

Abstract
Adrenoreceptor agonists induce a hypertrophic phenotype in vitro and in vivo. To investigate the molecular remodeling in chronic cardiac hypertrophy we infused adult male mice with vehicle, isoproterenol, phenylephrine or both agonists for 3, 7 or 14 days.
All drugs increased cardiac mass. After minipump removal cardiac mass regressed to control levels within 7 days after PE and ISO treatment whereas ISO+PE treated hearts were incompletely regressed.
ANF and β-MHC, but not α-MHC, expression were increased by agonists at all time points. GATA-4, Nkx-2.5, Egr-1, c-jun and c-fos expression were increased after 3, 7 and 14 days of treatment. Expression was greatest after ISO+PE>>ISO>PE>vehicle infusion suggesting a synergistic effect of adrenoreceptor stimulation and indicating a greater effect of β- than α-adrenergic action in vivo.
After PE or ISO drug withdrawal the HW/BW was normal and Egr-1, c-jun, c-fos and GATA-4, but not Nkx2.5, expression dropped to control levels. HW/BW regression was incomplete after ISO+PE and elevated levels of Egr-1, c-jun and Nkx2.5 expression remained.
A hydralazine-mediated reduction in blood pressure had no effect on the agonist-induced cardiac hypertrophy or gene expression.
In conclusion, we found that continued agonist stimulation, and not blood pressure, is responsible for the maintained increase in gene expression. Further, we found the decrease in gene expression in the regression after drug withdrawal was gene specific.
Keywords: Phenylephrine, isoproterenol, hydralazine, Egr-1, c-fos, c-jun, GATA-4, Nkx2.5, cardiac hypertrophy, regression
Introduction
Cardiomyocyte hypertrophy is the fundamental adaptation of this terminally differentiated cell type to stress. Despite the diverse stimuli that lead to cardiac hypertrophy, there is a prototypical final molecular response of cardiomyocytes to a hypertrophic signal that involves an increase in cell size and protein synthesis, enhanced sarcomeric organization and activation of foetal genes that were repressed or shut off during development (reviewed in Schneider & Parker, 1993; van Bilsen & Chien, 1993; Pollak, 1995).
In a number of pathophysiological conditions leading to cardiac hypertrophy, the activity of the sympathetic nervous system is enhanced. The myocardium contains α1- and β1-adrenoreceptors (Kolbilka, 1993) that respond to norepinephrine (NE), a primarily α1- and β1-receptor agonist (Ostman-Smith, 1981). Within hours of in vivo activation of adrenoreceptors there is a transient increase in some transcription factors such as c-jun, junB, early growth response gene 1 (Egr-1), c-fos and c-myc (Soonpaa & Field, 1994; Brand et al., 1993; Boluyt et al., 1995; Zierhut & Zimmer, 1989; reviewed in Pollak, 1995). These proteins are downstream targets of various signal transduction pathways and are thought to play an important role in the signal transduction pathways leading to cardiac hypertrophy (Hannan & West, 1991). Supporting this notion is that atrial natriuretic factor (ANF), α-myosin heavy chain (α-MHC) and several growth factor promoters, such as platelet derived growth factor, and other genes normally increased in cardiac hypertrophy (reviewed in Schneider & Parker, 1993; van Bilsen & Chien, 1993; Lee et al., 1988), are targets for transcription factors such as Fos and Jun (Kovacic-Milicojevic et al., 1996) and Egr-1 (Khachigian & Collins, 1997). Although they may be important for cardiac hypertrophy, ablation of the FOS, JUN or EGR-1 genes does not lead to abnormalities in cardiac development (Wang et al., 1992; Johnson et al., 1993; Lee et al., 1995) suggesting either that they are not absolutely necessary for cardiac gene expression or that redundant functions can mediate in their absence.
The cardiac-specific transcription factors, GATA-4 and Nkx-2.5, play important roles in cardiac development and may also be important in hypertrophy. Expression of GATA-4 is restricted to the heart and gonads and GATA-4 knockout mice die by 9.5 days postcoitum with profound defects in ventral morphogenesis (Arceci et al., 1993; Kuo et al., 1997; Molkentin et al., 1997). The GATA motif (5′-AGATAA-3′) is present in the promoter regions of α-MHC (Molkentin et al., 1994), cardiac troponin C (Ip et al., 1994), atrial and ventricular myosin light-chain 1 (Kurabayashi et al., 1990). GATA-4 is thought to form complexes with other transcription factors, such as AP-1, NF-AT3 and Nkx2.5 to activate foetal cardiac genes (Herzig et al., 1997; Durocher et al., 1997; Molkentin et al., 1998). During development expression of Nkx-2.5 precedes that of other known cardiac-specific genes, and mice with a targeted disruption of the Nkx-2.5 gene show arrested heart development at the looping stage and die in utero (Lyons et al., 1995). In addition to its interaction with GATA-4, Nkx-2.5 serves as a positive acting accessory factor for serum response factor and together, Nkx-2.5 and the serum response factor, provides strong transcriptional activation of the cardiac α-actin promoter (Chen et al., 1996; Sepulveda et al., 1998).
At least three successive steps occur in the genesis of adaptive cardiac hypertrophy. First, a greater work-load is placed on the heart. Second, there are intermediate links through which the extra work triggers events at the level of the cardiac muscle cell that leads, third, to the biochemical and structural changes inducing growth. The immediate biochemical alterations occurring within the myocardial cell in response to adaptive hypertrophy have been extensively studied; however, most experiments examined the role of transcriptions and growth factor expression within a few hours of the activating stimulus with little attention placed on the role of these factors at later times. The purpose of our study is to examine the role of the immediate early genes (c-fos, c-jun and Egr-1) as well as the role of the specific cardiac genes (GATA-4 and Nkx-2.5) in the induction, maintenance and regression of cardiac hypertrophy induced by adrenoreceptor agonists in adult male mice. To discriminate the contribution of the two families of adrenoreceptors to hypertrophy development in vivo, we administered phenylephrine (PE, a specific α1-agonist), isoproterenol (ISO, a non-selective β-agonist) or both (ISO+PE) to mice. To distinguish between the role of the agonists and the role of haemodynamic factors in cardiac hypertrophy, we investigated the effects of co-administration of hydralazine (HL), an antihypertensive drug, on induced cardiac hypertrophy and the effects of the blood pressure reduction on gene expression in comparison to drug withdrawal.
Methods
L-Ascorbic acid, isoproterenol (ISO), phenylephrine (PE), and hydralazine (HL) were purchased from Sigma.
Mouse Egr-1 cDNA, GenBank/EMBL J04089, and mouse c-jun cDNA, GenBank/EMBL J04115 were purchased from ATCC. c-Fos cDNA was a gift from Dr John Hiscott LDI/JGH, Montreal, Canada. ANF cDNA and Tubulin primer sequences were gifts from Dr Mona Nemer, IRCM, Montreal, Canada. Tubulin, Nkx-2.5 and GATA-4 cDNAs were generated by PCR, cloned into pBlueScript, and confirmed by sequencing.
Animal manipulation
Experimental studies were conducted in male C57B1/6 mice (22–26 g) according to the regulations of the Canadian Council of Animal Care and Animal Care Committee of the Lady Davis Institute for Medical Research. Agonists, dissolved in phosphate buffered saline (PBS) containing 0.5 mM ascorbic acid, were administered by continuous subcutaneous infusion for 3, 7 or 14 days via osmotic mini-pumps (model 2001 or 2002, Alza, Palo Alto, CA, U.S.A.). These pumps delivered ISO (30 mg kg−1 day−1) or PE (29 mg kg−1 day−1) or the combination ISO+PE (30 mg+29 mg kg−1 day−1). Control animals received vehicle loaded mini-pumps and were also treated for 3, 7, or 14 days. Mini-pumps were implanted into mice anaesthetized with 0.015 ml per g body weight of 2.5% avertin (tribromoethanol in tert-amyl-alcohol, Aldrich) in 1×PBS injected intraperitoneally. Anaesthesia was monitored using a toe pinch. Mini-pumps were implanted dorsally through a small 0.5 cm incision and the wound closed with Michel clips. Animals were housed in groups and given food and water ad libitum.
To explore the regression of cardiac hypertrophy, control and treated mice were anaesthetized as above and the mini-pumps removed 7 days after their implantation. The hearts were analysed after 7 days of recovery.
To examine the effect of an antihypertensive agent on induced cardiac hypertrophy, HL was added to the drinking water to achieve a dose of 82 mg day−1 to animals harbouring vehicle, ISO, PE or ISO+PE loaded mini-pumps as described above. The control group received tap water and vehicle or agonists as described above.
