

Abstract
The mechanisms and the subtypes of muscarinic receptors implicated in the cardiovascular effects of physostigmine were investigated in conscious normotensive and spontaneously hypertensive rats.
Intravenous (i.v.) physostigmine (50 μg kg−1) induced in both strains a long pressor response, accompanied by a bradycardia. This pressor response was larger in spontaneously hypertensive (+41±6 mmHg) than in Wistar-Kyoto (+25±2 mmHg) rats (P<0.05).
Pretreatment with atropine sulphate (0.4 mg kg−1 i.v.), completely abolished the physostigmine-induced pressor response in both normotensive and hypertensive rats. In both strains, the physostigmine pressor response was significantly reduced by the systemic administration of either an α1-adrenoceptor antagonist (prazosin, 1 mg kg−1) or a V1A-vasopressin receptor antagonist (AVPX, 20 μg kg−1). This physostigmine pressor effect was completely abolished in both strains when both antagonists were administered concomitantly.
In WKY rats, the pressor response to physostigmine (50 μg kg−1 i.v.) was inhibited in a dose-dependent manner by i.c.v. administration of atropine (ID50=3.70 nmoles), the M1 receptor antagonist pirenzepine (ID50=10.71 nmoles), the M2 receptor antagonist methoctramine (ID50=4.31 nmoles), the M3 receptor antagonist p-F-HHSiD (ID50=60.52 nmoles) and the M4 receptor antagonist tropicamide (ID50=214.20 nmoles). In the hypertensive strain, the ID50 were found to be significantly higher for atropine (7.34 nmoles), pirenzepine (21.60 nmoles) and p-F-HHSiD (139.50 nmoles) (P<0.05).
The present results indicate that physostigmine acts in normotensive and spontaneously hypertensive rats, through stimulation of both central M2 and M1 cholinoceptors to induce a rise in blood pressure mediated by an increase in plasma vasopressin and sympathetic outflow. Moreover, our results suggest that some modifications of the M1 receptor subtypes in terms of expression or affinity could be responsible for the hyper-responsiveness of the hypertensive strain to cholinomimetic agents.
Keywords: Physostigmine, blood pressure, noradrenaline, vasopressin, central cholinergic systems
Introduction
The spontaneously hypertensive rat (SHR) is the most widely used animal model of hypertension implicating the sympathetic nervous system. Although an exaggerated sympathetic activity seems to be implicated, the pathophysiological mechanisms inducing hypertension remains unknown. As with human hypertension, this rat strain is normotensive at birth and gradually develops hypertension with adulthood (see Birkenhager & Reid, 1984). In order to understand the origin of hypertension, a wide variety of peripheral as well as central neuromediators has been studied in this model and in normotensive Wistar-Kyoto (WKY) controls (see Birkenhager & Reid, 1984).
It has been known for many years that central cholinergic systems are involved in the central regulation of blood pressure (BP) in several species including man (see Brezenoff & Giuliano, 1982; Buccafusco, 1996). In humans, several individual case reports have described pressor responses with clinical applications of acetylcholinesterase (AChE) inhibitors (Cain, 1986; Allain et al., 1996). AChE inhibitors, by blocking the rapid inactivation of acetylcholine (ACh), prolong and enhance the action of the neurotransmitter released from cholinergic terminals. This pressor response is selectively antagonized by atropine but not by methylatropine, indicating that this is mediated by central muscarinic receptors (see Brezenoff & Giuliano, 1982). Moreover, studies performed in various species described choline acetyl-transferase-positive neurons and muscarinic receptors (Schwartz, 1986; Watson et al., 1986) within or nearby the brainstem area involved in the control of BP and heart rate (HR) (see Wainer et al., 1984). Central administration of ACh and cholinomimetic drugs induce a rise in BP (see Brezenoff & Giuliano, 1982) mediated by an increase in sympathetic tone (Krstic & Djurkovic, 1978; Buccafusco & Brezenoff, 1979) and the release of vasopressin (AVP) (Rascol et al., 1990). In the last decade, five muscarinic receptor genes (m1–5) encoding distinct muscarinic cholinoceptors have been cloned. Some authors have suggested the involvement of M1 (Hori et al., 1995), M2 (Özkutlu et al., 1993; Ally et al., 1995) or M3 (Martin, 1992) receptors in the central pressor response to cholinomimetics.
