

Abstract
NIH3T3 fibroblast cells transfected with the full-length coding region of the MT2 human melatonin receptor stably expressed the receptor that is coupled to a pertussis toxin-sensitive G protein and exhibits high affinity for melatonin (KI=261 pM).
The order of apparent affinity for selected compounds was: 4-phenyl-2-propionamidotetralin (4P-PDOT)>2-phenylmelatonin>2-iodomelatonin>2-bromomelatonin>6-chloromelatonin⩾melatonin>luzindole>N-acetyl-tryptamine⩾N-[(2-phenyl-1H-indol-3-yl)ethyl]cyclobutanecarboxamide (compound 6)>N-acetylserotonin.
4P-PDOT exhibited a very high selectivity (∼22,000 times) for the MT2 receptor with respect to the mt1 receptor subtype, as tested in comparative experiments with membrane preparations from NIH3T3 cells stably transfected with the human mt1 receptor.
MT2 melatonin receptors mediated incorporation of [35S]-GTPγS into isolated membranes via receptor catalyzed exchange of [35S]-GTPγS for GDP. The relative intrinsic activity and potency of the compounds were subsequently studied by using [35S]-GTPγS incorporation. The order of potency was equal to the order of apparent affinity. Melatonin and full agonists increased [35S]-GTPγS binding by 250% over basal (taken as 100%). Luzindole did not increase basal [35S]-GTPγS binding but competitively inhibited melatonin-stimulated [35S]-GTPγS binding, thus exhibiting antagonist action.
The other two mt1 antagonists used here, 4P-PDOT and N-[(2-phenyl-1H-indol-3-yl)ethyl]cyclobutanecarboxamide, behaved as partial agonists at the MT2 subtype, with relative intrinsic activities of 0.37 and 0.39, respectively.
These findings show, for the first time, important differences in the intrinsic activity of analogues between the human mt1 and MT2 melatonin receptor subtypes.
Keywords: Melatonin, MT2 receptor, melatonin, relative efficacy, melatonin analogues, melatonin antagonists, [35S]-GTPγS binding
Introduction
Melatonin is a hormone secreted rhythmically by the mammalian pineal gland. This hormone plays a central role in the regulation of many types of circadian and seasonal behaviour in mammals (Morgan et al., 1994). In man, melatonin elicits circadian, cardiovascular and hypnotic effects (Reppert et al., 1996; Dollins et al., 1994; Lewy et al., 1995; Cagnacci et al., 1998). Melatonin appears to evoke its effects through high affinity, G protein-coupled receptors (Morgan et al., 1994). Currently, two subtypes of human melatonin receptors have been identified: mt1 (preferentially expressed in the brain) and MT2 (preferentially expressed in the retina) (Reppert et al., 1994; 1995). Both native and recombinant melatonin receptors are negatively coupled to adenylyl cylase by pertussis toxin-sensitive G proteins (Morgan et al., 1994; Reppert et al., 1994; 1995). Little is known about the relative roles of the two receptor subtypes in the transduction of the melatonin signal. A recent study with mt1 receptor-deficient mice has suggested that the two melatonin effects on the mouse suprachiasmatic nuclei, inhibition of neuronal firing and entrainment of the circadian rhythm, could be independently mediated by the two receptor subtypes (Liu et al., 1998). The study of the different roles of the two receptor subtypes is further impeded by the lack of selective melatonin receptor agonists. Some melatonin receptor antagonists have been reported to be highly selective for the human MT2 receptor subtype (Dubocovich et al., 1997), but their efficacy has not yet been tested at the human melatonin receptor subtypes. For this reason we determined the affinity and efficacy of one of these analogues (4P-PDOT) at both human mt1 and MT2 receptors in the present study.
Recently we have studied the affinity and efficacy of a number of known melatonin analogues at the mt1 human melatonin receptor stably expressed in NIH3T3 fibroblast cells, using the agonist-mediated [35S]-GTPγS binding method for evaluation of the efficacy (Nonno et al., 1998). Agonist stimulation of G protein-coupled receptors induces conformational changes in their cognate G proteins, leading to activation of the G protein and effector activation. The essential step for G protein activation is the GDP/GTP exchange (Birnbaumer et al., 1990), that can be quantified by the use of [35S]-GTPγS (Hilf et al., 1989; Lorenzen et al., 1993). The amount of [35S]-GTPγS incorporation into membranes at maximal concentrations of agonist is related to the intrinsic activity of the agonist itself.
Studies on the efficacy of melatonin analogues at the human MT2 receptor are still lacking. Here we report the pharmacological characterization of the MT2 receptor stably expressed in NIH3T3 mouse fibroblast cells and the activity of known melatonin agonists, partial agonists and antagonists.
Methods
Cloning strategy and transfection
The human melatonin receptor MT2 gene has ben cloned by means of PCR with specific oligonucleotides designed on the GenBank sequence number U25341 (Human MT2 melatonin receptor mRNA, complete coding strand). Two pairs of primers have been used to clone the 5′ half and the 3′ half of the gene from human Hippocampus cDNA (Clontech): 10F (5′ GCG ATG TCA GAG AAC GGC TCC TT) and 535R (5′ ACT CCA GGG ACC CCA CAA AGA AGT) to amplify from nucleotide 9 (including the initiating methionine) to 559; 399F (5′ CAA TAT CAC TGC CAT CGC CAT TA) and 1081R (5′ CTA GAG AGC ATC TGC CTG GTG) to amplify from nucleotide 399–1101 (including the Amber codon). The PCR profile was as follows: 95°C for 5 min, (95°C for 1 min; 58°C for 1 min; 72°C for 1 min) for 5 cycles, (95°C for 1 min; 66°C for 1 min; 72°C for 1 min) for 30 cycles; 72°C for 7 min.