Haemodynamic evaluation in intact anaesthetized mice
Mice were anaesthetized as described above using 0.012 ml Avertin per gram body weight, placed supine on a thermostatically controlled 37°C table and a tracheal tube introduced. A PE-50 flame-stretched cannula was introduced through a cervical incision into the right carotid artery. After 20 min of equilibration, arterial systolic and diastolic blood pressures as well as heart rate were recorded on magnetic tape for 12 min. This record was low-pass filtered at 40 Hz and digitized at 96 Hz with 12 bit resolution. Drug and vehicle-treated animals were always studied simultaneously.
Heart to body weight ratios
Animals were killed by cervical dislocation. The mice were weighed, and their hearts were excised, rinsed in PBS, blotted on filter paper and weighed. Heart weight to body weight (HW/BW) ratios were calculated and are expressed as mg gm−1.
RNA preparation and analysis
RNA from ventricular tissue was extracted using the guanidium thiocyanate-cesium chloride centrifugation method (Chirgwin et al., 1979). The RNA pellets were resuspended in diethylpyrocarbonate-treated water, digested with RQ1 RNase-free DNase (Pharmacia Biotech), and treated with proteinase K (Sigma). The RNA concentration was spectrophotometrically determined at 260 nm.
Reverse transcription coupled to polymerase chain reactions (RT–PCR) were performed using 3 μg of total RNA. PCR for all genes analysed was performed on at least three independent RT reactions for each sample. The first strand cDNA was synthesized using random primers and SuperScript II RNase H− Reverse Transcriptase (Gibco BRL). When the reverse transcriptase was omitted, RNA samples did not generate amplified bands on PCR analyses indicating the absence of contaminating DNA (data not shown). Reverse transcription (RT) reactions proceeded according to instructions from the manufacturer. Ten per cent of the first strand reaction was used for polymerase chain reaction (PCR) using gene-specific primers, 200 μM of each of the four deoxynucleotide triphosphates (Pharmacia Biotech) and 1 U of Taq DNA polymerase. PCR reactions were optimized with respect to Mg2+ concentration, and annealing temperature. Positive control reactions contained genomic DNA as the substrate and negative control reactions contained all reagents except cDNA. These parameters, as well as primer sequences and the size of the amplified DNA fragments are given in Table 1. To verify that the PCR reactions were in the linear range of the assay, RT–PCR was performed using [α-32P]-dCTP incorporation in the PCR for all genes examined (Figure 1A and B). One μCi of [α-32P]-dCTP was added per PCR reaction and after the indicated cycle, 10 μl of the PCR product was harvested, electrophoresed through agarose gel, transferred to Gene Screen Plus membrane using the alkaline method and exposed to X-ray film (Figure 1A). Suitably exposed X-ray films were scanned and the area under the peak quantitated (Figure 1B). PCR reactions were linear to 30 cycles. All PCRs were performed for 25 cycles, 20–40 μl of the PCR mix electrophoresed through an agarose gel, stained with ethidium bromide, photographed and transferred to membranes. Membranes were prehybridized in 1% SDS/10% dextran sulphate/1 M sodium chloride at 65°C for 4 h, hybridized in the same buffer with radioactive gene-specific cDNA probes overnight, washed, and exposed to X-ray film for 2–10 min. Densitometry of suitably exposed X-ray film was performed using a HP ScanJet 5100C and HP Precision Scan software (Hewlett Packard). The areas under the peaks were quantitated using ScionImage Release Beta 3 Software (National Institutes of Health).
Table 1.
PCR conditions
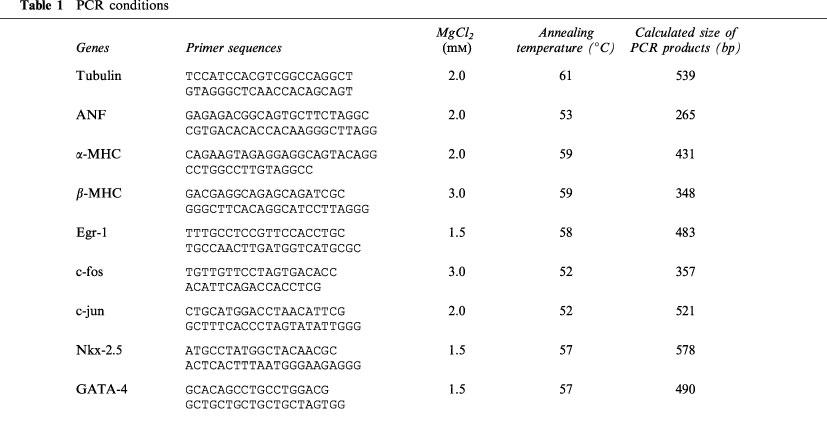
Figure 1.
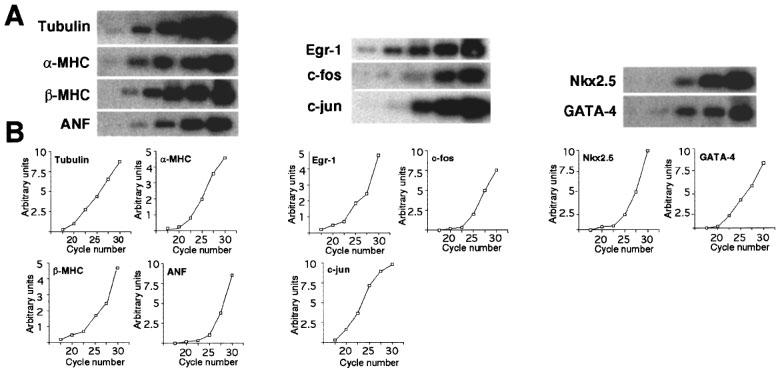
RT–PCR and [α-32P]-dCTP incorporation. One μCi of [α-32P]-dCTP was added to the PCR portion of the normal RT–PCR as described in Methods. (A) After the indicated cycle, an aliquot of the PCR product was harvested, electrophoresed through an agarose gel, transferred to membrane, and exposed to X-ray film. (B) The suitably exposed X-ray films were scanned and area under the peak quantified as described in Methods.
Histological analysis
Heart sections from half of the vehicle or treated mice were examined histologically. Horizontal slices of mid-ventricle heart were fixed by immersion in neutral buffered formalin, processed routinely, and embedded in paraffin. Sections were cut at 4 microns, stained with hematoxylin-eosin or Mason's trichrome then examined in a blinded fashion by light microscopy.
Statistical analysis
All values are expressed as mean±s.e.mean. Comparisons among three or more groups were made by one-way ANOVA followed by Dunnetts' modified t-test. A value of P<0.05 was considered statistically significant.
Results
Adrenergic induction of cardiac hypertrophy
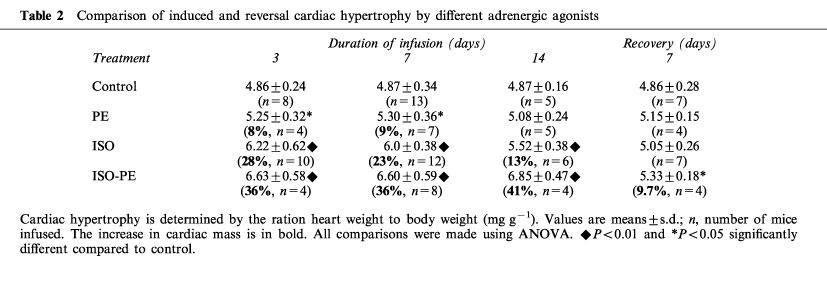
Stimulation of α- and/or β-adrenoreceptors resulted in a significant increase in the HW/BW in all groups of agonist-treated animals (Table 2). The body weight of the agonist or saline-treated mice did not differ by more than 10% throughout the time course. The HW/BW of vehicle-treated animals did not change throughout the time course studied. When compared to vehicle treated animals the HW/BW of PE-treated (P<0.05), ISO-treated (P<0.01) and ISO+PE-treated (P<0.01) animals were significantly increased after 3 or 7 days of treatment. After 14 days of continuous infusion, the cardiac mass was significantly increased (P<0.01) only in the ISO-treated and ISO+PE-treated groups when compared to control animals. There was no statistical difference between the HW/BW of the PE-treated mice when compared to the ISO-treated mice at any time point. ISO+PE-treated mice were significantly larger than PE-treated mice on day 3 and day 7 (P<0.05) and day 14 (P<0.01). ISO+PE-treated mice were significantly larger than the ISO-treated mice only on day 14 (P<0.05). Once enlarged on day 3 the HW/BW did not increase or decrease significantly with continued treatment, regardless of the stimulus.
Table 2.