However, the relative importance of the different muscarinic cholinoceptor subtypes remains unclear. Several experiments have demonstrated that the pressor response induced by cholinomimetic drugs is potentiated in SHR (see Buccafusco, 1996). A recent study reported a change in the expression of genes encoding for muscarinic receptor subtypes in this strain (Wei et al., 1995). However, to our best knowledge, no functional pharmacological experiment has yet studied the respective involvement of the different muscarinic receptor subtypes involved in the cholinergic hyper-responsiveness of SHR. The aims of the present study were (1) to investigate and compare the cardiovascular changes induced by the intravenous (i.v.) injection of physostigmine in both normotensive and hypertensive rats, (2) to characterize which mechanisms are involved at the peripheral level and (3) to determine the subtype(s) of central muscarinic cholinoceptors involved in the cardiovascular effects induced by the physostigmine.
Methods
General procedure
Experiments were performed on 12-week-old males WKY and SH rats weighing 250–300 g. These animals were obtained from Harlan (Gannat, France) and were maintained at 20–24°C with a 12 h light-dark cycle (light on 0800 h to 2000 h) at least 1 week before the experiment. Food pellets and tap water were available ad libitum. All animals procedure were conducted in strict compliance with approved French Agriculture Department for Animal Use for Research and Education groups.
Surgical preparation for intravenous drug administration
The rats were anaesthetized with sodium pentobarbitone (60 mg kg−1 i.p.). Animals were then placed on a blanket to avoid heat loss, body temperature was sensed by a rectal probe and maintained at 38°C (Harvard Apparatus, England). The right external jugular vein and the left carotid artery were cannulated with a polyethylene catheter (Plastimed, Denucath 3F, France), filled with heparinized saline (50 IU ml−1), to allow intravenous injections of drugs and direct BP recording.
Intracerebroventricular (i.c.v.) cannulation
Following anaesthesia, the rats were placed in a stereotaxic frame (Unimecanique, Paris, France) and a cannula was implanted into the right lateral cerebroventricle using the following coordinates relative to bregma: posterior 1.0 mm, lateral 1.5 mm, ventral 3.7 mm from the surface of the skull, on the basis of the atlas of Paxinos & Watson (1982). The guide cannula was anchored to the skull using mounting screws and dental caulk. Each animal was allowed at least 1 week recovery before being used for the first experiment. Between subsequent experiments, a 2 day recovery period was allowed for each animal. The rats were used for no more than four experiments. The arterial catheters were kept patent by flushing with 0.2 ml of heparinized saline (50 IU ml−1) daily.
The i.c.v. cannula was connected to a 25 μl Hamilton syringe with polyethylene (PE-50) tubing. The injection cannula was filled by backfilling with 10 μl of injectate. The syringe itself was filled with distilled water and an air bubble was left between the water and injection solution. The animals were not allowed access to food or water during the experiments. Methylene blue was injected i.c.v. after the fourth experiment for verification of the cannula placement and only proper i.c.v. placements were included in the study. The day of the experiment, the rat was placed in a rodent sampling cage and BP was continuously recorded via a pressure transducer (Abbott, Transpac IV, Ireland) and amplifier (Bionic Instruments, Qazap 94104, France) coupled to a MacLab hardware unit (ADInstruments, MacLab/4S, Australia) connected to a microcomputer (PowerMacintosh 6200, Apple, U.S.A.). HR was triggered by the BP signal and expressed in beat per min (b.p.m.). Respiratory rate was counted from the chest movements. An equilibration period of 30 min was allowed before the beginning of each experiment.
Experimental protocols
Intravenous administration
In the first part of this study, we assessed the effects of a bolus i.v. injection of physostigmine (50 μg kg−1, n=6) in freely moving rats of both strains (WKY and SHR). This physostigmine injection was preceded, 5 min before by an i.v. saline injection (0.3 ml) in order to test non-specific cardiovascular effects (Saline group). The effects of i.v. physostigmine were assessed on BP, HR and respiratory rate at three different times: 5, 10 and 15 min after administration according to the kinetic of the physostigmine induced cardiovascular changes (Brezenoff, 1973).
