

Abstract
The effect of angiotensin II, angiotensin III, angiotensin IV and angiotensin-(1–7) on the electrically induced release of noradrenaline was studied in preparations of mouse atria, spleen, hippocampus, occipito-parietal cortex and hypothalamus preincubated with [3H]-noradrenaline. The prejunctional angiotensin receptor type was investigated using the non-selective receptor antagonist saralasin (AT1/AT2) and the AT1 and AT2 selective receptor antagonists losartan and PD 123319, respectively.
In atrial and splenic preparations, angiotensin II (0.01 nM–0.1 μM) and angiotensin III (0.01 and 0.1 nM–1 μM) increased the stimulation-induced overflow of tritium in a concentration-dependent manner. Angiotensin IV, only at high concentrations (1 and 10 μM), enhanced tritium overflow in the atria, while angiotensin-(1–7) (0.1 nM–10 μM) was without effect in both preparations.
In preparations of hippocampus, occipito-parietal cortex and hypothalamus, none of the angiotensin peptides altered the evoked overflow of tritium.
In atrial and splenic preparations, saralasin (0.1 μM) and losartan (0.1 and 1 μM), but not PD 123319 (0.1 μM), shifted the concentration-response curves of angiotensin II and angiotensin III to the right.
In conclusion, in mouse atria and spleen, angiotensin II and angiotensin III facilitate the action potential induced release of noradrenaline via a prejunctional AT1 receptor. Only high concentrations of angiotensin IV are effective in the atria and angiotensin-(1–7) is without effect in both preparations. In mouse brain areas, angiotensin II, angiotensin III, angiotensin IV and angiotensin-(1–7) do not modulate the release of noradrenaline.
Keywords: Angiotensin, angiotensin receptors, noradrenaline release, mouse atria, mouse spleen, mouse hippocampus, mouse occipito-parietal cortex, mouse hypothalamus
Introduction
Prejunctional facilitation of noradrenaline release from postganglionic sympathetic axons is a classical effect of angiotensin II (see Zimmermann et al., 1972; Peach, 1977; Starke, 1977). The majority of studies carried out with selective AT1 and AT2 angiotensin receptor antagonists suggest that the effect is mediated by AT1 receptors (Wong et al., 1991; Suzuki et al., 1992; Brasch et al., 1993; Ohia & Jumblatt, 1993; Gironacci et al., 1994; Rump et al., 1995; Boicos et al., 1998). However, there are also reports at variance with this view. In the rat caudal artery, the facilitatory effect of angiotensin II on noradrenaline release was reduced by both the selective AT1 and AT2 receptor antagonists losartan and PD 123319, respectively (Cox et al., 1995; 1996a,1996b). Conversely, the effect was not altered by selective concentrations of either antagonist in the rat left ventricle (Moura et al., 1997) or canine mesenteric and pulmonary arteries (Guimarães et al., 1998). Thus, the prejunctional receptor for angiotensin II is not clearly defined.
The smaller naturally occurring peptide fragments of angiotensin II, such as angiotensin III, angiotensin IV (angiotensin (3–8)) and angiotensin (1–7), are also now considered active hormones of the renin-angiotensin system (see Jackson & Garrison, 1996). However, little is known about their prejunctional effects. Angiotensin III has been shown to enhance the release of noradrenaline in the human pulmonary artery and saphenous vein (Molderings et al., 1988) and rabbit aorta (Storgaard & Nedergaard, 1997), but the receptor involved has not been investigated. Angiotensin IV and angiotensin-(1–7) failed to alter the overflow of noradrenaline in rabbit aorta (Storgaard & Nedergaard, 1997), while angiotensin-(1–7) increased the overflow in rat isolated atria (Gironacci et al., 1994).
A renin-angiotensin system, including all of the components, exists within the brain (Ganten et al., 1984). An effect of angiotensin on transmitter release from central noradrenergic neurons remains controversial; some reports show an increase while others show no effect (see Discussion).
In the present study, we have investigated the effect of angiotensin II, angiotensin III, angiotensin IV and angiotensin-(1–7) on the release of noradrenaline from peripheral (atria and spleen) as well as central (hippocampus, occipito-parietal cortex and hypothalamus) neurons of the mouse. The prejunctional angiotensin receptor was examined using the selective receptor antagonists losartan and PD 123319. The mouse was chosen because nothing is known about prejunctional angiotensin receptors in this species, except for the occurrence of a prejunctional facilitatory effect of angiotensin II in the atria (Musgrave & Majewski, 1989; Rajanayagam et al., 1989), and also because of the possibility of gene manipulation which has made the mouse a particularly important species to study.