Comparison of induced and reversal cardiac hypertrophy by different adrenergic agonists
Reversal of induced-cardiac hypertrophy
To study the regression of induced-cardiac hypertrophy, mice were continually infused with vehicle, ISO, PE or ISO+PE for 7 days (7+) then the pumps were removed and the animals allowed to recover for 7 days before sacrifice, (7+/7−) (Table 2). The HW/BW decreased within 7 days after drug withdrawal in all treated animals. The HW/BW ratio of ISO-alone and PE-alone treated mice was similar to the ratio found in vehicle-treated control mice. In ISO+PE-treated mice the HW/BW ratio after recovery was decreased significantly compared to the HW/BW ratio found in ISO+PE-treated mice for 7 or 14 days, but was still slightly higher when compared to the HW/BW ratio found in vehicle-treated mice (P<0.05).
Histological analysis
After examination of multiple heart sections stained with H&E or with Mason's trichrome, we did not find areas of fibrosis in the left ventricles of control or PE-treated mice (Figure 2A). A single focus of fibrosis was found in the heart of a single ISO-treated mouse (data not shown). In comparison, multiple patches of fibrosis were noted in all animals treated with ISO+PE, in both short and long term experiments (Figure 2B). Pericardial inflammation was noted in one mouse treated with ISO-alone and in two mice treated with PE-alone.
Figure 2.
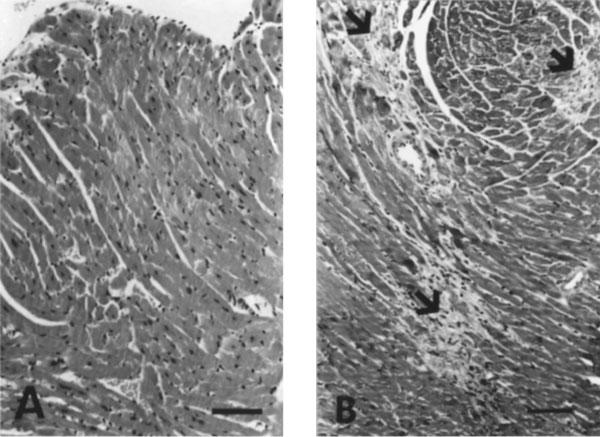
Histology of hearts. (A) Normal left ventricular myocardium from saline control treated mouse. (B) Multiple foci of myocardial fibrosis (arrow) in a mouse treated concurrently with ISO+PE. Sections were stained with hematoxylin and eosin (bar=150 μm).
Effect of antihypertensive compound on induced cardiac hypertrophy
We next studied the correlation between high blood pressure and cardiac hypertrophy induced by adrenoreceptor agonists. To reduce blood pressure, we used HL, an arterial vasodilator known to reduce blood pressure in rats (Reddy et al., 1996). During the 7 days of infusion with vehicle or drug, animals of each treatment type were separated in two groups, one group were supplied ad libitum with hydralazine supplemented drinking water (with HL) and the second group were supplied with regular drinking water (without HL).
We found that the mean arterial blood pressure of α- and β-agonist treated animals was higher compared to vehicle-treated animals and that treatment with HL led to a significant decrease (P<0.05) in mean blood pressure for all animals (Table 3). Control mice treated with HL for 7 days had a lower blood pressure and a 17% increase in heart rate compared to untreated control animals which did not receive HL. This result was expected as HL is known to increase heart rate in normotensive and hypertensive rats and humans (Mitchell et al., 1996). We also found ISO-, PE- and ISO+PE-treated animals had a higher heart rate compared to vehicle-treated animals. The heart rate increase induced by ISO+PE-treatment was intermediate between the heart rate increase induced by PE- or ISO-treatment (Table 3) and not significantly different from the ISO- or PE-treated mice. The ISO-treated mice heart rate was significantly greater (P<0.01) than the heart rate of the PE-treated mice. The addition of HL induced a significant decrease (P<0.05) in heart rate in PE-, ISO- and ISO+PE-treated animals. The decrease in heart rate in ISO-treated mice did not reach control levels (Table 3) and was significantly greater than that of the PE- (P<0.01) or ISO+PE-treated mice (P<0.01). The HW/BW ratio of the agonist-treated mice did not change after HL treatment, except for ISO-treated animals whose HW/BW significantly increased (P<0.05) after co-treatment with HL.
Table 3.
Effect of hydralazine on induced cardiac hypertrophy

Gene expression in induced cardiac hypertrophy
We analysed the expression of ANF, α-MHC, β-MHC, GATA-4, and Nkx-2.5, and the expression of the immediate early genes, Egr-1, c-jun and c-fos, in vehicle, ISO-, PE- and ISO+PE-treated mice by RT–PCR as previously described (Saadane et al., 1999). We chose tubulin expression as a control of gene expression to minimize loading differences (Saadane et al., 1999; Grepin et al., 1994).
Analysis of myosin isoform expression and induction of ANF
Stimulation of adrenoreceptors did not affect the expression of α-MHC. However, adrenoreceptor stimulation induced the expression of β-MHC. PE-alone and ISO-alone induced β-MHC (P<0.05 and P<0.01) after 3, 7 and 14 days of infusion (Figure 3A and C). ISO+PE infusion led to the largest increase in β-MHC expression (P<0.01) (Figure 3C). This induction was first detectable after 3 days of infusion and continued to increase after 7 and 14 days of infusion (respectively 4.7, 8.5 and 21 fold, Figure 3C). We found that the stimulation of PE-alone or ISO-alone resulted in an increase in ANF expression (P<0.05) (Figure 3A). The induction of ANF gene expression was modest in ISO- and in PE-treated mice after 3, 7 or 14 days of continuous administration. However, animals that received a combination of ISO+PE had a higher increase in ANF expression, and this induction increased with time of infusion (respectively 2.2, 3 and 4.5 fold, Figure 3C).
Figure 3.
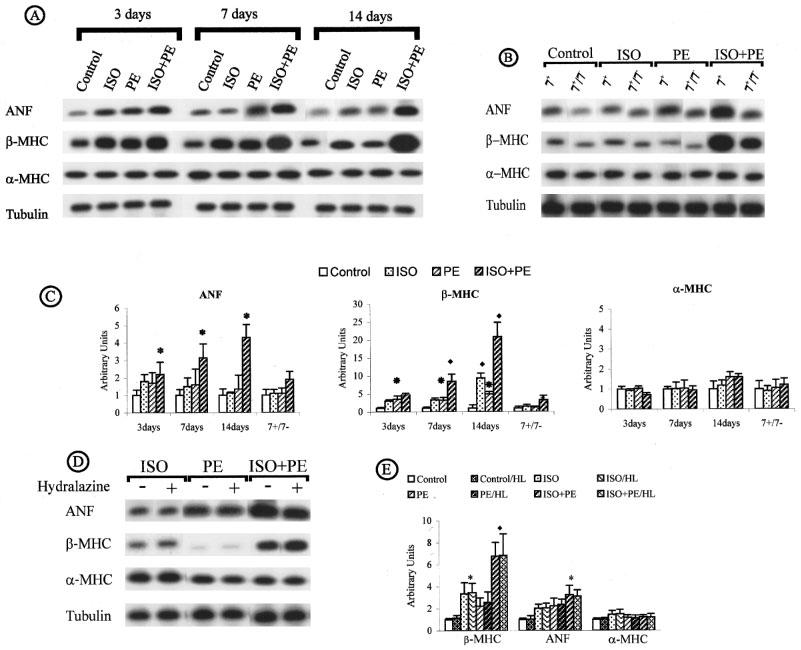
Specific cardiac gene expression in induced cardiac hypertrophy by adrenoreceptor stimulation. (A) Time course. (B) Reversal of induced cardiac hypertrophy. (C) ANF, β-MHC and α-MHC expression throughout the time course and regression. (D and E) Effect of hydralazine. cDNA prepared from RNA extracted from individual hearts of all control and treated animals described in Tables 2 and 3 were amplified by RT–PCR using the conditions shown in Table 1. PCR product, 20–40 μl, was electrophoresed through agarose gel and transferred to membrane. Membranes were hybridized with radioactive gene-specific cDNA probes. Data for gene expression are expressed in arbitrary densitometric units normalized to Tubulin and are plotted in relation to control expression levels (control=1). The bar graphs represent compilation of triplicate series of RT–PCR reactions from all animals in Tables 2 and 3. A P<0.05 is denoted by an asterisk and a P<0.01 is denoted by a filled diamond.
After removal of PE- or ISO-loaded pumps the HW/BW ratio was reduced to control levels (Table 2) as was expression of ANF and β-MHC (Figure 3B). However, after removal of the ISO+PE-loaded pumps HW/BW although reduced was still elevated in comparison to control samples (P<0.05). Expression of ANF and β-MHC remained increased after removal of the ISO+PE loaded pumps. Treatment with HL in combination with the adrenoreceptor agonists had no significant affect on the induced up-regulation of ANF or β-MHC (Figure 3D and E).