Four additional treatment protocols were designed in freely moving animals of both strains, in order to explain the mechanisms underlying the effects of i.v. physostigmine administration: (1) atropine+physostigmine (atropine group, n=6), (2) prazosin+physostigmine (prazosin group, n=6), (3) AVP antagonist+physostigmine (AVPX group, n=6), (4) prazosin+AVP antagonist+physostigmine (prazosin+AVPX group, n=6). Each antagonist was injected by i.v. route 5 min before the systemic administration of physostigmine. In the prazosin+AVPX group, prazosin was the first drug injected, followed by AVP antagonist and, 5 min later, physostigmine. BP, HR and respiratory rate were measured 1 min before and 3 min after the i.v. injection of the antagonists to assess their own effects on the measured parameters. All i.v. injections were administered in a volume of 0.25–0.30 ml.
Central administration
In order to determine the subtype of muscarinic cholinoceptor involved in the physostigmine-induced pressor response, saline and various muscarinic antagonists were administered by i.c.v. route, 10 min prior a physostigmine i.v. injection. At least three doses of each antagonist (atropine, pirenzepine, methoctramine, para-fluoro-HHSiD and tropicamide) were tested in at least six different freely moving rats. In order to confirm the involvement of muscarinic M2 receptors in the pressor response to physo-stigmine, a linear regression was calculated by plotting the Log ID50s calculated in this study, from the dose-response curves with each antagonist versus the Log Kis published in the literature (Martin, 1992; Lazareno & Birdsall, 1993; Caulfield & Birdsall, 1998). In order to confirm the involvement of muscarinic M1 receptors in the SHR hyper-responsiveness to cholinergic agents, thiopilocarpine, a muscarinic M1 receptor agonist (Eglen & Watson, 1996), was also injected by i.c.v. route.
Drugs
The following drugs were used: sodium pentobarbitone (Pentobarbital sodique®: Sanofi, Libourne, France), physo-stigmine salicylate salt, atropine sulphate, prazosin hydrochloride, [β-mercapto-β,β-cyclopenta-methylenepropionyl1, O-Me-Tyr2, Arg8]-vasopressin (Sigma-Aldrich, St. Quentin Fallavier, France). Pirenzepine, methoctramine, p-F-HHSiD (para-fluoro-hexahydro-sila-difenidol), tropicamide and 2-hydroxypropyl-β-cyclodextrin (Research Biochemicals Inc., Natick, U.S.A.). Thiopilocarpine was a gift from Novartis Pharma. The doses of atropine (0.4 mg kg−1), [β-mercapto-β,β-cyclopenta-methylenepropionyl1, O-Me-Tyr2, Arg8]-vasopressin (20 μg kg−1) and prazosin (1 mg kg−1) were chosen according to their ability to block in our experimental model the peripheral effects of ACh (2.5 μg kg−1), AVP (100 ng·kg−1) and phenylephrine (5 μg kg−1), respectively (data not shown). Such doses of ACh, AVP and phenylephrine induced BP changes in the range of the vascular effects induced by physostigmine (±50 mmHg). Prazosin was first dissolved in methanol and secondly added to physiological saline. Methanol represented 10% of the final volume. Tropicamide and p-F-HHSiD were dissolved in 2-hydroxypropyl-β-cyclodextrin (45% w v−1 solution in H2O). All vehicles were tested to make sure that they did not induce, by themselves, any effect on BP and HR. All other drugs were dissolved in physiological saline. All given doses of drugs refer to the free base.
Statistical analysis
The experimental data were obtained from six experiments for each treatment group, each experiment being performed in a different animal.
A one-way analysis of variance (ANOVA) was performed to compare the baseline means of the different parameters (BP, HR and respiratory rate) in each protocol in order to assess if there was any significant intergroup difference on baseline 5 min before any injection. The paired-sample Student's t-test was used to compare the means of the different parameters 1 min before and 2 min after i.v. antagonists in order to assess if these drugs induced any effects by themselves. According to the homogeneity of variances, a two-way multivariate ANOVA was used to compare the mean's variations of the different parameters in the different groups at different times in order to assess if there was any significant different effect of i.v. physostigmine in these groups. The Scheffe's test and the Dunnett's test were used as post hoc tests for intergroup and intragroup comparisons, respectively. Student's t-test was used to compare, for a given dose, the mean pressor response following treatment by muscarinic receptor antagonist or agonist, between normotensive and spontaneously hypertensive rats. Values are expressed as means±s.e.mean. The level of significance was accepted for P<0.05.