Methods
Preparations and protocols
Adult male NMRI mice were killed by exsanguination. The heart, spleen or brain was removed and placed in physiological salt solution (PSS) which had been bubbled with 95% CO2 and 5% O2 and stored on ice. The wall of each atrium was cut into three to four pieces. The spleen was cut into 14 pieces. Round slices, 0.3 mm thick, 2 mm diameter, six to seven per animal, were prepared from the occipito-parietal cortex, parallel to the surface, after a superficial layer of 0.2 mm had been removed. Fourteen slices, 0.3 mm thick, were prepared from one hippocampus. The hypothalamus was prepared as described by Glowinski & Iversen (1966) and divided into four equal sized pieces. The tissue pieces (12–14) were incubated in 3 ml of PSS containing (−)-[2,5,6-3H]-noradrenaline (46.8–56.1 Ci mmol−1, 0.1 μM) for 30 min at 37°C. Twelve preparations were then superfused in parallel in 12 superfusion chambers with PSS at a constant flow rate of 1.2 ml min−1. The preparations were subjected to seven periods of electrical field stimulation consisting of a train of square wave pulses of 1 ms width and 80 mA (peripheral tissues) or 60 mA (central tissues) current strength, yielding a voltage drop of 45 or 34 V cm−1, respectively, between the electrodes of each chamber. After 30 min of superfusion (t=30 min), a ‘priming' stimulation period (peripheral tissues: 180 p, 3 Hz; central tissues: 36 p, 3 Hz) was applied. Beginning at t=54 min, the preparations were subjected to six periods (S1–S6) of electrical field stimulation (peripheral tissues: 120 p, 3 Hz; central tissues: 36 p, 3 Hz), delivered 18 min apart. Consecutive 2-min samples of the superfusate were collected. At the end of the experiment the tissue was dissolved in 0.5 ml of Soluene (Packard, Frankfurt am Main, Germany) and tritium was determined in the superfusate samples and preparations.
Concentration-response curves to the angiotensin peptides were determined by introducing the peptide in increasing concentrations after S1, 12 min before S2, S3, S4, S5 and S6. To investigate the possibility of receptor desensitization, a single, maximally effective concentration of each peptide was introduced into the PSS 12 min before S3 and remained present for the duration of the experiment. The antagonists were introduced from the beginning of superfusion (t=0 min) and remained for the duration of the experiment. To determine an effect of antagonist alone, it was introduced into the PSS 12 min before S3 and remained present for the duration of the experiment. In each experiment with 12 preparations, at least one preparation was superfused throughout with drug-free PSS. In experiments investigating the effects of the antagonists, at least one preparation was superfused with the antagonist alone and at least one with the agonist alone.
Drugs and radiochemicals
The PSS had the following composition (mM): NaCl 118, KCl 4.8, CaCl2 2.5 (peripheral tissues) or 1.3 (central tissues), MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, glucose 11, ascorbic acid 0.57, ethylenediaminetetraacetic acid disodium salt 0.03 and desipramine 0.001. The PSS for incubation with 3H-noradrenaline contained 0.2 mM (peripheral tissues) or 1.3 mM (central tissues) CaCl2 and no desipramine.
The following drugs were used: angiotensin II (human), angiotensin III (human), angiotensin IV (human), angiotensin II fragments 1–7 (human, Asp-Arg-Val-Tyr-Ile-His-Pro), saralasin, S(+)-1-[(4-dimethylamino-3-methylphenyl)methyl]-5-(diphenylacetyl)-4,5,6,7-tetrahydro-1H-imidazo[4,5-c] pyridine-6-carboxylic acid di-trifluoroacetate (PD 123319), desipramine hydrochloride (Sigma, Deisenhofen, Germany), losartan (gift from Merck, Darmstadt, Germany), phentolamine hydrochloride (gift from Ciba-Geigy, Basel, Switzerland). (−)-[2,5,6-3H]-noradrenaline was supplied by New England Nuclear (Dreieich, Germany) with a specific activity of 46.8–56.1 Ci mmol−1 and a radioactive concentration of 1 mCi ml−1.
Analysis of data
The outflow of tritium was calculated as a fraction of the tritium content of the tissue at the onset of the respective collection period, per min. The overflow of tritium evoked by electrical field stimulation was calculated as the total tritium outflow during the collection period in which stimulation was applied and during the two collection periods thereafter, minus the estimated basal outflow. Basal outflow was assumed to decline linearly from the collection period before stimulation to the second collection period after stimulation. The evoked overflow was expressed as a percentage of the tritium content of the tissue at the time of stimulation. Overflow ratios (Sn/S1) were then determined for each period of stimulation. Percentage changes of Sn/S1 ratios caused by a drug added after S1 were calculated for each preparation, taking as reference value the average corresponding Sn/S1 ratio in control experiments, in the absence of the drug. In experiments in which angiotensin antagonists (from t=0 min) were combined with agonists (after S1), the percentage changes of Sn/S1 ratios caused by the agonists added after S1 were also calculated for each preparation, taking as reference value the average corresponding Sn/S1 ratio in experiments in which the antagonist was present alone. Effects of drugs added after S1 on basal tritium outflow were calculated in the same manner, based on samples collected immediately before stimulation.
Data are expressed as means±s.e.mean; n denotes the number of preparations. The statistical significance of differences between groups was determined by Mann-Whitney test followed by Bonferroni correction. In all cases probability levels less than 0.05 (P<0.05) were taken to indicate significant differences.