The PCR products have both been cloned (pGEM-T Vector System, Promega) and sequenced and they have been found to be identical to the U25341 sequence.
The 10F–535R sequence overlaps for 160 bp with the 399F–1081R sequence so that they share a Cfr10 I restriction site that is unique to the entire gene sequence. The two PCR products have been consequently cut by means of Cfr10 I restriction enzyme (Boehringer Mannheim) and ligated at their protruding ends. The final product was cloned in pcDNA3 expression vector (Invitrogen) and transfected in NIH3T3 cells by using liposomal transfection (DOTAP, Boehringer Mannheim) according to the manufacturer's instructions. The NIH3T3 cells were plated at a density yielding approximately 60% confluence at the time of transfection. Cells were cultured in Dulbecco's Modified Eagle's Medium containing high glucose (4.5 g l−1), 10% calf serum, 1 mM sodium pyruvate, in 5% CO2/95% air at 37°C. Selection with G418 (1 mg ml−1) was started 48 h after transfection. Transformed NIH3T3 cells were isolated and the single colonies were selected by using 2-[125I]-iodomelatonin binding (with a radioligand concentration of 100 pM). Colonies expressing apparent Bmax values higher than 150 fmol mg−1 of total cellular protein were plated in 150 cm2 flasks.
Membrane preparation
NIH3T3 cells stably expressing the cloned human MT2 receptor were grown to confluence. On the day of assay, cells were detached from flasks with 4 mM EDTA in 50 mM Tris-HCl (pH 7.4 at room temperature) and centrifuged at 1000×g for 10 min at 4°C. The cells were then resuspened in 2 mM EDTA/50 mM Tris-HCl, homogenized in 10–15 volumes of ice-cold 2 mM EDTA/50 mM Tris-HCl with Ultra-Turrax and centrifuged at 50,000×g at 4°C for 25 min. The final pellet was then resuspended in ice-cold 50 mM Tris-HCl assay buffer.
In experiments with pertussis toxin, cells were treated with 100 ng ml−1 of pertussis toxin in culture medium for the 24 h preceding the day of assay, and then prepared as described above. Membrane protein levels were determined according to the method of Bradford (1976).
2-[125I]-iodomelatonin binding
The final membrane concentration was 2–4 mg ml−1 and the protein concentration was 5–10 μg per tube. The binding conditions were described in detail elsewhere (Stankov et al., 1991). The incubation time was 90 min. In preliminary experiments for the selection of colonies and in competition experiments, the total 2-[125I]-iodomelatonin concentration was 100 pM. In saturation studies 2-[125I]-iodomelatonin was added to achieve a concentration range of 10 up to 1000 pM. Nonspecific binding was measured in presence of 0.1 μM cold 2-iodomelatonin.
[35S]-GTPγS binding
Agonist-stimulated [35S]-GTPγS binding was studied by using a modification of previously published methods (Hilf et al., 1989; Lorenzen et al., 1993). The final pellet, obtained as described above, was resuspended in ice-cold 50 mM Tris-HCl assay buffer to give a final membrane concentration of 20–30 mg ml−1. Then membranes (30–45 μg of protein per tube) were incubated for 30 min at 30°C, with and without various drugs, in assay buffer containing 0.3–0.5 nM [35S]-GTPγS, 50 μM GDP, 100 mM NaCl and 3 mM MgCl2.
The final incubation volume was 100 μl. Basal binding was assessed in the absence of drug and nonspecific binding was measured in presence of 10 μM GTPγS. In preliminary experiments, carried out to determine optimal conditions to study basal and melatonin-stimulated [35S]-GTPγS binding to NIH3T3MT2 membranes, GDP 0.1–100 μM and NaCl 1–100 mM were used. The incubation was terminated by adding 1 ml of ice-cold Tris-HCl buffer, pH 7.4, followed by rapid filtration under vacuum through Whatman GF/B glass fibre filters and by three washes with 3 ml of ice-cold Tris-HCl buffer, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry after overnight extraction in 4 ml Filter-Count scintillation fluid.
Data analysis
Data are reported as means±s.e.mean of at least three independent experiments that were each performed in duplicate (2-[125I]-iodomelatonin binding experiments) or triplicate ([35S]-GTPγS binding experiments). The IC50, EC50 and KD values were determined by using nonlinear curve fitting strategies. Saturation curves were analysed using the one-site model compared with the two-site model. A two-site model was accepted only when the 'goodness-of-fit' was significantly (P<0.05) improved by this model, as tested using a partial F-test procedure (De Lean et al., 1978). Hill transformation of the data has been performed to calculate Hill slope values in saturation experiments. KI values were calculated from the IC50 values using the Cheng-Prusoff equation (Cheng & Prusoff, 1973). The data from [35S]-GTPγS binding experiments are given as percentage of basal binding, where the basal binding was fixed as 100%. The relative intrinsic activity values are expressed as a fraction of melatonin maximal net stimulation. The analysis of competitive interaction between melatonin and luzindole was performed by using the equation:
where DR is the ratio of agonist IC50 values with or without antagonist, and [B] is the antagonist concentration. When n (the slope of the corresponding Schild plot) is not significantly different from 1, the pKB (instead of pA2) was calculated by nonlinear fitting by using the following equation, as proposed by Lew & Angus (1995):
where the agonist pEC50 in the presence of a given antagonist concentration is plotted versus the antagonist concentration [B] and allows the calculation of the pKB value as a fitted parameter. The parameter −logc is the difference between the antagonist pKB and the agonist pEC50.