Activation of immediate early genes
Egr-1 expression was induced after 3 days of infusion in the ISO-, PE- and ISO+PE-treated animals. The combination of ISO+PE induced the highest expression, ISO-alone mediated an intermediate increase and PE-alone the smallest increase when compared to vehicle-treated samples (Figure 4A). The maximum Egr-1 induction was reached after 3 days of ISO+PE-infusion (P<0.05) (5 fold) and remained high in 7 and 14 day treated animals (P<0.01) (Figure 4C). PE-or ISO-treated mice showed no c-jun induction after 3, 7 or 1 day (Figure 4A). In ISO+PE-treated animals, c-jun induction was evident but not of significance until after 7 day treatment and reached significance only after 14 days treatment (4 fold, P<0.05) (Figure 4C). Upregulation of c-fos was induced in all groups of treated animals. However, the infusion with ISO+PE exhibited a remarkable increase of c-fos expression. This induction was maintained after 3, 7 and 14 day treatment and was respectively 34, 31 and 35 fold compared to c-fos expression in control animals (P<0.01) (Figure 4C). After removal of PE- or ISO-loaded pumps the HW/BW ratio was reduced to control levels (Table 2) as was expression of Egr-1, c-jun, c-fos (Figure 4B). Expression of Egr-1 and c-jun remained increased after removal of the ISO+PE loaded pumps. Egr-1, c-jun and c-fos (Figure 4D and E) expression was unaffected by the reduction in blood pressure.
Figure 4.
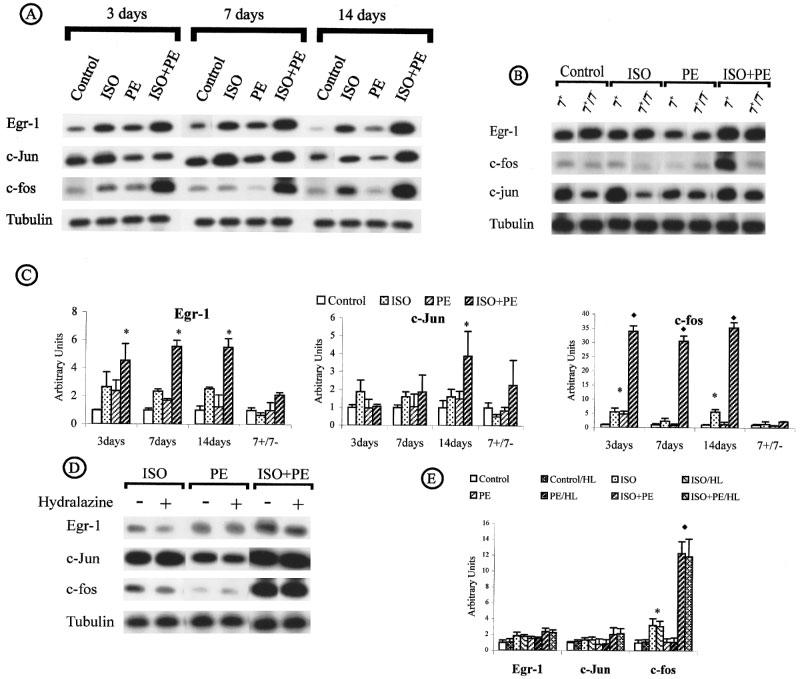
Immediate early gene expression in induced cardiac hypertrophy by adrenoreceptor stimulation. (A) Time course. (B) Reversal of induced cardiac hypertrophy. (C) Egr-1, c-jun and c-fos expression throughout the time course and regression. (D and E) Effect of hydralazine. (For the conditions see legend of Figure 3).
Induction of GATA-4 and Nkx-2.5 genes
Stimulation of α- and β-adrenoreceptors induced overexpression of GATA-4 and Nkx-2.5 in all treated animals (Figure 5A). A high level of Nkx-2.5 was found after 3, 7 and 14 days of ISO+PE induction. ISO-treatment initially caused a slight increase (3 fold) that became more evident after 7 and 14 days infusion (respectively 25 and 33.8 fold). Similarly, PE-treatment led to a slight increase at 3 days (4.7 fold) that was further increased at 7 days and 14 days (respectively 33 and 35 fold). Nkx2.5 expression, regardless of the initiating stimulus, decreased significantly after drug withdrawal, but remained 7 times higher when compared to control (Figure 5B). When GATA-4 was measured stimulation by ISO, PE and ISO+PE was equal and no synergism was evident when single versus both agonists were present (P<0.01) (Figure 5C). GATA-4 expression dropped from 10 fold to 7 fold after 7 and 14 day infusion, respectively. ISO+PE induced a massive upregulation of Nkx-2.5 expression with an average increase of 40 fold after 3 days of infusion (P<0.01). GATA-4 expression was at the control level after removal of the ISO+PE loaded pumps. GATA-4 and Nkx-2.5 (Figure 5D and E) expression was unaffected by the reduction in blood pressure.
Figure 5.
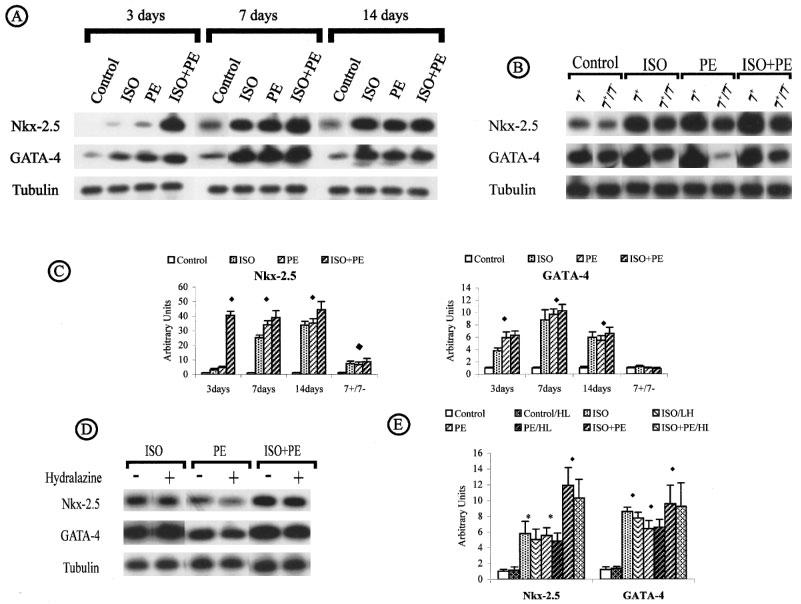
Nkx-2.5 and GATA-4 expression in induced cardiac hypertrophy by adrenoreceptor stimulation. (A) Time course. (B) Reversal of induced cardiac hypertrophy. (C) Nkx-2.5 and GATA-4 expression throughout the time course and regression. (D and E) Effect of hydralazine. (For the conditions see legend of Figure 3).
Discussion
Stimulation of adrenoreceptors in mice induces an increase in cardiac mass unaffected by a reduction in afterload
Hypertrophy can result from pressure overload with increased wall stress, volume overload or catecholamine activation. Our study shows that the long term stimulation of α1- and/or β-adrenoreceptors induces a cardiac hypertrophy in adult male mice. ISO is a non-selective β-agonist acting primarily via the PKA pathway to up-regulate cyclic AMP and PE is a selective α1 agonist acting primarily via the PKC pathway through changes in Ca2+, phospholipids and diacylglycerol (Kobilka, 1993). We found the cardiac hypertrophy most pronounced when both α1- and β-adrenoreceptors are activated suggesting synergism when both PKA and PKC pathways are induced. ISO+PE-induced cardiac hypertrophy is more pronounced (P<0.05) than PE-induced cardiac hypertrophy throughout the time course studied whereas ISO-induced and ISO+PE-induced hypertrophy are not significantly different. A similar effect was seen in young piglets exposed to NE, an α1-agonist, a strong β1 agonist and weak activator of β2-adrenoreceptors, when compared to ISO or PE (Kolbilka, 1993; Buu et al., 1993; Cassidy et al., 1997). In this porcine study NE>ISO>PE on their measured indices of contractility and circulatory function. Thus, it appears that activation of the PKA pathway, rather than the PKC pathway is of more consequence in the in vivo generation of cardiac hypertrophy.
The extent of ISO-induced cardiac hypertrophy as measured by the heart to body weight ratio, did not decrease significantly within the time course. When both α- and β-adrenoreceptors are stimulated, as in the ISO+PE-treated mice, the increased cardiac mass is maintained throughout the time course. In a similar study, that also used osmotic minipumps to deliver the drugs, the HW/BW remained elevated in NE-treated versus saline-treated rats even when a reduced positive response to acutely given NE was detected (Laycock et al., 1995). The reduced acute response was attributed to a reduction in the number of β-adrenoreceptors in the NE-treated group. Downregulation of β1-adrenoreceptors was also shown in transgenic rats harbouring the mouse Ren-2d gene and in spontaneously hypertensive rats (Bohm et al., 1994; 1995). Although not proven, it is likely that a reduction in β-adrenoreceptors may have occurred in the ISO- and/or ISO+PE-treated animals despite the maintenance of an elevated HW/BW.