Results
Basal outflow
In the absence of drugs, except for desipramine which was always present throughout superfusion, the basal outflow of tritium preceding the first period of stimulation averaged 0.0020±0.0001 min−1 in atrial (n=33), 0.0032±0.0001 min−1 in splenic (n=29), 0.0019±0.0002 min−1 in hippocampal (n=9), 0.0019±0.0002 min−1 in occipito-parietal cortex (n=10) and 0.0021±0.0001 min−1 (n=6) in hypothalamic preparations. There was a small decline in the basal outflow of tritium during the course of the experiments. None of the agonists, nor any of the antagonists, caused any change of basal tritium efflux (data not shown).
Evoked tritium overflow: atria
Electrical field stimulation produced an overflow of tritium which amounted to 0.979±0.073% of total tissue tritium (S1, n=33).
The effect of angiotensin II on tritium overflow from a single preparation is illustrated in Figure 1 (upper panel). Angiotensin II and angiotensin III increased the overflow of tritium with similar maximal enhancements of about 80% (Figure 2). The EC50 values were determined as the concentrations causing half-maximal enhancement, i.e. 40%, and amounted to 0.17 nM for angiotensin II and 2.2 nM for angiotensin III (interpolated by hand from the nearest points of the concentration-response curves). A single concentration of angiotensin II or angiotensin III (0.1 μM), when introduced into the PSS 12 min before S3, increased the evoked overflow, an effect which was maintained in the continued presence of the peptide and thus, was not subject to desensitization (n=4, data not shown). Angiotensin IV enhanced the stimulation-induced overflow of tritium only at concentrations of 1 and 10 μM (P<0.05, Figure 2). Angiotensin-(1–7) was without effect (Figure 2).
Figure 1.
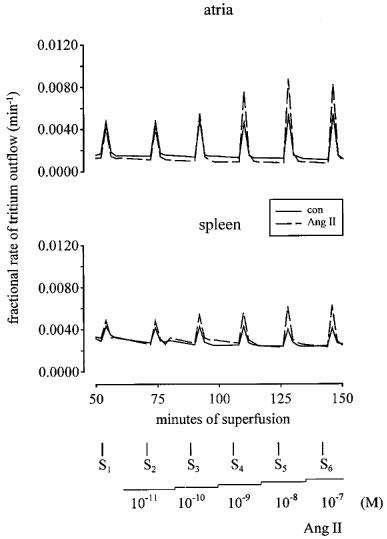
Tritium efflux from mouse atrial and splenic preparations in the absence and presence of angiotensin II (Ang II). Each preparation was stimulated for six periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Solid lines represent the control (con) experiments, in the absence of drugs, and dashed lines, the effect of Ang II when introduced into the PSS in increasing concentrations (0.01 nM–0.1 μM), 12 min before S2, S3, S4, S5 and S6. Each line represents the outflow of tritium from a single preparation.
Figure 2.
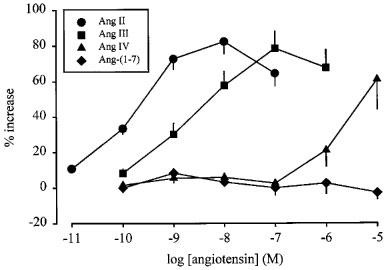
Effect of angiotensin II (Ang II), angiotensin III (Ang III), angiotensin IV (Ang IV) and angiotensin-(1–7) (Ang-(1–7)) on the stimulation-induced overflow of tritium from mouse atria. The preparations were stimulated for six periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Angiotensin was introduced into the PSS in increasing concentrations, 12 min before S2, S3, S4, S5 and S6. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of agonists. The vertical lines represent the s.e.mean from 4–27 preparations.
Little enhancement by angiotensin II (Figure 3) and angiotensin III (Figure 4) remained in the presence of the non-selective antagonist saralasin. The AT1 selective receptor antagonist losartan shifted the concentration-response curve of angiotensin II (Figure 3) and angiotensin III (Figure 4) to the right. The pKB values of losartan were calculated from the increase in EC50 concentrations, assuming that the maximal effect of the agonists, i.e. an 80% increase, was not changed by losartan (see Table 1). The AT2 selective receptor antagonist PD 123319 was without effect on the concentration-response curves to angiotensin II and angiotensin III (Figures 3 and 4).
Figure 3.
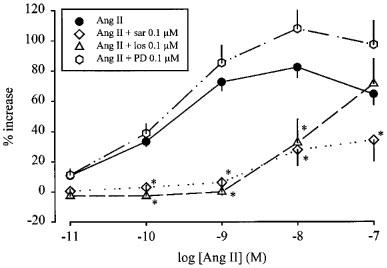
Effect of angiotensin II (Ang II), in the absence and presence of saralasin (sar), losartan (los) and PD 123319 (PD), on the stimulation-induced overflow of tritium from mouse atria. The preparations were stimulated for six periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Ang II was administered either alone or in the presence of sar, los or PD. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of Ang II. The vertical lines represent the s.e.mean, from 4–27 preparations. Asterisks represent significant differences from Ang II given alone; P<0.05.
Figure 4.