In order to test the possible presence of 'spare' receptors in our system (defined as the fraction of the total receptor pool not required for maximal G protein activation in NIH3T3 cells) we used the equation proposed by Venter (1997):
where H is the height of the concentration effect curve, Φ is a fixed concentration ratio [antagonist]/[agonist] and eES is an effect-stimulus parameter, related to efficacy, that may be defined as eES=hm/Hm, that is maximum height of the concentration stimulus curve/maximum height of the concentration effect curve. An eES=1 indicates the absence of 'spare' receptors.
Statistical significance of the data was determined by analysis of variance followed by the nonpaired two-tailed Student's t-test.
Drugs
2-[125I]-iodomelatonin (specificity activity=2000 Ci mmol−1) and [35S]-GTPγS (specific activity 1070 Ci mmol−1) were purchased from Amersham (Buckinghamshire, U.K.). Melatonin, N-acetylserotonin, pertussis toxin, GDP and GTPγS were from Sigma Chemical Co. (St. Louis, MO, U.S.A.). 2-Iodomelatonin was obtained from RBI (Natick, MA, U.S.A.). 6-chloromelatonin was a gift from Ely Lilly laboratories (Indianapolis, IN, U.S.A.). 2-Bromomelatonin, 2-phenylmelatonin and N-[(2-phenyl-1H-indol-3-yl)ethyl]cyclobutanecarboxamide (compound 6) were synthesized as described elsewhere (Duranti et al., 1992; Spadoni et al., 1993; Garratt et al., 1995). Luzindole and 4-phenyl-2-propionamidotetralin (4P-PDOT) were from Tocris Cookson (Bristol, U.K.). Geneticin (G418) was purchased from GIBCO (Grand Island, NY, U.S.A.). General laboratory reagents including TrisHCl, Calf Serum, Dulbecco's Modified Eagle's medium were from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Filter-Count scintillation fluid was from Packard (Downers Grove, IL, U.S.A.).
Results
Binding of 2-[ 125I]-iodomelatonin to the MT2 receptor and competition studies
A number of clones were obtained and tested for MT2 receptor expression with 2-[125I]-iodomelatonin (100 pM). The clone expressing the highest 2-[125I]-iodomelatonin binding (single point assay) was chosen for subsequent studies, in order to ensure a good effect-to-noise ratio in the G protein stimulation assay.
Saturation experiments show that, in the absence of sodium ions and GTPγS, 2-[125I]-iodomelatonin binds to a single class of high affinity binding sites (Hill slope=1.01±1.02), with a pKD of 10.28±0.05 and a Bmax of 502±38 fmol mg−1 protein. In order to investigate receptor-G protein coupling, a series of saturation experiments conducted in the presence of GTPγS and NaCl was performed. In the presence of GTPγS (100 μM) 2-[125I]-iodomelatonin binds to a single class of high affinity binding sites (Hill slope=0.98±0.02), with a pKD of 10.1±0.07 (not significantly different from the pKD value obtained in absence of GTPγS, t=2.092, df=4, P=0.0523) and a Bmax of 585±21 fmol mg−1 protein, showing little difference to the binding profile in absence of GTPγS. In the presence of 500 mM NaCl, 2-[125I]-iodomelatonin binds to a single class of high affinity binding sites (Hill slope=0.97±0.03), with a pKD of 9.92±0.04 (significantly different from the pKD value obtained in absence of GTPγS, t=5.44, df=4, P<0.05) and a Bmax of 592±33 fmol mg−1 protein. On the contrary, the saturation curves obtained in the presence of both 500 mM NaCl and 100 μM GTPγS were significantly better described by the two-site model (Hill slope= 0.80±0.04, significantly different from unity, t=4.33, df=3, P<0.05), with 20% of receptors being in the high affinity state, pKD=10.25±0.04, and 80% in the low affinity state, with a pKD of 9.38±0.04.
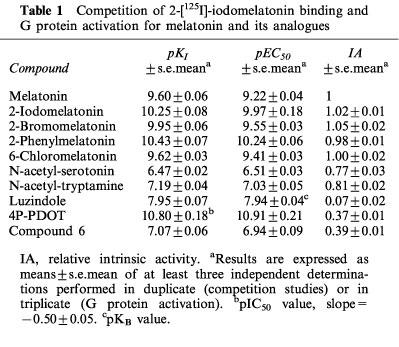
The pharmacological profile of the transfected MT2 receptor was studied in competition experiments with a number of known melatonin analogues (Figures 1 and 3). Melatonin showed a pKI of 9.60±0.06 (Table 1). The rank order of apparent affinity was: 4P-PDOT>2-phenylmelatonin≈thinsp;4 2-iodomelatonin>2-bromomelatonin>6-chloromelatonin⩾melatonin>luzindole>N-acetyl-tryptamine⩾N-[(2-phenyl-1H-indol-3-yl)ethyl]cylobutanecarboxamide (we will refer to this compound as compound 6)>N-acetylserotonin (pKI values are shown in Table 1). 4P-PDOT competed with 2-[125I]-iodomelatonin binding (Figure 3) with a slope of −0.50±0.03 (significantly different from unity, t=18.51, df=4, P<0.0001), thus not allowing KI calculation. The Hill slope values of all other compounds tested were between −0.9 and −1, and they were not significantly different from unity.