Results
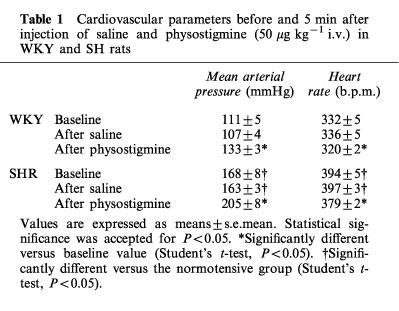
Baseline cardiovascular and respiratory parameters were not significantly different, in each strain, before i.v. injections. BP and HR were significantly higher in SHR than in WKY (Table 1). Saline induced no significant change in any parameters after i.v. (Table 1) or i.c.v. injection at any time.
Table 1.
Cardiovascular parameters before and 5 min after injection of saline and physostigmine (50 μg kg−1 i.v.) in WKY and SH rats
Physostigmine cardiovascular responses (saline groups)
Physostigmine i.v. injections induced yawning, motor agitation, micturition, defecation and licking in most of the normotensive and hypertensive rats.
In both strains, physostigmine (50 μg kg−1) i.v. injection induced, within 1 min, a significant increase in mean arterial pressure (MAP) (Table 1). The effect was maximal after 5 min (Table 2) and the magnitude of this BP increase was significantly greater in SH than in WKY rats (P<0.05) (Figure 1). In each strain, the pressor response remained significant over 15 min and then slowly returned to basal value within 30 min. For both normotensive and hypertensive rats, a short lasting but significant bradycardia accompanied the pressor effect (WKY: −16±2 b.p.m., P<0.05–SHR: −18±2 b.p.m., P<0.05) (Table 1).
Table 2.
Mean arterial pressure values 5, 10 and 15 min following physostigmine (50 μg kg−1 i.v.) administered in Wistar Kyoto (WKY) and spontaneously hypertensive rats (SHR)
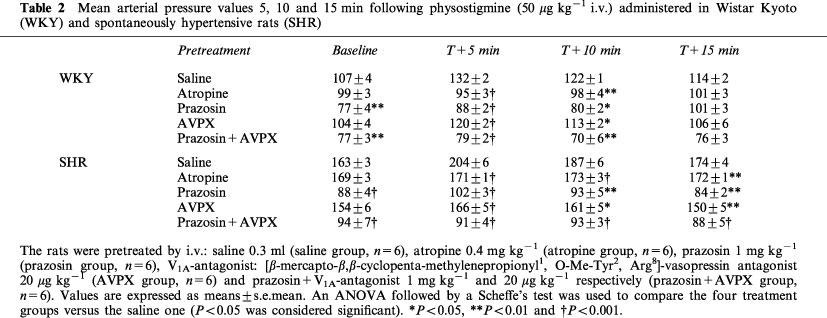
Figure 1.
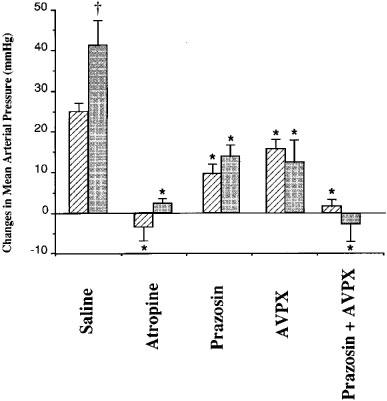
Changes in mean arterial pressure elicited by intravenous (i.v.) injection of physostigmine (50 μg kg−1) 5 min after i.v. pretreatment with saline (0.3 ml, n=6), atropine (0.4 mg kg−1, n=6), prazosin (1 mg kg−1, n=6), V1A-vasopressin receptor antagonist: [β-mercapto-β,β-cyclopenta-methylenepropionyl1, O-Me-Tyr2, Arg8]-vasopressin (AVPX, 20 μg kg−1, n=6) and prazosin+V1A-vasopressin receptor antagonist (prazosin+AVPX, 1 mg kg−1 and 20 μg kg−1 respectively, n=6) in freely moving Wistar-Kyoto (hatched columns) and spontaneously hypertensive rats (shaded columns). Values are expressed as variation of the means±s.e.mean. Statistical significance (ANOVA) versus the saline group: *P<0.05 and versus the normotensive group: †P<0.05.