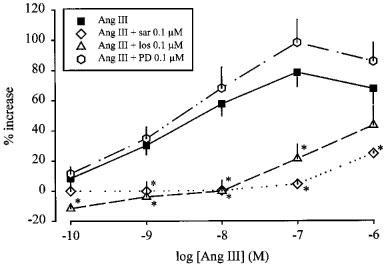
Effect of angiotensin III (Ang III), in the absence and presence of saralasin (sar), losartan (los) and PD 123319 (PD), on the stimulation-induced overflow of tritium from mouse atria. The preparations were stimulated for 6 periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Ang III was administered either alone or in the presence of sar, los or PD. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of Ang III. The vertical lines represent the s.e.mean from 4–19 preparations. Asterisks represent significant differences from Ang III given alone; P<0.05.
Table 1.
pKB values of losartan at prejunctional angiotensin receptors in mouse atria and spleen
Evoked tritium overflow: spleen
Electrical field stimulation produced an increased overflow of tritium which amounted to 0.376±0.035% of total tissue tritium (S1, n=29).
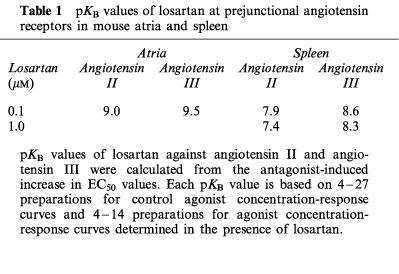
Angiotensin II and angiotensin III increased the stimulation-induced overflow of tritium. The maximal enhancements again were about 80% and the EC50 values, calculated as described above, were 0.25 and 1.5 nM, respectively (Figure 5). Figure 1 (lower panel) illustrates the effect of angiotensin II on tritium overflow from a single preparation. Angiotensin IV and angiotensin-(1–7) were without effect (Figure 5).
Figure 5.
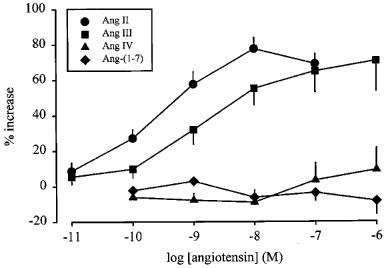
Effect of angiotensin II (Ang II), angiotensin III (Ang III), angiotensin IV (Ang IV) and angiotensin-(1–7) (Ang-(1–7)) on the stimulation-induced overflow of tritium from mouse spleen. The preparations were stimulated for six periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Angiotensin was introduced into the PSS in increasing concentrations, 12 min before S2, S3, S4, S5 and S6. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of agonists. The vertical lines represent the s.e.mean from 4–19 preparations.
Saralasin again strongly antagonized the effects of angiotensin II (Figure 6) and angiotensin III (Figure 7). Losartan produced a concentration-dependent rightward shift of the concentration-response curve of both angiotensin II (Figure 6) and angiotensin III (Figure 7). The pKB values are given in Table 1. PD 123319 did not significantly alter the facilitatory effect of angiotensin II (Figure 6) or angiotensin III (Figure 7).
Figure 6.
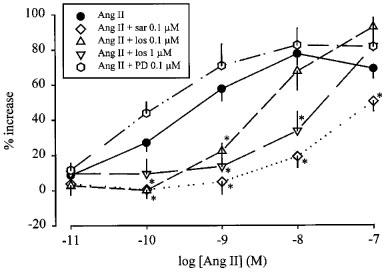
Effect of angiotensin II (Ang II), in the absence and presence of saralasin (sar), losartan (los) and PD 123319 (PD), on the stimulation-induced overflow of tritium from mouse spleen. The preparations were stimulated for six periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Ang II was administered either alone or in the presence of sar, los or PD. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of Ang II. The vertical lines represent the s.e.mean from 4–27 preparations. Asterisks represent significant differences from Ang II given alone; P<0.05.
Figure 7.
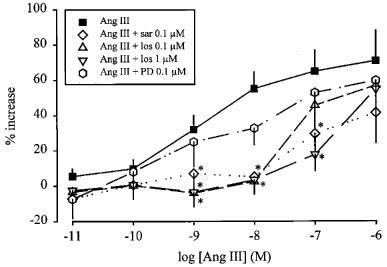
Effect of angiotensin III (Ang III), in the absence and presence of saralasin (sar), losartan (los) and PD 123319 (PD), on the stimulation-induced overflow of tritium from mouse spleen. The preparations were stimulated for six periods (S1–S6, 120 p, 3 Hz), delivered 18 min apart. Ang III was administered either alone or in the presence of sar, los or PD. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of Ang III. The vertical lines represent the s.e.mean from 4–19 preparations. Asterisks represent significant differences from Ang III given alone; P<0.05.
Evoked tritium overflow: hippocampus, occipito-parietal cortex and hypothalamus
The overflow of tritium evoked by the first stimulation period, S1, amounted to 2.08±0.17, 3.40±0.49 and 0.44±0.05% of total tissue tritium, in the hippocampus, occipito-parietal cortex and hypothalamus, respectively (n=9, 10 and 6). In the hypothalamus, the α adrenoceptor antagonist phentolamine, when added before S3, at a concentration of 1 μM, increased the evoked overflow of noradrenaline by 397±26% (n=4).