Figure 1.
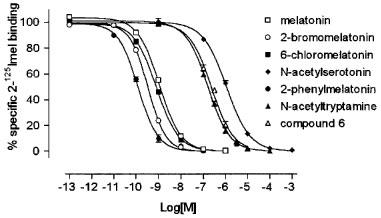
Competition curves of melatonin and its analogues for 2-[125I]-iodomelatonin (2-125Imel) binding to NIH3T3MT2 membranes. The data are representative of a single experiment with each point determined in duplicate. The rank order of apparent affinity was 2-phenylmelatonin> 2-bromomelatonin> 6-chloromelatonin⩾melatonin>N-acetyl-tryptamine⩾compound 6>N-acetylserotonin. The experiment was carried out as described in Methods, for 90 min at 37°C. Each point is the mean of duplicate determinations; the error bars indicate the standard deviation.
Figure 3.
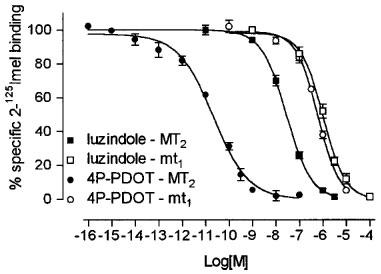
Comparison of competition curves for 4P-PDOT and luzindole to 2-[125I]-iodomelatonin (2-125Imel) binding to NIH3T3MT2 (closed symbols) and NIH3T3mt1 (open symbols) membranes. The data are representative of a single experiment in which each time point is determined in duplicate. Both 4P-PDOT and luzindole show higher affinity for the MT2 receptor subtype. Note the slope of the competition curve of 4P-PDOT (−0.58) in NIH3T3MT2 membranes with respect to the slope of luzindole (−0.91) in NIH3T3MT2 membranes and to the slope of 4P-PDOT (−0.96) and luzindole (−0.94) in NIH3T3mt1 membranes. The experiment was carried out as described in Methods, for 90 min at 37°C. Each point is the mean of duplicate determinations; the error bars indicate the standard deviation.
Table 1.
Competition of 2-[125I]-iodomelatonin binding and G protein activation for melatonin and its analogues
G protein activation by melatonin and its analogues
The basal [35S]-GTPγS binding to NIH3T3MT2 membranes was 80±5 fmol mg−1 protein. Melatonin caused a dose-dependent increase of the basal binding, to reach a plateau at ≈250±9% with a pEC50 value of 9.22±0.04 (Figure 2; pEC50 and relative intrinsic activity data are reported in Table 1). 2-Bromomelatonin, 2-iodomelatonin, 2-phenylmelatonin and 6-chloromelatonin also increased basal binding with a similar maximum (262±6, 225±9, 245±3 and 250±2%), pEC50 values being 9.55±0.03, 9.97±0.18, 10.24±0.06 and 9.41±0.03, respect-ively. N-acetyl-serotonin, N-acetyl-tryptamine, compound 6 and 4P-PDOT behaved as partial agonists with relative intrinsic activities ranging from 0.37 to 0.81 (see Table 1 and Figure 1). Luzindole was without any effect on basal [35S]-GTPγS binding with the exception of a very little stimulation over basal (0.07±0.02 relative to melatonin) at the highest concentrations tested.
Figure 2.
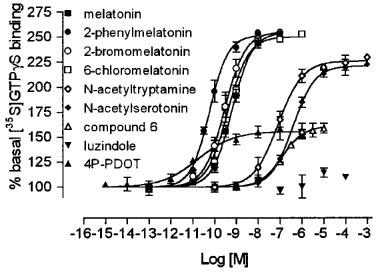
Comparison of the stimulation of [35S]-GTPγS binding to NIH3T3MT2 membranes by melatonin and its analogues. The data are representative of a single experiment with each point determined in triplicate. The order of agonist potency (with relative intrinsic activities obtained in this experiment reported in brackets) was: 4P-PDOT (0.37)>2-phenylmelatonin (0.98)>2-bromomelatonin (1.03)>6-chloromelatonin (0.97)>melatonin (1)>N-acetyl-tryptamine (0.82)>compound 6 (0.41)>N-acetylserotonin (0.78). Luzindole was without any effect on basal [35S]-GTPγS binding. The experiment was carried out as described in Methods, for 30 min at 37°C. Each point is the mean of triplicate determinations; the error bars indicate the standard error. Values represent percentage of basal binding, defined as 100%.