Effects of antagonists pretreatment
Atropine (1.5–15 nmoles i.c.v.), AVPX (20 μg kg−1 i.v.), pirenzepine (6–50 nmoles i.c.v.), methoctramine (1.4–14 nmoles i.c.v.), p-F-HHSiD (10–100 nmoles i.c.v.) and tropicamide (26–260 nmoles i.c.v.) induced no significant changes in any measured parameters. Although the behavioural parameters were not precisely assessed, the central administration of muscarinic receptor antagonists did not influence significant behavioural changes versus the physo-stigmine group. Atropine (0.4 mg kg−1 i.v.) did not modify the pressure values in WKY and SHR, however, it induced a slight increase of HR in normotensive rats (+9±4 b.p.m., P<0.05) which did not reach a significant level in hypertensive rats (+3±7 b.p.m.). Prazosin (1 mg kg−1 i.v.) induced a significant decrease in MAP (−30±3 and −75±3 mmHg, P<0.01) in WKY and SHR, respectively. In both strains, this BP decrease was not accompanied by a significant increase in HR (WKY: +6±4 b.p.m.–SHR: +14±9 b.p.m.).
Cardiovascular changes in the different i.v. protocols: peripheral mechanisms involved in the pressor response (Table 2 and Figure 1)
The two-way ANOVA showed that there was a significant time by group interaction (P<10−3). Five minutes after physostigmine administration (T+5), the post hoc analysis showed that the mean physostigmine-induced increases of BP were significantly different among the five groups (P<10−3) in both normotensive and hypertensive rats. The physostigmine-induced increase in MAP was abolished in the ‘atropine' and ‘prazosin+AVPX' groups (P<10−3 vs ‘saline' group). When compared with the ‘saline' group, the pressor effect was significantly and similarly reduced in the ‘prazosin' and ‘AVPX' groups (P<10−3). At T+10 min, MAP in the ‘saline' groups of both strains was significantly higher than in the ‘atropine', ‘prazosin', ‘AVPX' and ‘prazosin+AVPX' groups (P<0.05). At T+15 min, in normotensive rats, there were no significant differences between the five treatment groups. Conversely, in hypertensive rats, MAP in the ‘saline' group remained significantly increased compared with the four other groups (P<0.05).
Despite a slight bradycardia following systemic injection of physostigmine, HR was not significantly different, at any time, between the various groups.
Cardiovascular changes in the different i.c.v. protocols: central muscarinic receptors subtypes involved in the pressor response to physostigmine (Table 3 and Figures 2 and 3)
Table 3.
ID50 (or ED50) obtained in WKY and SHR on the physostigmine-induced pressor response with various muscarinic receptor antagonists
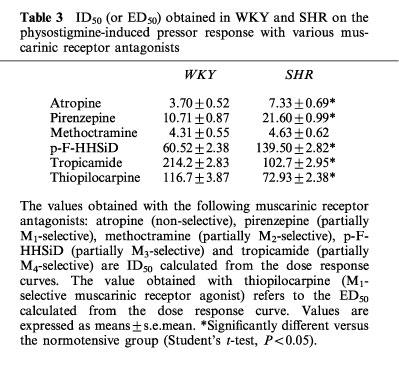
Figure 2.