Angiotensin II, angiotensin III, angiotensin IV and angiotensin-(1–7) (each 0.1 nM–0.1 μM), when introduced in increasing concentrations, 12 min before S2, S3, S4, S5 and S6, did not significantly alter the the stimulation-induced overflow of tritium (data not shown, n=4–6). Figure 8 illustrates the lack of effect of angiotensin II in a single preparation from the hippocampus, occipito-parietal cortex and hypothalamus.
Figure 8.
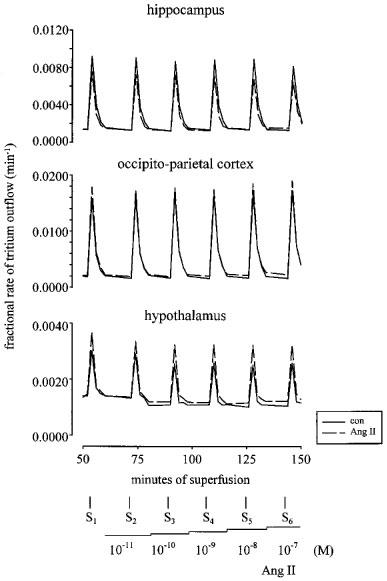
Tritium efflux from mouse hippocampal, occipito-parietal cortex and hypothalamic preparations in the absence and presence of angiotensin II (Ang II). Each preparation was stimulated for six periods (S1–S6, 36 p, 3 Hz), delivered 18 min apart. Solid lines represent the control (con) experiments, in the absence of drugs, and dashed lines, the effect of Ang II when introduced into the PSS in increasing concentrations (0.01 nM–0.1 μM), 12 min before S2, S3, S4, S5 and S6. Each line represents the outflow of tritium from a single preparation.
Discussion
In mouse hippocampus, occipito-parietal cortex and hypothalamus, angiotensin II, angiotensin III, angiotensin IV and angiotensin-(1–7) all failed to alter the electrically-induced release of noradrenaline. Previous studies investigating the effects of angiotensin on noradrenaline release in central tissues have revealed mixed findings. In agreement with the present study, angiotensin II failed to alter the depolarization-evoked release of noradrenaline in rat occipital cortex (Taube et al., 1977), hypothalamus (Taube et al., 1977; Meldrum et al., 1984; Meldrum & Westfall, 1986) and locus coeruleus (Huang et al., 1987). Other authors have reported that angiotensin enhances depolarization-evoked noradrenaline release in rabbit (Garcia-Sevilla et al., 1979) and rat hypothalamus (Sato et al., 1993) and rat parietal cortex (Huang et al., 1987). The present negative findings with angiotensin contrast with the expected release-enhancing effect of phentolamine in the hypothalamus. In the mouse occipito-parietal cortex and hippocampus, the α2 adrenoceptor antagonist rauwolscine also, in contrast to the angiotensins, increases release of noradrenaline (unpublished observations). In view of the findings to date, we are unable to identify the reason for the discrepant results on the effects of angiotensin in brain preparations.
Of the angiotensin peptide fragments studied, angiotensin IV and angiotensin-(1–7) had little or no effect in the peripheral tissues as well: only angiotensin IV, when given at high concentrations (1 and 10 μM), produced facilitation in the atria. Similarly, Storgaard & Nedergaard (1997) found that the two fragments did not alter the release of noradrenaline in the rabbit aorta, although angiotensin-(1–7) enhanced release in rat isolated atria (Gironacci et al., 1994). Overall, the effects of the two peptides on noradrenaline release seem to be minor.
Major, potent, and concentration-dependent facilitatory effects in both atria and spleen, however, were obtained with angiotensin II and III. In mouse atria, the observations with angiotensin II confirm previous findings by Musgrave & Majewski (1989) and Rajanayagam et al. (1989). Enhancement by angiotensin of noradrenaline release in the spleen apparently has not been shown previously. In the whole perfused cat spleen, angiotensin II did not alter the nerve stimulation-evoked overflow of noradrenaline (Hertting & Suko, 1966). This negative finding could be due to a species difference or to the vasoconstrictor effect of angiotensin II, which reduced flow through the spleen (Hertting & Suko, 1966) and in this way may have favoured retention of released noradrenaline (Hertting & Schiefthaler, 1963) and thus concealed the facilitatory effect of angiotensin.
Angiotensin III was approximately 13 and six times less potent than angiotensin II in the atria and spleen respectively. Previous studies investigating the pre- (Molderings et al., 1988; Trachte, 1988; Storgaard & Nedergaard, 1997) and postjunctional (Moore et al., 1976; Pendleton et al., 1991; Li et al., 1995; 1996) effects of angiotensin II and angiotensin III have generally observed a lower potency of angiotensin III compared to angiotensin II.