In our study, treatment with ISO, PE or ISO+PE increased mean arterial blood pressure to about the same level. Because the mice had been treated with the adrenergic agonists continuously for 7 days it is unlikely that any of the immediate acting pressure control mechanisms are operable (Guyton, 1992). We found that a reduction in mean arterial blood pressure by HL did not prevent the agonist-induced increase in cardiac mass. Similar results were found in ISO-induced cardiac hypertrophy (Golomb et al., 1994), in spontaneously hypertensive rat (Mitchell et al., 1996) or pressure overload rats (Susic et al., 1996). Thus, it appears that a reduction of blood pressure to a near normal level is insufficient to reduce ventricle weight in the presence of a second stimulus for hypertrophy, such as an excess of catecholamine. We and others have found that ISO-induced cardiac hypertrophy could be prevented by co-treatment with propranolol (β-adrenoreceptor antagonist) (Boluyt et al., 1995 and data not shown). We found that when the mini-pumps were removed a decrease in the HW/BW ratio to normal or approaching normal for all agonists occurred. These findings indicate that the maintenance of cardiac hypertrophy, although linked, is not solely dependent on afterload, but may be influenced by catecholamine excess. Further, these studies show that reversal of the hypertrophy can only be achieved by blockage or removal of agonist action suggesting that the catecholamine-induced hypertrophy results independent of agonist effects on blood pressure.
Concomitant with the agonist-induced increase in blood pressure was an increase in heart rate with the highest heart rate in ISO>ISO+PE>PE-treated mice. The increases in heart rate do not correlate with the increase in HW/BW or gene expression changes. The higher rate (P<0.01) in ISO-treated versus PE-treated mice was expected because of the known chronotropic effect of ISO acting on the β1-receptors in the myocardium and the increase in heart rate associated with constitutive expression of β2-adrenergic receptors in transgenic mice (Rockman et al., 1997; Milano et al., 1994). The heart rate in the ISO- versus ISO+PE-treated mice without HL was similar indicating no or undetectable effect of additional PE when ISO was infused. The heart rate increase in control mice treated with HL was expected because unopposed HL is known to cause an increase in heart rate in humans. Treatment with HL reduced the heart rate in PE and ISO+PE-treated mice to control levels, but was unable to reduce the heart rate to normal in ISO-treated mice. This is similar to the decrease in blood pressure, but not heart rate, found after captopril treatment of NE-induced hypertrophy in rats (Laycock et al., 1996). Our data suggest that unloading the heart is not sufficient to cause a reduction in heart rate if β-receptors alone are stimulated.
ANF and β-MHC induction
In agreement with most models of in vivo cardiac hypertrophy (reviewed in Schneider & Parker, 1993; van Bilsen & Chien, 1993; Pollak, 1995; Boluyt et al., 1995), our study shows induction of ANF by ISO, PE and ISO+PE, with the most pronounced induction by ISO+PE. Indeed, upregulation of ANF and β-MHC gene expression is a highly conserved event in ventricular hypertrophy both in vitro and in vivo in virtually all animals examined. We found ANF induction is closely correlated with cardiac mass increases, is maintained while agonists are present and further that ANF is decreased during reversal of cardiac hypertrophy. ANF with its vasodilator effect is thought to play a role in reducing haemodynamic load imposed on the heart and ANF knockout mice had a significantly increased blood pressure when compared to control littermates (John et al., 1995). However, we did not find lower arterial blood pressures in agonist-treated groups with the highest ANF levels suggesting that the higher ANF is insufficient to counteract the catecholamine action. Upregulation of β-MHC also correlated with cardiac mass increases, persists in the chronic phase of hypertrophy and is decreased after drug withdrawal. In physiological terms, the overexpression of the foetal isoforms may be a beneficial long-term adaptation to haemodynamic overload because β-MHC is thought to be bioenergetically more efficient than the adult isoform (Argentin et al., 1994). In our study, β-MHC expression is upregulated, but no change was detectable in α-MHC expression.
Neither ANF nor β-MHC expression decreased after an HL-mediated reduction of afterload in the PE- or ISO+PE-treated mice. Co-treatment of ISO-alone plus HL reduced the amount of β-MHC in comparison with PE-alone, but this level was still higher than in control mice. β-MHC expression was unaltered in pressure over-loaded rats treated with HL (Ardati & Nemer, 1993). These data suggest that upregulation of ANF or β-MHC levels is not closely tied to blood pressure, but rather is linked to agonist action, contractility and/or heart rate. In contrast, expression of α-MHC appears to be unaffected by either agonist action or the reduction in afterload caused by HL co-treatment. This is contradictory to findings in rats where α-MHC usually decreases in concert with an increase in β-MHC expression to result in a reversal of the α-MHC/β-MHC ratio (Schneider & Parker, 1993). We found a reduction in ANF and β-MHC, again without a change in α-MHC expression, only when the agonists were eliminated by removal of the minipumps. This result indicates that adjustments to a pre-treatment pattern of gene expression can only occur after agonist action on the heart itself is eliminated. These data suggest that control of α-MHC and β-MHC expression is different in mice compared to rats and that, whereas ANF and β-MHC are regulatable by agonists and blood pressure in mice, α-MHC expression is refractory.
Immediate early and muscle-specific transcription factor gene induction
Many studies have examined transient induction of transcription factors in the initiation of hypertrophy (Brand et al., 1993; Hannan & West, 1991; Schneider & Parker, 1993), but none of these studies examined their role in hypertrophy maintenance and regression. The present study shows that agonist-induced cardiac hypertrophy is accompanied by a continued high level of expression of c-fos, Egr-1, and, to a lower extent, c-jun. Thus, the data suggests that immediate early genes can be activated by both α- or β-adrenoreceptor, that activation of α- or β-agonists results in the highest levels in vivo and that the level of the immediate early genes is maintained by continuous infusion. The induction of their gene expression is well correlated with the extent of cardiac hypertrophy.
By comparing the pattern of expression after agonist stimulation, we observed that in adult cardiac hypertrophy, ISO, and to a lesser extent PE, induced c-fos and Egr-1, and that in ISO-PE-treatment the increase in c-jun, c-fos and Egr-1 expression was synergistic. The synergistic induction of Egr-1 and c-fos expression in our model suggests that both PKA and PKC pathways are involved in their induction. In other studies using aortic constriction to produce an acute pressure overload, c-fos induction was coupled to increases in cyclic AMP content and PKA activity in addition to a rise in PKC activity (Osaki et al., 1997) further implicating activation of both pathways in c-fos gene regulation in heart. In vitro binding of c-fos/c-jun heterodimers to ANF gene sequences has already been shown and it was suggested that the heterodimer plays a role in the regulation of ANF gene transcription in vivo (Rauscher et al., 1988; Rosenzweig et al., 1991; Barka et al., 1987). Correlation of the continued increase in ANF, parallel to increases in c-fos and c-jun expression, further implicates AP-1 activity in the maintenance of the high levels of ANF.
Egr-1 is a transcription factor that binds to a conserved GC-rich element found in such genes as ANF and α-MHC, and competes with Sp1 for DNA binding (Gashler & Sukhatme, 1995; Huang et al., 1997). We had previously found Egr-1 increased in every hypertrophied heart examined in our transgenic line expressing polyomavirus large T-antigen in heart (Holder et al., 1995). In the present study Egr-1 expression was increased primarily in ISO+PE and ISO-treated mice, with the smallest increase in PE-treated mice, at all times examined. Our results must be differentiated from the results of experiments performed by Brand et al. (1993) and Iwaki et al. (1990). Brand et al. (1993) treated isolated rat hearts for only 30 min to 1 h with ISO, PE or NE and found that PE and NE, but not ISO induced Egr-1 expression. The short time period of his experiments and the lack of a vascular component are likely to be significant differences affecting the outcome of the two studies. Iwaki et al. (1990) found Egr-1 upregulated in neonatal rat cardiomyocytes only after PE stimulation. However, significant differences exist between the gene expression in neonate versus adult cardiomyocytes and indicate that simple transfer of results from the neonatal cultures to adult heart can be problematic (Kitsis & Leinwand, 1992). Thus, differences may exist between the response of mouse and rat to adrenoreceptor stimulation and/or between cardiac hypertrophy induced by short stimulation (0.5–1 h) compared to cardiac hypertrophy induced by longer stimulation (3–14 days) or between neonatal cardiomyocyte culture in vitro and adult heart in vivo. Gupta et al. (1991) found Egr-1 regulated expression of α-MHC in neonatal rat cardiomyocytes. It is unclear if this activity is also present in adulthood as we found no change in α-MHC expression when Egr-1 levels were increased. This suggests either that Egr-1 does not control α-MHC expression in the adult or that other elements negate any Egr-1-mediated increase with no observable change in α-MHC expression as the end-result. Supporting the idea that Egr-1 does not control α-MHC expression was the finding of beating cardiomyocytes in differentiated embryonic stem cells deficient of Egr-1 and that adult mice lacking Egr-1 have apparent normal heart architecture (Lee et al., 1995).