Luzindole was further able to dose-dependently shift the melatonin concentration-effect curve when added at three different concentrations (10, 100 nM and 1 μM), showing competitive and surmountable antagonism. The slope of the Schild plot was not significantly different from unity therefore suggesting simple competitive antagonism. Analysis of these data with the equation proposed by Lew & Angus (1995) allowed calculation of a pKB value of 7.94±0.07 (Table 1). Using the equation proposed by Venter (1997), in order to investigate the presence of 'spare' receptors in our system, we calculated an eES value of 0.99, consistent with the absence of 'spare' receptors, and a KA/KB value of 0.04.
Binding affinity and efficacy of 4P-PDOT and luzindole at the human mt1 receptor
In previous work (Nonno et al., 1998) we characterized the human mt1 melatonin receptor with the same analogues used in this study with the exception of luzindole and 4P-PDOT, so we also studied the interaction of these two compounds with the mt1 receptor for comparison with MT2 (Figure 3). 4P-PDOT and luzindole inhibited 2-[125I]-iodomelatonin binding to membrane preparation obtained from NIH3T3 cells stably expressing 600 fmol mg−1 protein of the mt1 human receptor, with pKI values of 6.98±0.05 and 6.72±0.04, respectively; comparison of the competition curves obtained with mt1 and MT2 membrane preparation reveals a high selectivity for 4P-PDOT (IC50mt1/IC50MT2=22,000) and, to a lesser extent, also for luzindole (KImt1/KIMT2=16.6).
Both analogues behaved as antagonists in the G protein activation assay, being unable to significantly affect basal [35S]-GTPγS binding to NIH3T3mt1 membranes at any concentration tested. In these experiments melatonin (100 nM) increased [35S]-GTPγS binding to 350±10% over basal.
Pertussis toxin sensitivity
A 24 h pretreatment of NIH3T3MT2 cells with 1 μg ml−1 pertussis toxin (PTX) completely abolished the melatonin induced increase in [35S]-GTPγS binding. Basal [35S]-GTPγS binding was significantly lower in membranes prepared from PTX-pretreated cells (45±6%) than in membranes prepared from control cells (100%). Melatonin stimulated binding was 245±12% in control cells while it was not significantly different from basal values in PTX-pretreated cells (the basal [35S]-GTPγS binding to PTX-pretreated membranes was 37±3 fmol mg−1 protein and the melatonin-stimulated binding was 40±2 fmol mg−1 protein).
Discussion
MT2 human melatonin receptors stably expressed in NIH3T3 cells bound melatonin and its analogues with high affinity and mediated incorporation of [35S]-GTPγS into isolated membranes via receptor-catalyzed exchange of [35S]-GTPγS for GDP. G protein activation was completely abolished by pertussis toxin treatment, showing Gi/o coupling with MT2 receptors.
2-[125I]-iodomelatonin bound to a single class of high affinity binding sites and this binding was resistant to modulation by GTPγS. Both GTPγS and NaCl, when not coincubated, reduced the affinity of 2-[125I]-iodomelatonin for the MT2 melatonin receptor, probably by inducing a partial dissociation of R/G complexes, but the Hill slopes (0.98 and 0.97, respectively) calculated from these saturation curves indicate a homogeneous receptor population. On the contrary, receptor/G protein complexes were efficiently dissociated in presence of both GTPγS and sodium chloride (Hill slope of 0.8 and significant improvement of the 'goodness of fit' with the two-site model). These data indicate that most of the melatonin receptors expressed in NIH3T3MT2 cells were coupled with G proteins in the presence of a full agonist (radiolabeled 2-iodomelatonin), suggesting that in our system the G protein concentration is not limiting; this conclusion is supported by the finding that no spare receptors were present in our system, as measured with the method proposed by Venter (1997). GTPγS-insensitivity indicates that the receptors form very stable receptor/G protein complexes. A GTPγS-insensitivity has been only reported for the melatonin receptors expressed in bovine (Nonno et al., 1995) and in the ovine hippocampus (Barrett et al., 1994), but not in other native tissues that express melatonin binding sites (Morgan et al., 1994), nor in the human cerebellum (Fauteck et al., 1994) that expresses the mt1 subtype (Mazzucchelli et al., 1996). We recently reported insensitivity to modulation of receptor coupling to G proteins by GTPγS also for the mt1 subtype stably expressed in NIH3T3 cells (Nonno et al., 1998), while both recombinant mt1 and MT2 subtypes were sensitive to guanine nucleotides when transiently expressed in COS-7 cells (Dubocovich et al., 1997). In that sense, the GTPγS-sensitivity of melatonin receptors seems to be tissue- or system-dependent. Similar findings have been reported for other PTX-sensitive G protein-coupled receptors, such as A1 adenosine (Nanoff et al., 1995) and the human 5HT1a receptor (Varrault et al., 1992). Furthermore, Nanoff and coworkers (1997) reported the partial purification of a membrane protein which stabilizes a tight receptor/Gi protein coupling mode of the A1 adenosine receptor; this protein, 'coupling cofactor', is responsible for the resistance of R/G complexes to modulation by guanine nucleotides. Wreggett & De Lean (1984) have also suggested that stability of R/G complexes could be a common feature for many Gi/o-coupled receptors.