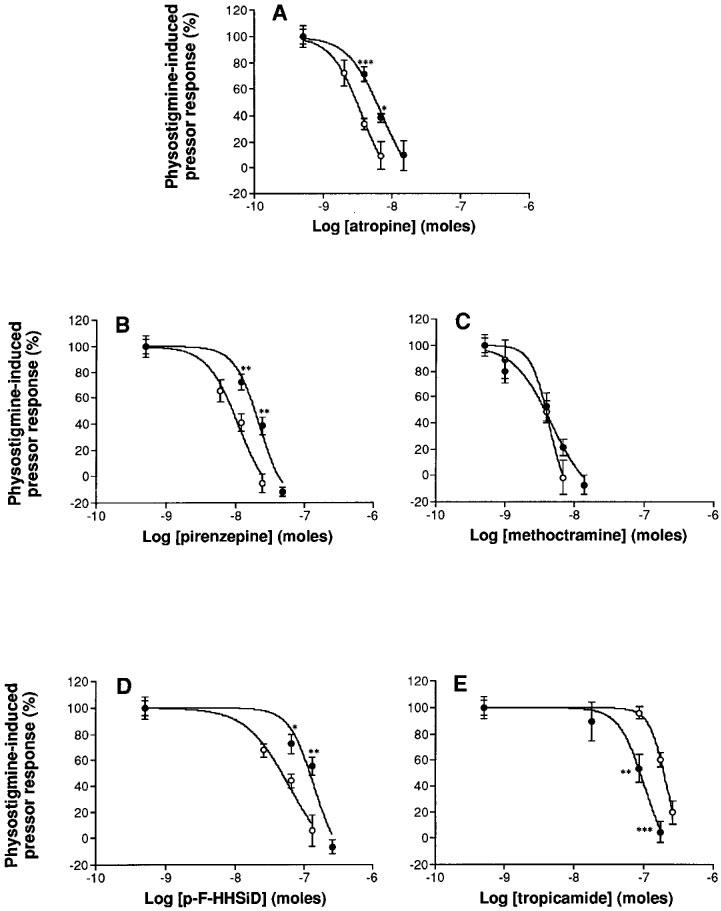
Inhibition of the physostigmine-induced pressor response in normotensive Wistar-Kyoto (open circles) and spontaneously hypertensive (closed circles) rats by various doses of the non-selective muscarinic receptor antagonist atropine (2A, 1.5–15 nmoles i.c.v., n=6) and the selective muscarinic receptor antagonists: pirenzepine (2B, 6–50 nmoles i.c.v., n=6), methoctramine (2C, 1.4–14 nmoles i.c.v., n=6), p-F-HHSiD (2D, 10–100 nmoles i.c.v., n=6) and tropicamide (2E, 1.5–15 nmoles i.c.v., n=6). Values are expressed as means±s.e.mean. Statistical significance versus the normotensive group: *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
Figure 3.
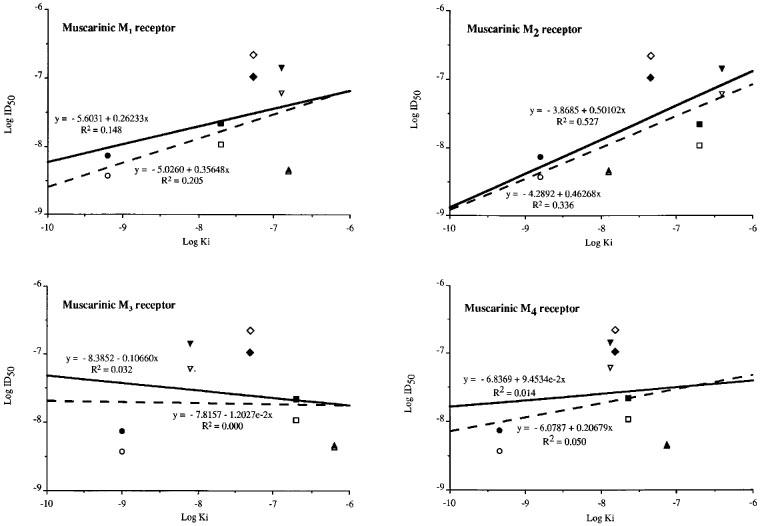
Correlation of the Log of the ID50s calculated from the dose-response curves shown in Figure 2 with the Log of the Kis published for the muscarinic M1, M2, M3 and M4 receptor subtypes for the muscarinic receptor antagonists used in this study. The Log Kis were obtained from Martin, 1992; Lazareno & Birdsall, 1993; Caulfield & Birdsall, 1998. The full lines refer to the linear regressions obtained for the hypertensive rats and the dotted lines refer to the linear regressions obtained for the normotensive rats. The drugs are: atropine (•), pirenzepine (▪), methoctramine (▴), p-F-HHSiD (▾) and tropicamide (⧫). The symbols are open for normotensive rats and closed for hypertensive rats.