The function of sustained expression of c-fos, c-jun and Egr-1 in heart pathology is unknown. It has been suggested that high levels of c-fos, c-jun and Egr-1 in terminally differentiated cells is correlated with growth arrest, progression and maintenance of the differentiation program (Gandarillas & Watt, 1995; Lord et al., 1993; Panterne et al., 1992; Liu et al., 1998; reviewed in Liebermann & Hoffman, 1998). In support of this idea, mice containing a fos-lacZ transgene show high levels of fos-driven lacZ in the most differentiated cells of the epidermis, hair follicle and epiphyseal plate with sustained expression preceding cell death (Smeyne et al., 1993). In another study high levels of c-fos induced growth inhibition of keratinocyte cell lines and increased their sensitivity to apoptosis (Mils et al., 1997). These studies suggest a correlation between high c-fos expression and bone and keratinocyte differentiation. However, overexpression of c-fos in transgenic mice led to dysregulation of bone growth and high expression in other tissues, including heart, led to no overt phenotype (Ruther et al., 1989; Bachiller and Ruther, 1990). Mice null for c-fos expression are viable, form a normal epidermis and did not form malignant papillomas when stimulated (Saez et al., 1995). Egr-1 deficient mice are also viable and display no obvious phenotype except for female infertility (Lee et al., 1995). Thus, the available transgenic and null expression results are in contrast to in vitro data. It may be that the other members of the Fos, Jun and Egr-1 families are able to overcome the defects in the transgene and null mutations. Adult cardiomyocytes have not been shown to proliferate to any great extent so it is unlikely that sustained expression of these genes is linked to increased proliferation in heart. It is more likely that, sustained expression of c-fos, c-jun and Egr-1 is involved in cardiomyocyte differentiation and/or apoptosis.
We show, for the first time, that the induction of Nkx-2.5 and GATA-4 can be produced by adrenereceptor stimulation with the highest increase seen when both α- and β-adrenoreceptors are stimulated. We suspect that GATA-4 expression can be equally initiated and maintained by either PKA or PKC activated mechanisms because GATA-4 expression was similar regardless of agonist stimulus and no synergism was detected if both pathways were activated simultaneously. High levels of Nkx-2.5 were found initially when both PKA and PKC pathways were activated in the ISO+PE-treated mice, but at later times Nkx2.5 was increased after 7 days of PE- or ISO-stimulation and this high level was maintained after 14 days. This indicates that co-stimulation by ISO and PE may serve to accelerate the time course for initiation of the high level of Nkx-2.5. The upregulation of GATA-4 and Nkx2.5 in our models of cardiac hypertrophy combined with its upregulation in aortic constriction and pressure overload, suggests that in increase in these genes may be a general feature of cardiac hypertrophy. They most likely play a role in initiating and maintaining the process of cardiac hypertrophy by activating cardiac promoters (Chen et al., 1996; Grepin et al., 1994; Durocher et al., 1996; Sepulveda et al., 1998).
Regression
Drug withdrawal after PE- or ISO-treatments is accompanied by a significant decrease in ANF, β-MHC, Egr-1, c-fos, c-jun and GATA-4 expression and a decrease in the HW/BW ratio to control levels. This implies that whereas continued expression of these genes is found when agonists are present once the heart has regressed to its normal weight maintenance of their expression is not required. Support for the notion that agonist stimulation is the important stimulant for their elevated expression is that when the agonist-treated animals were given HL and blood pressure was normalized the expression of these genes remained increased.
Heart regression was incomplete after ISO+PE stimulation as the HW/BW ratio remained elevated. In these hearts, ANF, β-MHC, Egr-1, c-jun, but not c-fos or GATA-4 remained elevated. This result suggests that Egr-1 and c-jun may be required for complete regression of heart weight to occur whereas continued expression of c-fos or GATA-4 is not necessary. Interestingly, GATA-4 and Nkx2.5 expression is very different after drug withdrawal. Nkx2.5 expression, although decreased, persists in the withdrawal after drug treatment regardless of the initiating stimulus and regardless of whether the HW/BW indicates complete or incomplete regression. In contrast, GATA-4 was decreased even when the HW/BW indicated incomplete regression. The implication is that regression might be a dynamic process that requires the expression of a subset of transcription factors. Further, the data also suggests that although HW/BW ratios and determination of ANF or β-MHC levels might indicate that complete regression has occurred considerable molecular changes might still be underway that require specific transcription factors.
Conclusions
Recognition of the central role that transcription regulatory genes play in gene expression led us to search for specific gene expression changes in the hypertrophying heart. How the different transcription factors involved in cardiogenesis operate in the entire regulatory cascade of cardiac hypertrophy has yet to be fully delineated. We found that Egr-1, c-fos, c-jun, Nkx2.5 and GATA-4 are upregulated in chronic-induced cardiac hypertrophy, that the induction is not transient, but persists through the time course. We found that Egr-1, c-fos, c-jun, Nkx2.5 and GATA-4 expression does not decrease when the blood pressure is reduced suggesting that direct drug action of adrenoreceptor agonists on cardiomyocyte is more important in inducing gene expression than afterload reduction. Regression after drug withdrawal appears to be an active process that requires a subset of specific transcription factors. To investigate the role of c-fos and Egr-1 in response to hypertrophic stimuli and to dissect the mechanism of cardiac hypertrophy, we are extending our study to include NGFI-A−/− (null mutant mice for Egr-1 gene) (Lee et al., 1995) and/or c-fos−/− (null mutant mice for c-fos gene) (Wang et al., 1992) as subjects for these in vivo models.
Acknowledgments
We are grateful to David Ajikobi for helping us to measure the arterial blood pressure and heart rate in mice, Dr William Cupples for his helpful comments and Dr David Langleben for critical reading of the manuscript. This work was supported by grants from the Medical Research Council of Canada (MT-13111) and the Heart and Stroke Foundation of Quebec to L. Chalifour.