The pharmacological profile obtained with competition studies reveals that melatonin and 6-chloromelatonin share the same apparent affinity for the MT2 receptor, that is different from the findings with the mt1 subtype, to which melatonin binds with a 6–7 times higher affinity than 6-chloromelatonin (Nonno et al., 1998), thus confirming previous reports (Reppert et al., 1994; 1995; Dubocovich et al., 1997). 4P-PDOT was the compound with the highest affinity to the MT2, while it showed only a modest affinity for the mt1 subtype, thus revealing a very high mt1/MT2 selectivity (∼22,000 fold). Studies of affinity of melatonin analogues for recombinant human mt1 and MT2 receptor subtypes are limited. Dubocovich et al. (1997) studied the affinity of a number of melatonin agonists, partial agonists and antagonists for both the mt1 and MT2 human melatonin receptor expressed in COS-7 cells. They reported that 4P-PDOT and some structurally-related analogues bound with ∼300 times higher affinity to the MT2 subtype, 4P-PDOT pKI values being 8.8 at MT2 and 6.3 at the mt1 subtype, while in the present study 4P-PDOT showed a pIC50 of 10.8 for the MT2 receptor and a pKI value of 6.98 for the mt1 subtype. We cannot exclude that the MT2 affinity of 4P-PDOT reported in the present work could be overestimated due to the very low slope value of its competition curves, accounting in part for the differences observed between the two studies and for the very high selectivity that we measured. In the case of luzindole, in contrast, our results for both mt1 and MT2 subtypes (pKI values of 6.72 and 7.95, respectively) were very similar to those reported by Dubocovich et al. (1997) (pKI values of 6.8 and 8).
To our knowledge the present work is the first study that reports the efficacy of melatonin and its analogues for the MT2 human melatonin receptor. The results of the G protein activation assay show that the human MT2 melatonin receptor expressed in NIH3T3 cells is efficiently coupled to a PTX-sensitive G protein. Melatonin induced [35S]-GTPγS binding in a dose-dependent manner with a pEC50 value of 9.22. This value, similar to the other pEC50 values reported here for agonists and partial agonists (see Table 1), is slightly lower than the pKI value. This finding probably reflects the different binding conditions of the two assays: in fact the [35S]-GTPγS binding experiments were performed in the presence of 50 μM GDP and 100 mM NaCl, that appears to induce the shift of a portion of the melatonin receptor to the low-affinity state, while the membrane preparations used for 2-[125I]-iodomelatonin binding experiments appear to contain virtually only high-affinity binding sites (see data from 2-[125I]-iodomelatonin saturation binding isotherms). This interpretation is supported by the fact that, according to the ternary complex model (Costa et al., 1992), the differences induced by the presence of GDP and NaCl are higher for full agonists than for partial agonists and antagonist (the pKI value and the pKB value of luzindole are identical).
Luzindole was the only antagonist at the MT2 subtype, while other putative melatonin antagonists such as compound 6 and 4P-PDOT behaved as partial agonists (with relative intrinsic activities of 0.39 and 0.37), the last showing very high potency (pEC50=10.91). In a previous study we reported that compound 6 showed antagonist activity at the mt1 receptor stably transfectd in NIH3T3 cells (KB=67 nM) (Nonno et al., 1998). In the present work we show that luzindole and 4P-PDOT are also mt1 antagonists as well. Luzindole and compound 6 are generally considered low affinity melatonin antagonists (Takaki et al., 1997; Garratt et al., 1995). In a recent work Dubocovich et al. (1997) studied the efficacy of 4P-PDOT and some structually related compound in the rabbit retina and found them to be high-affinity antagonists. However, the above considerations are based on efficacy studies performed in tissues or cells that express native melatonin receptors and may contain a heterogeneous receptor population. Therefore, they do not lend themselves to the unequivocal characterization of an analogue action at a specific receptor subtype. For example, the antagonist activity of compound 6 has been tested on pigment aggregation in Xenopus laevis melanophores (Garratt et al., 1995) that contain the Mel1c receptor, a melatonin receptor subtype that is not present in mammals (Reppert et al., 1996), while luzindole antagonizes the functional responses to melatonin in the calcium-dependent dopamine release assay from the rabbit retina (Dubocovich, 1988) and in the pigment aggregation in Xenopus laevis melanophores (Sugden, 1992). The receptor subtype composition of the rabbit retina is not definitively known. Comparison of the antagonist affinity constants obtained from their ability to inhibit dopamine release mediated by melatonin with their affinity constants (2-[125I]-iodomelatonin binding) determined in COS-7 cells expressing the human MT2 receptors, suggested that the rabbit retina may contain MT2 melatonin receptors (Dubocovich et al., 1997). The difference between the results obtained with 4P-PDOT in the rabbit retina and the present data (obtained in NIH3T3 cells stably expressing human MT2 melatonin receptors) could be due to diversity between the human and the rabbit MT2 melatonin receptors; another possible explanation is that 4P-PDOT expresses residual intrinsic activity in our system because of the high receptor number, and that such activity could not be detected in native tissue.
On the basis of the data obtained with the human mt1 and MT2 receptors expressed in the same cell type (NIH3T3) and characterized with the same method for the evaluation of relative intrinsic activities (stimulation of [35S]-GTPγS binding to membranes) we are able to draw some conclusions regarding the properties of compounds considered melatonin receptor antagonists, as tested in our system: (i) luzindole appears to antagonize both the human melatonin receptor subtypes, with low affinity to the mt1 and moderate affinity for the MT2; (ii) compound 6 behaves as a low-affinity mt1-antagonist and as a low-affinity MT2-partial agonist and (iii) 4P-PDOT is a low-affinity mt1-antagonist and a high-affinity MT2-partial agonist. These conclusions indicate that there are different requirements for antagonist action between the two human melatonin receptor subtypes and that those differences must be taken into account when studying the analogue's action in tissues for which the subtype composition is unknown.
