118,551 on left atrial isometric tension of TG35 mice chronically treated with ICI-118,551. Left atria were isolated from ICI-118,551 treated mice and mounted in the organ bath as previously described. A concentration response curve to ICI-118,551 was performed 96 h after implantation of the subcutaneous mini-pump. Shown are mean values±s.e.mean.
Discussion
We have shown that chronic treatment of TG35 mice with βAR ligands displaying inverse agonism enhance baseline atrial tension more effectively than neutral antagonists do. These mice provide a model for chronic adrenoceptor activation because the mice possess a greater number of β2ARs in constitutively active conformation (R*). TG35 mice exhibit increases in the amounts of the myocardial GRK2 and G proteins, Gsα, and Giα analogous to the effects observed with agonist-dependent overstimulation of adrenoceptors. Chronic treatment with the inverse agonists, ICI-118,551, carvedilol and propranolol increased the baseline atrial tension in TG35 mice. However, ligand treatment did not increase the maximal atrial tension in response to isoprenaline. In parallel experiments, inverse agonist treatment increased myocardial Gsα proteins. ICI-118,551 and propranolol restored the Giα in TG35 mice hearts to normal or wild type levels. Carvedilol, which increased the baseline tension comparably to other inverse agonists, had a modest effect on the GRK2 and G proteins. Based on these data, we suggest three possible mechanisms for the improvement in basal atrial contractility after inverse agonist treatment, first, due to the upregulation of the receptors in R* conformation; second, the upregulation of Gsα; third, the restoration of Giα levels to normal.
The increase in baseline tension after chronic treatment with inverse agonists is may be due to the upregulation of the β2ARs in the R*-conformation. Inverse agonists are thought to bind to the receptors in inactive (R) conformation and shift the R←→R* equilibrium to reduce the number of receptors in R* conformation (constitutively active receptors). This reduction in R* during the treatment period may relieve the system from the adverse effects of adrenoceptor overstimulation, such as restoring the Giα protein levels to normal. After the cessation of the treatment, the number of R* may return to pretreatment levels or may be upregulated. This upregulated spontaneously active conformation of β2ARs (R*) may have promoted formation of more pre-coupled R*-Gsα which translated into an increased baseline contractility. Upregulation of the constitutively active mutant β2AR levels is observed following sustained treatment with the inverse agonist, betaxolol in neuroblastoma X glioma hybrid cells (MacEwan & Milligan, 1996). The upregulation of R* in our study is suggested by an increase in baseline tension (in absence of the agonist) without augmenting the agonist-stimulated maximal tension and indirectly by the increase in GRK2 protein in mice treated with the inverse agonists. Additional support for this interpretation is that the GRK2 increase is proportional to the increasing order of inverse agonist activity exhibited by the ligands (ICI-118,551>Propranolol⩾Carvedilol). However, this increase in GRK2 is in contrast to other studies which reported decreases in β-ARK (GRK2) activity after chronic treatment with βAR antagonists (Ping et al., 1995, Iaccarino et al., 1997). This discrepancy could be due to the fact that GRK2 protein restoration to normal levels may require longer exposure to ligands as these previous studies used 25 and 14 days exposure time respectively, vs 4 days in our study. The neutral antagonist, alprenolol treatment did not change the baseline contractility possibly due to its non-preferential binding to both R and R* and subsequent lack of effect on the R←→R* equilibrium.
The second factor that may contribute to the increase in atrial tension is the upregulation of myocardial Gαs in TG35 mice treated with inverse agonists. Since the β2AR number is 50 fold above that found in normal mice hearts, the accessibility of the receptor to the more abundant Gsα protein may have increased. This R*-Gs coupling further stimulates adenylate cyclase to enhance baseline atrial tension. We observed an upregulation of Gsα protein after ICI-118,551 in WT mice but no increase in baseline atrial tension which suggests that both the receptor density and the amount of Gsα is important. Despite starting at a higher baseline tension, the atria from mice treated with inverse agonists showed no increase in the maximal tension in response to isoprenaline. These results are consistent with previous findings that overexpression of Gsα in NG108-15 cells did not produced an increase in maximal agonist responses, but did increase baseline adenylate cyclase activity (Mullaney et al., 1996). An increase in expression of adenylate cyclase was required to produce increases in maximal adenylate cyclase activity suggesting the limiting step was the quantity of the enzyme (MacEwan et al., 1996). Furthermore, this ceiling effect on the agonist-stimulated myocardial contractility is also observed in TG4 mice hearts which possess a 200 fold increase in the total βAR density due to overexpression of β2ARs (Bond et al., 1995).