Abbreviations
- ANF
atrial natriuretic factor
- α-MHC
alpha-myosin heavy chain
- β-MHC
beta-myosin heavy chain
- cyclic AMP
cyclic adenosine monophosphate
- Egr-1
early growth response gene 1
- HL
hydralazine
- HW/BW
heart weight in mg divided by body weight in grams
- NE
norepinephrine
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PE
phenylephrine
- ISO
isoproterenol
- PKA
phosphorylase kinase A
- PKC
phosphorylase kinase C
- ISO+PE
isoproterenol plus phenylephrine
- RT–PCR
reverse transcription followed by the polymerase chain reaction
References
- ARCECI R.J., KING A.A.J., SIMO M.C., ORKIN S.H., WILSON D.B. Mouse GATA-4: a retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol. Cell. Biol. 1993;13:2235–2246. doi: 10.1128/mcb.13.4.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARDATI A., NEMER M. A nuclear pathway for α1-adrenergic receptor signaling in cardiac cells. EMBO J. 1993;12:5131–5139. doi: 10.1002/j.1460-2075.1993.tb06208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARGENTIN S., ARDATI A., TREMBLAY S., LIHRMANN I., ROBITAILLE L., DROUIN J., NEMER M. Development stage-specific regulation of atrial natriuretic factor gene transcription in cardiac cells. Mol. Cell. Biol. 1994;14:777–790. doi: 10.1128/mcb.14.1.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BACHILLER D., RUTHER U. Inducible expression of the proto-oncogene c-fos in transgenic mice. Archiv fur Geschwulstforschung. 1990;60:357–360. [PubMed] [Google Scholar]
- BARKA T., VAN DER NOEN H., SHAW P.A. Proto-oncogene fos (c-fos) expression in the heart. Oncogene. 1987;1:439–443. [PubMed] [Google Scholar]
- BÖHN M., CASTELLANO M., AGABITI-ROSEI E., FLESCH M., PAUL M., ERDMANN E. Dose-dependent dissociation of ACE-inhibitor effects on blood pressure, cardiac hypertrophy, and β-adrenergic signal transduction. Circ. 1995;92:3006–3013. doi: 10.1161/01.cir.92.10.3006. [DOI] [PubMed] [Google Scholar]
- BÖHM M., MOLL M., SCHMID B., PAUL M., GANTEN D., CASTELLANO M., ERDMANN E. β-adrenergic neuroeffector mechanisms in cardiac hypertrophy of renin transgenic rats. Hypertension. 1994;24:653–662. doi: 10.1161/01.hyp.24.6.653. [DOI] [PubMed] [Google Scholar]
- BOLUYT M.O., LONG X., ESCHENHAGEN T., MENDE U., SCHMITZ W., CROW M.T., LAKATTA E.G. Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. Am. J. Physiol. 1995;269:H638–H647. doi: 10.1152/ajpheart.1995.269.2.H638. [DOI] [PubMed] [Google Scholar]
- BRAND T., SHARMA H.S., SCHAPER W. Expression of nuclear proto-oncogenes in isoproterenol-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 1993;25:1325–1337. doi: 10.1006/jmcc.1993.1145. [DOI] [PubMed] [Google Scholar]
- BUU N.T., HIU R.T., FALARDEAU P. Norepinephrine in neonatal rat ventricular myocytes: Association with the cell nucleus and binding to nuclear α1- and β-adrenergic receptors. J. Mol. Cell. Cardiol. 1993;25:1037–1046. doi: 10.1006/jmcc.1993.1116. [DOI] [PubMed] [Google Scholar]
- CASSIDY S.C., MCGOVERN J.J., CHAN D.P., ALLEN H.D. Effects of commonly used adrenergic agonists on left ventricular function and systemic vascular resistance in young piglets. Am. Heart J. 1997;133:174–183. doi: 10.1016/s0002-8703(97)70206-0. [DOI] [PubMed] [Google Scholar]
- CHEN C.Y., CROISSANT J., MAJESKY M., TOPOUZ S., MCQUINN T., FRANKOVSKY M.J., SCHWARTZ R.J. Activation of the cardiac alpha-actin promoter depends upon serum response factor, Tinman homologue, Nkx2-5, and intact serum response elements. Dev. Genet. 1996;19:119–130. doi: 10.1002/(SICI)1520-6408(1996)19:2<119::AID-DVG3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- CHIRGWIN J.M., PRZYBULA A.E., MACDONALD R.J., RUTTER W.J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochem. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- DUROCHER D., CHARRON F., WARREN R., SCHWARTZ R.J., NEMER M. The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J. 1997;16:5687–5696. doi: 10.1093/emboj/16.18.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUROCHER D., CHEN C.-Y., ARDATI A., SCHWARTZ R.J., NEMER M. The atrial natriuretic factor promoter is a downstream target for Nkx-2.5 in the myocardium. Mol. Cell. Biol. 1996;16:4648–4655. doi: 10.1128/mcb.16.9.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GANDARILLAS A., WATT F.M. Changes in expression of members of the fos and jun families and myc network during terminal differentiation of human keratinocytes. Oncogene. 1995;11:1403–1407. [PubMed] [Google Scholar]
- GASHLER A., SUKHATME V.P. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog. Nucl. Acids Res. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- GOLOMB E., ABASSI Z.A., CUDA G., STYLIANOU M., PANCHEL V.R., TRACHEWSKY D., KEISER H.R. Angiotensin II maintains, but does not mediate, isoproterenol-induced cardiac hypertrophy in rats. Am. J. Physiol. 1994;267:H1496–1506. doi: 10.1152/ajpheart.1994.267.4.H1496. [DOI] [PubMed] [Google Scholar]
- GRÉPIN C., DAGNINO L., ROBITAILLE L., HABERSTROH L., ANTAKLY T., NEMER M. A hormone-encoding gene identifies a pathway for cardiac but not skeletal muscle gene transcription. Mol. Cell. Biol. 1994;14:3115–3129. doi: 10.1128/mcb.14.5.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUPTA M.P., GUPTA M., ZAK R., SUKHATME V.P. Egr-1, a serum-inducible zinc finger protein, regulates transcription of the rat cardiac α-myosin heavy chain gene. J. Biol. Chem. 1991;266:12813–12816. [PubMed] [Google Scholar]
- GUYTON A.C. Kidneys and fluids in pressure regulation; small volume but large pressure changes. Hyperten. 1992;19:12–18. doi: 10.1161/01.hyp.19.1_suppl.i2. [DOI] [PubMed] [Google Scholar]
- HANNAN R.D., WEST A.K. Adrenergic agents, but not triiodo-1-thyronine induce c-fos and c-myc expression in the heart. Basic Res. Cardiol. 1991;86:154–164. doi: 10.1007/BF02190548. [DOI] [PubMed] [Google Scholar]
- HERZIG T.C., JOBE S.M., AOKI H., MOLKENTIN J.D., COWLEY A.W., JR, ARKHAM B.E. Angiotensin II type1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7543–7548. doi: 10.1073/pnas.94.14.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLDER E.L., AL MOUSTAFA A.-E., CHALIFOUR L.E. Molecular remodeling in hypertrophied hearts from polyomavirus large T-antigen transgenic mice. Mol. Cell. Biochem. 1995;152:131–141. doi: 10.1007/BF01076075. [DOI] [PubMed] [Google Scholar]
- HUANG R.P., FAN Y., NI Z., MERCOLA D., ADAMSON E.D. Reciprocal modulation between Sp1 and Egr-1. J. Cell. Biochem. 1997;66:489–499. [PubMed] [Google Scholar]
- IP H.S., WILSON D.B., HEIKINHEIMO M., TANG Z., TING C.N., SIMON M.C., LEIDEN J.M., PARMACEK M.S. The GATA-4 transcription factor transactivates the cardiac muscle-specific troponin C promoter-enhancer in nonmuscle cells. Mol. Cell. Biol. 1994;14:7517–7526. doi: 10.1128/mcb.14.11.7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IWAKI K., SUKHATME V.P., SHUBEITA H.E., CHIEN K.R. Alpha- and beta-adrenergic stimulation induced distinct patterns of immediate early gene expression in neonatal rat myocardial cells. Fos/jun expression is associated with sarcomere assembly; Egr-1 induction is primarily an alpha 1-mediated response. J. Biol. Chem. 1990;265:13809–13817. [PubMed] [Google Scholar]
- JOHN S.W.M., KREGE J.H., OLIVER P.M., HAGAMAN J.R., HODGIN J.B., PANG S.C., FLYNN T.G., SMITHIES O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995;267:679–681. doi: 10.1126/science.7839143. [DOI] [PubMed] [Google Scholar]
- JOHNSON R.S., LINGEN B.V., PAPAIOANNOU V., SPIEGELMAN B.M. A null mutation at the c-Jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 1993;7:1309–1317. doi: 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- KHACHIGIAN L.M., COLLINS T. Inducible expression of Egr-1 dependent genes. A paradigm of transcriptional activation in vascular endoethelium. Circ. Res. 1997;81:457–461. doi: 10.1161/01.res.81.4.457. [DOI] [PubMed] [Google Scholar]
- KITSIS R.N., LEINWAND L.A. Discordance between gene regulation in vitro and in vivo. Gene Expression. 1992;2:313–317. [PMC free article] [PubMed] [Google Scholar]
- KOBILKA B.Adrenergic and muscarinic receptors of the heart Molecular Basis of Cardiology 1993London: Blackwell Scientific Publications; 211–238.ed. Roberts, R. pp [Google Scholar]