The facilitatory effect of angiotensin II on transmitter noradrenaline release in the mouse atria and spleen was blocked by the non selective angiotensin receptor antagonist saralasin and the selective AT1 receptor antagonist losartan, but was not altered by the AT2 receptor antagonist PD 123319. This finding is in accord with the majority of studies in a number of tissues from different species, including guinea-pig (Brasch et al., 1993) and rat atria (Gironacci et al., 1994), rat trachea (Boicos et al., 1998), rabbit iris ciliary body (Ohia & Jumblatt, 1993), and human (Rump et al., 1995) and dog (Wong et al., 1991; Suzuki et al., 1992) kidney. In most studies, full concentration-response curves to angiotensin in the presence of the receptor antagonists were not performed and hence, little is known about the pKB values for losartan against the facilitatory effect of angiotensin. Guimarães et al. (1998) reported very low pKB values for losartan against the facilitation of noradrenaline release by angiotensin II in canine mesenteric (pKB=5.24) and pulmonary (pKB=5.40) arteries. In the present study, the pKB estimates for losartan against angiotensin II were similar to those previously reported for the antagonism by losartan of the contractile effects of angiotensin II (6.9–9.5; Zhang et al., 1994; Boulanger et al., 1995; Li et al., 1997; Guimarães et al., 1998; de Gasparo et al., 1998). The pKB estimates for losartan in the present study are also similar to its pIC50 for inhibition of [125I]-angiotensin II (0.05 nM) binding to AT1 receptors (Chiu et al., 1989).
In addition to confirming the (predominant) AT1 character of the prejunctional receptor for angiotensin II, our experiments show that the same receptor also mediates the effect of angiotensin III, both in the atria and in the spleen: in either tissue, the pKB value of losartan against angiotensin III was similar to that against angiotensin II. Postjunctionally, angiotensin II and angiotensin III also act on the same AT1 receptor (Li et al., 1997). The reason for the relatively large difference (greater than 1 log unit) in the pKB values of losartan between the atria and spleen (Table 1) is not known, but could be due to tissue differences. Nevertheless, all values lie within the range of published pKB values at the AT1 receptor, as mentioned above.
In conclusion, apart from confirmation of the facilitatory effect of angiotensin II on action potential-evoked noradrenaline release in mouse atria, our study presents the following new findings: the analogous facilitatory effect of angiotensin II in mouse spleen and of angiotensin III in both peripheral tissues, the AT1 character of the prejunctional receptor for angiotensin II and angiotensin III, the lack of, or at most weak, effect of angiotensin IV and angiotensin-(1–7) in atria and spleen, and the lack of any effect of all four peptides in the hippocampus, occipito-parietal cortex and hypothalamus.
Acknowledgments
S.L. Cox is a recipient of an Alexander von Humboldt-Stiftung and their support is greatly appreciated.
Abbreviations
- Ang
angiotensin
- con
control
- los
losartan
- PD
PD 123319
- PD 123319
S(+)-1-[(4-dimethylamino-3-methylphenyl)methyl]-5-(diphenylacetyl)-4,5,6,7-tetrahydro-1H-imidazo[4,5-c] pyridine-6-carboxylic acid di-trifluoroacetate
- PSS
physiological salt solution
- sar
saralasin
References
- BOICOS K.M., COX S.L., STORY D.F. Modulation of noradrenergic transmission in rat isolated trachea by angiotensin I and angiotensin II. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358 supp 1:R293. [Google Scholar]
- BOULANGER C.M., CAPUTO L., LÉVY B.I. Endothelial AT1-mediated release of nitric oxide decreases angiotensin II contractions in rat carotid artery. Hypertension. 1995;26:752–757. doi: 10.1161/01.hyp.26.5.752. [DOI] [PubMed] [Google Scholar]
- BRASCH H., SIEROSLAWSKI L., DOMINIAK P. Angiotensin II increases norepinephrine release from atria by acting on angiotensin subtype 1 receptors. Hypertension. 1993;22:699–704. doi: 10.1161/01.hyp.22.5.699. [DOI] [PubMed] [Google Scholar]
- CHIU A.T., HERBLIN W.F., MCCALL D.E., ARDECKY R.J., CARINI D.J., DUNCIA J.V., PEASE L.J., WONG P.C., WEXLER R.R., JOHNSON A.L., TIMMERMANS P.B.M.W.M. Identification of angiotensin II receptor subtypes. Biochem. Biophys. Res. Comm. 1989;165:196–203. doi: 10.1016/0006-291x(89)91054-1. [DOI] [PubMed] [Google Scholar]