Alternatively, in vivo, cross-sensitization of other myocardial G-protein coupled receptors may explain the enhanced baseline atrial tension. For example, chronic β1AR antagonism also enhanced atrial contractility in human tissue by sensitizing the Gs-coupled, histamine receptors (Sanders et al., 1996) and 5-HT4 receptors (Sanders et al., 1995). Albeit, we have not examined the responses mediated by the H2 or 5-HT4 receptors in enhancing the basal tension in TG35 atria after inverse agonist treatment. If this is the mechanism for improving myocardial contractility, it appears that inverse agonist activity is required for cross-sensitization of other receptors because alprenolol, the neutral antagonist did not produce the same effect on contractility. Also arguing against cross-sensitization as the mechanism for the increase in baseline contractility in these studies is that the increase in baseline atrial contractility observed in vitro could be completely reversed by ICI-118,551 (Figure 4), thus suggesting the increase was mediated by β2-adrenoceptors.
Furthermore, inverse agonist treatment restored the Giα levels to normal. Cardiac β2ARs have been shown to couple to both Gs and Gi (Xiao et al., 1995). The functional coupling of β2AR-Gi has been shown to not only inhibit adenylate cyclase but may also promote hypertrophy by stimulating the MAPK pathway (Daaka et al., 1997). We propose that the inverse agonist treatment probably resulted in β2AR coupling showing further preferential coupling to Gs rather than Gi protein. The increase in the baseline atrial tension and the elimination of the potentiation of the isoprenaline response after pertussis toxin treatment in TG35 mice atria (unpublished data) demonstrates that Giα levels may have been restored to normal and no longer contributing substantially to the β2AR-mediated signalling. However, since the partial inverse agonist, carvedilol, did not decrease the Giα but increased the baseline atrial tension, it is possible that upregulation of R* and Gsα are more important contributors to the beneficial effects of inverse agonists.
Inverse agonists enhance baseline atrial contractility
In this study, we provide evidence to show that during chronic treatment with βAR ligands exhibiting inverse agonism enhance baseline contractile function more than neutral antagonists. The inverse agonists used in this study also increase the cardiac Gsα/Giα ratio and this may be the reason for the observed enhanced cardiac contractility.
Acknowledgments
This work was supported by the National Institutes of Health (No. 5 R29 GM54805-02 and DK30577). The authors wish to thank Mr Tuong Huynh for his excellent technical assisstance and Dr Douglas Eikenburg for his rigorous review of the manuscript.