- KOVACIC-MILIVOJEVIC B., WONG V.S.H., GARDNER D.G. Selective regulation of the atrial natriuretic peptide gene by individual components of the activator protein-1 complex. Endocrinology. 1996;137:1108–1117. doi: 10.1210/endo.137.3.8603581. [DOI] [PubMed] [Google Scholar]
- KUO C.T., MORRISEY E.E., ANANDAPPA R., SIGRIST K., LU M.M., PARMACEK M.S., SOUDAIS C., LEIDEN J.M. GATA-4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- KURABAYASHI M., KOMURO I., SHIBASAKI Y., TSUCHIMOCHI H., TAKAKU F., YAZAKI Y. Functional identification of the transcriptional regulatory elements within the promoter region of the human ventricular myosin alkali light chain gene. J. Biol. Chem. 1990;265:19271–19278. [PubMed] [Google Scholar]
- LAYCOCK S.K., KANE K.A., MCMURRAY J., PARRATT J.R. Captopril and norepinephrine-induced hypertrophy and haemaodynamics in rats. J. Cardiovasc. Pharmacol. 1996;27:667–672. doi: 10.1097/00005344-199605000-00008. [DOI] [PubMed] [Google Scholar]
- LAYCOCK S.K., MCMURRAY J., KANE K.L., PARRAT J.R. Effects of chronic norepinephrine administration on cardiac function in rats. J. Cardiovasc. Pharmacol. 1995;26:584–589. doi: 10.1097/00005344-199510000-00012. [DOI] [PubMed] [Google Scholar]
- LEE R.T., BLOCH K.D., PFEFFER J.M., PFEFFER M.A., NEER E.J., SEIDMAN C.E. Atrial natriuretic factor gene expression in ventricles of rats with spontaneous biventricular hypertrophy. J. Clin. Invest. 1988;81:431–434. doi: 10.1172/JCI113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE S.L., TOURTELLOTTE L.C., WESSELSCHMIDT R.L., MILBRANDT J. Growth and differentiation proceeds normally in cells deficient in the immediate early gene NGFI-A. J. Biol. Chem. 1995;270:9971–9977. doi: 10.1074/jbc.270.17.9971. [DOI] [PubMed] [Google Scholar]
- LIEBERMANN D.A., HOFFMAN B. MyD genes in negative growth control. Oncogene. 1998;17:3319–3329. doi: 10.1038/sj.onc.1202574. [DOI] [PubMed] [Google Scholar]
- LIU C., RANGNEKAR V.M., ADAMSON E., MERCOLA D. Suppression of growth and transformation and induction of apoptosis by Egr-1. Cancer Gene Therapy. 1998;5:3–18. [PubMed] [Google Scholar]
- LORD K.A., ABDOLLAHI A., HOFFMANN-LIEBERMANN B., LIEBERMANN D.A. Proto-oncogenes of the fos/jun family of transcripton factors are positive regulators of myeloid differentiation. Mol. Cell. Biol. 1993;13:841–851. doi: 10.1128/mcb.13.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYONS I., PARSONS L.M., HARTLEY L., LI R., ANDREWS J.E., ROBB L., HARVEY R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx-2.5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- MILANO C.A., ALLEN L.F., ROCKMAN H.A., DOLBER P.C., MCMINN T.R., CHIEN K.R., JOHNSON T.D., BOND R.A., LEFKOWITZ R.J. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- MILS V., PIETTE J., BARETTE C., VEYRUNE J-L, , TESNIERE A., ESCOT C., GUILHOU J-J., BASSET-SEQUIN N. The proto-oncogene c-fos increases the sensitivity of keratinocytes to apoptosis. Oncogene. 1997;14:1555–1561. doi: 10.1038/sj.onc.1200991. [DOI] [PubMed] [Google Scholar]
- MITCHELL G.F., PFEFFER M.A., FINN P.V., PFEFFER J.M. Equipotent antihypertensive agents variously affect pulsatile hemodynamics and regression of cardiac hypertrophy in spontaneously hypertensive rats. Circ. 1996;94:2923–2929. doi: 10.1161/01.cir.94.11.2923. [DOI] [PubMed] [Google Scholar]
- MOLKENTIN J.D., KALVAKOLANU D.V., MARKHAM B.E. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the α-myosin heavy chain gene. Mol. Cell. Biol. 1994;14:4947–4957. doi: 10.1128/mcb.14.7.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLKENTIN J.D., LIN Q., DUNCAN S.A., OLSON E.N. Requirement of the transcription factor GATA-4 for heart tube formation and morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- MOLKENTIN J.D., LU J.-R., ANTOS C.L., MARKHAM B., RICHARDSON J., ROBBINS J., GRANT S.R., OLSON E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSAKI J., HANEDA T., SAKAI H., KIKUCHI K. cAMP-mediated c-fos expression in pressure-overloaded acceleration of protein synthesis in adult rat heart. Cardio. Res. 1997;33:631–640. doi: 10.1016/s0008-6363(97)00005-9. [DOI] [PubMed] [Google Scholar]
- ÖSTMAN-SMITH I. Cardiac sympathetic nerves as the final common pathway in the induction of adaptive cardiac hypertrophy. Clin. Science. 1981;61:265–272. doi: 10.1042/cs0610265. [DOI] [PubMed] [Google Scholar]
- PANTERNE B., HATZFELD J., BLANCHARD J.-M., LEVESQUE J.-P., BERTHIER R., GINSBURG M., HATZFELD A. c-Fos mRNA constitutive expression by mature human megakaryocytes. Oncogene. 1992;7:2341–2344. [PubMed] [Google Scholar]
- POLLAK P.S. Proto-oncogenes and the cardiovascular system. Chest. 1995;107:826–835. doi: 10.1378/chest.107.3.826. [DOI] [PubMed] [Google Scholar]
- RAUSCHER F.J., III, VOULALAS P.J., FRANZA B.R., JR, CURRAN T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 1988;2:1687–1699. doi: 10.1101/gad.2.12b.1687. [DOI] [PubMed] [Google Scholar]
- REDDY D.S., SINGH M., GANGULY N.K. Effects of long-term hydralazine treatment on myocardial structure and expression of myosin isogenes in cardiac pressure overload in rats. Exp. Clin. Pharmacol. 1996;18:261–271. [PubMed] [Google Scholar]
- ROCKMAN H.A., KOCH W.J., LEFKOWITZ R.J. Cardiac function in genetically engineered mice with altered adrenergic receptor signalling. Am. J. Physiol. 1997;272:H1553–H1559. doi: 10.1152/ajpheart.1997.272.4.H1553. [DOI] [PubMed] [Google Scholar]
- ROSENWEIG A., HALAZONETIS T.D., SEIDMAN J.G., SEIDMAN C.E. Proximal regulatory domains of rat atrial natriuretic factor gene. Circ. 1991;84:1256–1265. doi: 10.1161/01.cir.84.3.1256. [DOI] [PubMed] [Google Scholar]
- RUTHER U., KOMITOWSKI D., SCHUBERT F.R., WAGNER E.F. C-fos expression induces bone tumors in transgenic mice. Oncogene. 1989;4:861–865. [PubMed] [Google Scholar]
- SAADANE N., ALPERT L., CHALIFOUR L.E. TAFII250, Egr-1, and D-type cyclin expression in mice and neonatal rat cardiomyocytes treated with doxorubicin. Am. J. Physiol. 1999;276:H803–H814. doi: 10.1152/ajpheart.1999.276.3.H803. [DOI] [PubMed] [Google Scholar]
- SAEZ E., RUTBERG S.E., MUELLER E., OPPENHEIM H., SMOLUK J., YUSPA S.H., SPIEGELMAN B.M. c-Fos is required for malignant progression on skin tumors. Cell. 1995;82:721–732. doi: 10.1016/0092-8674(95)90469-7. [DOI] [PubMed] [Google Scholar]
- SCHNEIDER M.D., PARKER T.G.Molecular mechanisms of cardiac growth and hypertrophy: Myocardial growth factors and proto-oncogenes in development and disease Molecular Basis of Cardiology 1993London: Blackwell Scientific Publications; 113–134.Roberts, R. pp [Google Scholar]
- SEPULVEDA J.L., BELAGULI N., NIGAM V., CHEN C.-Y., NEMER M., SCHWARTZ R.J. GATA-4 and Nkx2.5 coactive Nkx-2 DNA binding targets: role for regulating early cardiac gene expression. Mol. Cell. Biol. 1998;18:3405–3415. doi: 10.1128/mcb.18.6.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMEYNE R.J., VENDRELL M., HAYWARD M., BAKER S.J., MIAO G.G., SCHILLING K., ROBERTSON L.M., CURRAN T., MORGAN J.L. Continuous c-fos expression precedes programmed cell death in vivo. Nature. 1993;363:166–169. doi: 10.1038/363166a0. [DOI] [PubMed] [Google Scholar]
- SOONPAA M.H., FIELD L.J. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am. J. Physiol. 1994;266:H1439–H1445. doi: 10.1152/ajpheart.1994.266.4.H1439. [DOI] [PubMed] [Google Scholar]
- SUSIC D., NUÑEZ E., FROHLICH E.D., PRAKASH O. Angiotensin II increases left ventricular mass without affecting myosin isoform mRNAs. Hypertension. 1996;28:265–268. doi: 10.1161/01.hyp.28.2.265. [DOI] [PubMed] [Google Scholar]
- VAN BILSEN M., CHEIN K.R. Growth and hypertrophy of the heart: towards an understanding of cardiac specific and inducible gene expression. Cardiovasc. Res. 1993;27:1140–1149. doi: 10.1093/cvr/27.7.1140. [DOI] [PubMed] [Google Scholar]
- WANG Z.-Q., OVITT C., GRIGORIADIS A.E., MÖHLE-STEINLEIN U., RÜTHER U., WAGNER E.F. Bone and hematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- ZIERHUT W., ZIMMER H.-G. Significance of myocardial α- and β-adrenoreceptors in catecholamine-induced cardiac hypertrophy. Cir. Res. 1989;65:1417–1425. doi: 10.1161/01.res.65.5.1417. [DOI] [PubMed] [Google Scholar]