- COX S.L., BEN A., STORY D.F., ZIOGAS J. Evidence for the involvement of different receptor subtypes in the pre- and postjunctional actions of angiotensin II at rat sympathetic neuroeffector sites. Br. J. Pharmacol. 1995;114:1057–1063. doi: 10.1111/j.1476-5381.1995.tb13313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX S.L., STORY D.F., ZIOGAS J. Angiotensin II receptors involved in the enhancement of noradrenergic transmission in the caudal artery of the spontaneously hypertensive rat. Br. J. Pharmacol. 1996a;119:965–975. doi: 10.1111/j.1476-5381.1996.tb15766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX S.L., STORY D.F., ZIOGAS J. Multiple prejunctional actions of angiotensin II on noradrenergic transmission in the caudal artery of the rat. Br. J. Pharmacol. 1996b;119:976–984. doi: 10.1111/j.1476-5381.1996.tb15767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE GASPARO M., CATT K.J., INAGAMI T.Angiotensin receptors The IUPHAR Compendium of Receptor Characterization and Classification 1998Cambridge: London: IUPHAR Media; 80–86.ed. Girdlestone, D. pp [Google Scholar]
- GANTEN D., LANG R.E., LEHMANN E., UNGER T. Brain angiotensin: on the way to becoming a well-studied neuropeptide system. Biochem. Pharmacol. 1984;33:3523–3528. doi: 10.1016/0006-2952(84)90132-1. [DOI] [PubMed] [Google Scholar]
- GARCIA-SEVILLA J.A., DUBOCOVICH M.L., LANGER S.Z. Angiotensin II facilitates the potassium-evoked release of 3H-noradrenaline from the rabbit hypothalamus. Eur. J. Pharmacol. 1979;56:173–176. doi: 10.1016/0014-2999(79)90449-7. [DOI] [PubMed] [Google Scholar]
- GIRONACCI M.M., ALDER-GRASCHINSKY E., PEÑA C., ENERO M.A. Effects of angiotensin II and angiotensin-(1-7) on the release of [3H] norepinephrine from rat atria. Hypertension. 1994;24:457–460. doi: 10.1161/01.hyp.24.4.457. [DOI] [PubMed] [Google Scholar]
- GLOWINSKI J., IVERSEN L. Regional studies of catecholamines in the rat brain-I The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J. Neurochem. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- GUIMARÃES S., PAIVA M.Q., MOURA D. Different receptors for angiotensin II at pre- and postjunctional level of the canine mesenteric and pulmonary arteries. Br. J. Pharmacol. 1998;124:1207–1212. doi: 10.1038/sj.bjp.0701959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERTTING G., SCHIEFTHALER T. Beziehung zwischen Durchflussgrösse and Noradrenalinfreisetzung bei Nervenreizung der isolierten durchströmten Katzenmilz. Naunyn-Schmiedebergs Arch. exp. Pathol. Pharmakol. 1963;246:13–14. [Google Scholar]
- HERTTING G., SUKO J. Influence of angiotensin, vasopressin or changes in flow rate on vasoconstriction, changes in volume and [3H]-noradrenaline release following postganglionic sympathetic nerve stimulation in the isolated cat spleen. Br. J. Pharmacol. 1966;26:686–696. doi: 10.1111/j.1476-5381.1966.tb01848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG Y., ROGERS J., HENDERSON G. Effects of angiotensin II on [3H]noradrenaline release and phosphatidylinositol hydrolysis in the parietal cortex and locus coeruleus of the rat. J. Neurochem. 1987;49:1541–1549. doi: 10.1111/j.1471-4159.1987.tb01025.x. [DOI] [PubMed] [Google Scholar]
- JACKSON E.K., GARRISON J.C.Renin and angiotensin The Pharmacological Basis of Therapeutics 1996New York: McGraw-Hill; 733–758.Ed. 9, ed. Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W. & Goodman Gilman, A. pp [Google Scholar]
- LI Q., FEENSTRA M., PFAFFENDORF M., EIJSMAN L., VAN ZWIETEN P.A. Comparative vasoconstrictor effects of angiotensin II, III, and IV in human isolated saphenous vein. J. Cardiovasc. Pharmacol. 1997;29:451–456. doi: 10.1097/00005344-199704000-00004. [DOI] [PubMed] [Google Scholar]
- LI Q., ZHANG J., PFAFFENDORF M., VAN ZWIETEN P.A. Comparative effects of angiotensin II and its degradation products angiotensin III and angiotensin IV in rat aorta. Br. J. Pharmacol. 1995;116:2963–2970. doi: 10.1111/j.1476-5381.1995.tb15951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI Q., ZHANG J., PFAFFENDORF M., VAN ZWIETEN P.A. Direct positive chronotropic effects of angiotensin II and angiotensin III in pithed rats and in rat isolated atria. Br. J. Pharmacol. 1996;118:1653–1658. doi: 10.1111/j.1476-5381.1996.tb15588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELDRUM M.J., WESTFALL T.C. Angiotensin facilitation of 3H-norepinephrine release in central tissue of spontaneously hypertensive rats but not in Wistar-Kyoto rats: effects of sodium depletion. J. Cardiovasc. Pharmacol. 1986;8:582–587. [PubMed] [Google Scholar]
- MELDRUM M.J., XUE C.-S., BADINO L., WESTFALL T.C. Angiotensin facilitation of noradrenergic neurotransmission in central tissues of the rat: Effects of sodium restriction. J. Cardiovasc. Pharmacol. 1984;6:989–995. [PubMed] [Google Scholar]