Abbreviations
- βAR
β-adrenoceptors
- CHF
congestive heart failure
- CHO
Chinese hamster oocytes
- GRK2
G protein receptor kinase-2 or β-adrenergic receptor kinase-1
- NTX
untreated
- WT
wild type
References
- BERG K.A., STOUT B.D., CROPPER J.D., MAAYANI S., CLARKE W.P.Novel actions of inverse agonists on 5-HT2C receptor systems Mol. Pharmacol. 1999in press [PubMed]
- BOND R.A., LEFF P., JOHNSON D.T., MILANO C.A., ROCKMAN H.A., MCMINN T.R., APPASUNDARAM S., HYEK H.F., KENAKIN T.P., ALLEN L.F., LEFKOWITZ R.J. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of β2-Adrenoceptor. Nature. 1995;374:272–276. doi: 10.1038/374272a0. [DOI] [PubMed] [Google Scholar]
- CASTELLANO M, BOHM M. The cardiac beta-adrenoceptor mediated signaling pathway and its alterations in hypertensive heart disease. Hypertension. 1997;29:715–722. doi: 10.1161/01.hyp.29.3.715. [DOI] [PubMed] [Google Scholar]
- DAAKA Y., LUTTRELL L.M., LEFKOWITZ R.J. Switching of the coupling of the β2-adrenoceptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- IACCARINO G., LEFKOWITZ R.J., KOCH W.J.Reciprocal regulation of myocardial βARK1 by β-adrenergic agonists and antagonists Circulation 199796I405(abstract) [Google Scholar]
- KRUM H. β-adrenoceptor blockers in chronic heart failure-a review. Br. J. Clin. Pharmacol. 1997;44:111–118. doi: 10.1046/j.1365-2125.1997.00659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACEWAN D.J., MILLIGAN G. Inverse agonist-induced up-regulation of the human beta2-adrenoceptor in transfected neuroblastoma x glioma hybrid cells. Mol. Pharm. 1996;50:1479–1486. [PubMed] [Google Scholar]
- MACEWAN D.J., GUN-DO K., MILLIGAN G. Agonist regulation of adenylate cyclase activity in neuroblastoma x glioma hybrid NG108-15 cells transfected to co-express adenylate cyclase type II and the β2-Adrenoceptor. Biochem. J. 1996;318:1033–1039. doi: 10.1042/bj3181033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILANO C.A., ALLEN L.F., ROCKMAN H.A., DOLBER P.C., MCMINN T.R., CHIEN K.R., JOHNSON T.D., BOND R.A., LEFKOWITZ R.J. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G., BOND R.A. Inverse agonism and the regulation of receptor number. Trends Pharmacol. Sci. 1997;18:468–474. doi: 10.1016/s0165-6147(97)01139-5. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G., BOND R.A., LEE M. Inverse agonism: Pharmacological curiosity or potential therapeutic strategy. Trends Pharmacol. Sci. 1995;16:10–13. doi: 10.1016/s0165-6147(00)88963-4. [DOI] [PubMed] [Google Scholar]
- MULLER F.U., BOHELER K.R., ESCHENHAGEN T., SCHMITZ W., SCHOLZ H. Isoprenaline stimulates gene transcription of the inhibitory G protein α-subunit giα2 in rat heart. Circ. Res. 1993;72:696–700. doi: 10.1161/01.res.72.3.696. [DOI] [PubMed] [Google Scholar]
- MULLANEY I., CARR C., MILLIGAN G. Overexpression of Gsa in NG108-15, neuroblastoma X glioma cells: effects on receptor regulation of the stimulatory adenylyl cyclase cascade. FEBS Lett. 1996;397:325–330. doi: 10.1016/s0014-5793(96)01208-2. [DOI] [PubMed] [Google Scholar]
- PING P., GELZER-BELL R., ROTH D.A., KIEL D., INSEL P.A., HAMMOND H.K. Reduced beta-adrenergic receptor activation decreases G-protein expression and beta-adrenergic receptor kinase activity in porcine heart. J. Clin. Inv. 1995;95:1271–1280. doi: 10.1172/JCI117777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANDERS L., LYNHAM J.A., BOND B., DEL MONTE F., HARDING S.E., KAUMANN A.J. Sensitization of human atrial 5-HT4 receptors by chronic beta-blocker treatment. Circ. 1995;92:2526–2539. doi: 10.1161/01.cir.92.9.2526. [DOI] [PubMed] [Google Scholar]
- SANDERS L., LYNHAM J.A., KAUMANN A.J. Chronic beta1-adrenoceptor blockade sensitizes the H1 and H2 receptor systems in human atrium: role of cyclic nucleotides. Naunyn-Schmiedebergs. Arch. Pharmacol. 1996;353:661–670. doi: 10.1007/BF00167185. [DOI] [PubMed] [Google Scholar]
- XIAO R-P., JI X., LAKATTA E.G. Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol. Pharmacol. 1995;47:322–329. [PubMed] [Google Scholar]
- ZHOU Y., FRIEDMAN E., ROBERTS J., JOHNSON M.D. Modulation of aortic and cardiac G protein alpha subunits and their mRNAs during norepinephrine infusion in rats. J. Vasc. Res. 1995;32:16–23. doi: 10.1159/000159073. [DOI] [PubMed] [Google Scholar]