In summary, agonist-stimulated [35S]-GTPγS binding in NIH3T3 cells is a sensitive method to study the human MT2 receptor activation of G proteins. MT2 receptor activation is measured at a point preceding second messenger generation and allows one to distinguish compounds of differing intrinsic activity and potency.
Abbreviations
- 4P-PDOT
4-phenyl-2-propionamidotetralin
- Compound 6
N-[(2-phenyl-1H-indol-3-yl)ethyl]cyclobutanecarboxamide
References
- BARRETT P., MACLEAN A., MORGAN P.J. Evidence for multiple forms of melatonin receptor-G-protein complexes by solubilization and gel electrophoresis. J. Neuroendocrinol. 1994;6:509–515. doi: 10.1111/j.1365-2826.1994.tb00613.x. [DOI] [PubMed] [Google Scholar]
- BIRNBAUMER L., ABRAMOWITZ J., BROWN A.M. Receptor-effector coupling by G proteins. Biochim. Biophys. Acta. 1990;1031:163–224. doi: 10.1016/0304-4157(90)90007-y. [DOI] [PubMed] [Google Scholar]
- BRADFORD M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CAGNACCI A., ARANGINO S., ANGIOLUCCI M., MASCHIO E., MELIS G.B. Influences of melatonin administration on the circulation of women. Am. J. Physiol. 1998;274:R335–R338. doi: 10.1152/ajpregu.1998.274.2.R335. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COSTA T., OGINO Y., MUNSON J.P., ONGUN ONARAN H., RODBARD D. Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol. Pharmacol. 1992;41:549–560. [PubMed] [Google Scholar]
- DE LEAN A., MUNSON P.J., RODBARD D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- DOLLINS A.B., ZHDANOVA I.V., WURTMAN R.J., LYNCH H.J., DENG M.H. Effect of inducing nocturnal serum melatonin concentrations in daytime on sleep, mood, body temperature, and performance. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1824–1828. doi: 10.1073/pnas.91.5.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBOCOVICH M.L. Luzindole (N-0774): a novel melatonin receptor antagonist. J. Pharmacol. Exp. Ther. 1988;246:902–910. [PubMed] [Google Scholar]
- DUBOCOVICH M.L., MASANA M.I., IACOB S., SAURI D.M. Melatonin receptor antagonists that differentiate between the human Mel1a and Mel1b recombinant subtypes are used to assess the pharmacological profile of the rabbit retina ML1 presynaptic heteroceptor. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;355:365–375. doi: 10.1007/pl00004956. [DOI] [PubMed] [Google Scholar]
- DURANTI E., STANKOV B., SPADONI G., DURANTI A., LUCINI V., CAPSONI S., BIELLA G., FRASCHINI F. 2-Bromomelatonin: synthesis and characterization of a potent melatonin agonist. Life Sci. 1992;51:479–485. doi: 10.1016/0024-3205(92)90024-j. [DOI] [PubMed] [Google Scholar]
- FAUTEK J.-D., LERCHL A., BERGMANN M., MOLLER M., FRASCHINI F., WITTKOVSKI W., STANKOV B. The adult human cerebellum is a target of the neuroendocrine system involved in the circadian timing. Neurosci. Lett. 1994;179:60–64. doi: 10.1016/0304-3940(94)90935-0. [DOI] [PubMed] [Google Scholar]
- GARRATT P.J., JONES R., TOCHER D.A. Mapping the melatonin receptor. 3. Design and synthesis of melatonin agonists and antagonists derivd from 2-phenyltryptamines. J. Med. Chem. 1995;38:1132–1139. doi: 10.1021/jm00007a010. [DOI] [PubMed] [Google Scholar]
- HILF G., GIERSHIK P., JACOBS K.H. Muscarinic acetylcholine receptor-stimulated binding of guanosine 5′-O-(3-thiotriphosphate) to guanine nucleotide-binding proteins in cardiac membranes. Eur. J. Biochem. 1989;186:725–731. doi: 10.1111/j.1432-1033.1989.tb15266.x. [DOI] [PubMed] [Google Scholar]
- LEW M.J., ANGUS J.A. Analysis of competitive agonist-antagonist interactions by nonlinear regression. Trends Pharmacol. Sci. 1995;16:328–337. doi: 10.1016/s0165-6147(00)89066-5. [DOI] [PubMed] [Google Scholar]
- LEWY A.J., SACK R.L., AHMED S., BAUER V.K., BLOOD M.L.The influence of melatonin on the human circadian clock The Pineal Gland and Its Hormones: Fundamentals and Clinical Perspectives 1995Nato Asi Series. New York: Plenum Press; 173–182.ed. Fraschini, F., Reiter, R.J. & Stankov, B. pp [Google Scholar]
- LIU C., WEAVER D.R., JIN X., SHEARMAN L.P., PIESCHL R.L., GRIBKOFF V.K., REPPERT S.M. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron. 1998;19:91–102. doi: 10.1016/s0896-6273(00)80350-5. [DOI] [PubMed] [Google Scholar]