- MOLDERINGS G.J., LIKUNGU J., HENTRICH F., GÖTHERT M. Facilitatory prejunctional angiotensin receptors on the sympathetic nerves of the human saphenous vein and pulmonary artery. Potential involvement in β-adrenoceptor-mediated facilitation of noradrenaline release. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;338:228–233. doi: 10.1007/BF00173392. [DOI] [PubMed] [Google Scholar]
- MOORE A.F., HALL M.H., KHAIRALLAH P.A. A comparison of the effects of angiotensin II and heptapeptide on smooth muscle (vascular and uterine) Eur. J. Pharmacol. 1976;39:101–107. doi: 10.1016/0014-2999(76)90117-5. [DOI] [PubMed] [Google Scholar]
- MOURA D., VAZ-DA-SILVA M., MAGINA S., PINHEIRO H., GUIMARÃES S. Involvement of angiotensin II receptors in the facilitatory effect of bradykinin on noradrenaline release from the rat left ventricle. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:R51. [Google Scholar]
- MUSGRAVE I.F., MAJEWSKI H. Effect of phorbol ester and pertussis toxin on the enhancement of noradrenaline release by angiotensin II in mouse atria. Br. J. Pharmacol. 1989;96:609–616. doi: 10.1111/j.1476-5381.1989.tb11859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHIA S.E., JUMBLATT J.E. Prejunctional receptors and second messengers for angiotensin II in the rabbit iris-ciliary body. Exp. Eye Res. 1993;57:419–425. doi: 10.1006/exer.1993.1143. [DOI] [PubMed] [Google Scholar]
- PEACH M.J. Renin-angiotensin system: biochemistry and mechanisms of action. Physiol. Rev. 1977;57:313–370. doi: 10.1152/physrev.1977.57.2.313. [DOI] [PubMed] [Google Scholar]
- PENDLETON R.G., GESSNER G., HORNER E. Comparative effects of angiotensin II and angiotensin III in rabbit adrenal and aortic tissues. J. Pharmacol. Exp. Ther. 1991;256:614–620. [PubMed] [Google Scholar]
- RAJANAYAGAM M.A.S., MUSGRAVE I.F., RAND M.J., MAJEWSKI H. Facilitation of noradrenaline release by isoprenaline is not mediated by angiotensin II in mouse atria and rat tail artery. Arch. Int. Pharmacodyn. 1989;299:185–199. [PubMed] [Google Scholar]
- RUMP L.C., BOHMANN C., SCHAIBLE U., SCHULTZE-SEEMAN W., SCHOLLMEYER P.J. β-adrenergic, angiotensin II, and bradykinin receptors enhance neurotransmission in human kidney. Hypertension. 1995;26:445–451. doi: 10.1161/01.hyp.26.3.445. [DOI] [PubMed] [Google Scholar]
- SATO K., OKUMURA F., GOSHIMA Y., MIYAMAE T., MISU Y. L-Dopa potentiates presynaptic inhibitory α2-adrenoceptor- but not facilitatory angiotensin II receptor-mediated modulation of noradrenaline release from rat hypothalamic slices. Jap. J. Pharmacol. 1993;62:119–122. doi: 10.1254/jjp.62.119. [DOI] [PubMed] [Google Scholar]
- STARKE K. Regulation of noradrenaline release by presynaptic receptor systems. Rev. Physiol. Biochem. Pharmacol. 1977;77:1–124. doi: 10.1007/BFb0050157. [DOI] [PubMed] [Google Scholar]
- STORGAARD T., NEDERGAARD O.A. Prejunctional modulation by angiotensins of noradrenaline release from sympathetic neurons in isolated rabbit aorta. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:706–711. doi: 10.1007/pl00005109. [DOI] [PubMed] [Google Scholar]
- SUZUKI Y., MATSUMURA Y., EGI Y., MORIMOTO S. Effects of losartan, a nonpeptide angiotensin II receptor antagonist, on norepinephrine overflow and antidiuresis by stimulation of renal nerves in anesthetized dogs. J. Pharmacol. Exp. Ther. 1992;263:956–963. [PubMed] [Google Scholar]
- TAUBE H.D., STARKE K., BOROWSKI E. Presynaptic receptor systems on the noradrenergic neurones of rat brain. Naunyn-Schmiedeberg's Arch. Pharmacol. 1977;299:123–141. doi: 10.1007/BF00498554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRACHTE G.J. Angiotensin effects on vas deferens adrenergic and purinergic neurotransmission. Eur. J. Pharmacol. 1988;146:261–269. doi: 10.1016/0014-2999(88)90301-9. [DOI] [PubMed] [Google Scholar]
- WONG P.C., HART S.D., TIMMERMANS P.B.M.W.M. Effect of angiotensin II antagonism on canine renal sympathetic nerve function. Hypertension. 1991;17:1127–1134. doi: 10.1161/01.hyp.17.6.1127. [DOI] [PubMed] [Google Scholar]
- ZHANG J., PHAFFENDORF M., ZHANG J.S., VAN ZWIETEN P.A. A non-competitive type of angiotensin-receptor antagonism by losartan in renal artery preparations. Eur. J. Pharmacol. 1994;252:337–340. doi: 10.1016/0014-2999(94)90183-x. [DOI] [PubMed] [Google Scholar]
- ZIMMERMAN B.G., GOMER S.K., LIAO J.C. Action of angiotensin on vascular adrenergic nerve endings: Facilitation of norepinephrine release. Fed. Proc. 1972;31:1344–1350. [PubMed] [Google Scholar]