- LORENZEN A., FUSS M., VOGT H., SCHWABE U. Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding. Mol. Pharmacol. 1993;44:115–123. [PubMed] [Google Scholar]
- MAZZUCCHELLI C., PANNACCI M., NONNO R., LUCINI V., FRASCHINI F., STANKOV B.M. The melatonin receptor in the human brain: cloning experiments and distribution studies. Mol. Brain Res. 1996;39:117–126. doi: 10.1016/0169-328x(96)00017-4. [DOI] [PubMed] [Google Scholar]
- MORGAN P.J., BARRET P., HOWELL H.E., HELLIWELL R. Melatonin receptors: localization, molecular pharmacology and physiological significance. Neurochem. Int. 1994;24:101–146. doi: 10.1016/0197-0186(94)90100-7. [DOI] [PubMed] [Google Scholar]
- NANOFF C., MITTERAUER T., ROKA F., HOHENEGGER M., FREISSMUTH M. Species differences in A1 adenosine receptor/G protein coupling: Identification of a membrane protein that stabilizes the association of the receptor/G protein complex. Mol. Pharmacol. 1995;48:806–817. [PubMed] [Google Scholar]
- NANOFF C., WALDHOER M., ROKA F., FREISSMUTH M. G protein coupling of the rat A1-adenosine receptor Partial purification of a protein which stabilizes the receptor-G protein association. Neuropharmacology. 1997;36:1211–1219. doi: 10.1016/s0028-3908(97)00135-4. [DOI] [PubMed] [Google Scholar]
- NONNO R., LUCINI V., PANNACCI M., MAZZUCCHELLI C., ANGELONI D., FRASCHINI F., STANKOV B.M. Pharmacological characterization of the human melatonin Mel1a receptor following stable transfection into NIH3T3 cells. Br. J. Pharmacol. 1998;124:485–492. doi: 10.1038/sj.bjp.0701860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NONNO R., LUCINI V., STANKOV B., FRASCHINI F. 2-[125I]Iodomelatonin binding sites in the bovine hippocampus are not sensitive to guanine nucleotides. Neurosci. Lett. 1995;194:113–116. doi: 10.1016/0304-3940(95)11742-f. [DOI] [PubMed] [Google Scholar]
- REPPERT S.M., GODSON C., MAHLE C.D., WEAVER D.R., SLAUGENHAUPT S.A., GUSELLA J.F. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8734–8738. doi: 10.1073/pnas.92.19.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REPPERT S.M., WEAVER D.R., EBISAWA T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron. 1994;13:1177–1185. doi: 10.1016/0896-6273(94)90055-8. [DOI] [PubMed] [Google Scholar]
- REPPERT S.M., WEAVER D.R., GOODSON C. Melatonin receptors step into the light. Trends Pharmacol. Sci. 1996;17:100–102. doi: 10.1016/0165-6147(96)10005-5. [DOI] [PubMed] [Google Scholar]
- SPADONI G., STANKOV B., DURANTI A., BIELLA G., LUCINI V., SALVATORI A., FRASCHINI F. 2-Substituted 5-methoxy-N-acyltryptamines: synthesis, binding affinity for the melatonin receptor, and evaluation of the biological activity. J. Med. Chem. 1993;36:4069–4074. doi: 10.1021/jm00077a010. [DOI] [PubMed] [Google Scholar]
- STANKOV B., COZZI B., LUCINI V., FUMAGALLI P., SCAGLIONE F., FRASCHINI F. Characterization and mapping of melatonin receptors in the brain of three mammalian species: rabbit, horse and sheep. Neuroendocrinology. 1991;53:214–221. doi: 10.1159/000125721. [DOI] [PubMed] [Google Scholar]
- SUGDEN D. Effect of putative melatonin receptor antagonists on melatonin induced pigment aggregation in isolated Xenopus laevis melanophores. Eur. J. Pharmacol. 1992;213:405–408. doi: 10.1016/0014-2999(92)90629-i. [DOI] [PubMed] [Google Scholar]
- TAKAKI K.S., MAHLE C.D., WATSON A.J. Melatoninergic ligands: pharmaceutical development and clinical applications. Curr. Pharmacol. Design. 1997;3:429–438. [Google Scholar]
- VARRAULT A., JOURNOT L., AUDIGIER Y., BOCKAERT J. Transfection of human 5-hydroxytryptamine1A receptors in NIH3T3 fibroblasts: effects of increasing receptor density on the coupling of 5-hydroxytryptamine1A receptors to adenylyl cyclase. Mol. Pharmacol. 1992;41:999–1007. [PubMed] [Google Scholar]
- VENTER D.P. Efficacy I: a new method for estimating the relative efficacy of full agonists via a newly defined efficacy related parameter. Eur. J. Pharmacol. 1997;320:223–231. doi: 10.1016/s0014-2999(96)00899-0. [DOI] [PubMed] [Google Scholar]
- WREGGETT K.A., DE LEAN A. The ternary complex model. Its properties and application to ligand interactions with the D2-dopamine receptor of the anterior pituitary gland. Mol. Pharmacol. 1984;26:214–227. [PubMed] [Google Scholar]