Pretreatment of the rats by an i.c.v. injection of 1.5–15 nmoles of atropine resulted in a dose-dependent inhibition of the peak physostigmine effects on BP in both normotensive and SH rats. Dose-response curves analysis showed that atropine had significantly different ID50 values between the two strains (normotensive: 3.70 and SH rats: 7.34 nmoles, P<0.05) (Figure 2A).
Pretreatment with pirenzepine (6–50 nmoles i.c.v.) also inhibited in a dose-dependent way the BP response to 50 μg kg−1 i.v. of physostigmine in both strains. The ID50 calculated from the dose-response curves was equal to 10.71 nmoles in WKY and 21.60 nmoles in hypertensive rats (P<0.05) (Figure 2B).
Methoctramine pretreatment (1.4–14 nmoles i.c.v.) inhibited in a dose-dependent way the increase in BP evoked by physostigmine with the same ID50 in both strains (4.31 nmoles in WKY and 4.63 nmoles in SHR) (Figure 2C).
The physostigmine-induced pressor response was also dose-dependently inhibited by p-F-HHSiD (10–100 nmoles i.c.v.) in both strains. The ID50 was smaller in WKY (60.52 nmoles) than in SH rats (139.50 nmoles) (P<0.05) (Figure 2D).
Pretreatment with tropicamide (26–260 nmoles i.c.v.) also inhibited in a dose-dependent way the BP response to 50 μg kg−1 i.v. of physostigmine in both strains. The ID50 calculated from the dose-response curves was larger in WKY (214.20 nmoles) than in hypertensive rats (102.70 nmoles) (P<0.05) (Figure 2E).
The correlations between these Log ID50s and the published Log Kis were: r2=0.205 and 0.148, respectively in WKY and SHR for the M1 receptor subtype; r2=0.336 and 0.527, respectively in WKY and SHR for the M2 receptor subtype; r2=0.000 and 0.032, respectively in WKY and SHR for the M3 receptor subtype and r2=0.050 and 0.014, respectively in WKY and SHR for the M4 receptor subtype.
The potency order for the participation of muscarinic receptors in the pressor response to i.v. physostigmine in normotensive as well as in hypertensive rats was thus: M2>M1>>M3>>M4. This potency order was also confirmed by the Log ID50s vs Log Kis correlations (Figure 3).
The physostigmine-induced bradycardia, observed after saline pretreatment (WKY: −18±2 b.p.m.–SHR: −16±2 b.p.m., P<0.05) was not significantly altered by the highest doses of atropine (WKY: −15±6 b.p.m.–SHR: −21±7 b.p.m., P<0.05), pirenzepine (WKY: −17±2 b.p.m. – SHR: −29±8 b.p.m., P<0.05), methoctramine (WKY: −20±2 b.p.m.–SHR: −25±10 b.p.m., P<0.05), p-F-HHSiD (WKY: −16±4 b.p.m.–SHR: −19±3 b.p.m., P<0.05) and tropicamide (WKY: −28±10 b.p.m.–SHR: −21±7 b.p.m., P<0.05).
Thiopilocarpine cardiovascular responses (Figure 4)
Figure 4.
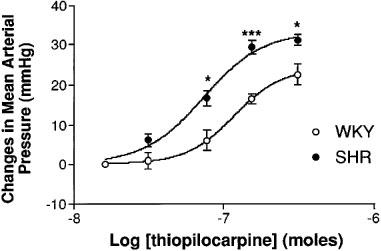
Changes in mean arterial pressure following i.c.v. thiopilocarpine (31–310 nmoles, n=6) in freely moving Wistar-Kyoto (open circles) and spontaneously hypertensive (closed circles) rats. Values are expressed as variation of the means±s.e.mean. Statistical significance versus the normotensive group: *P<0.05; ***P<0.001 (Student's t-test).
Injections i.c.v. of the M1 receptor agonist thiopilocarpine induced, like physostigmine, yawning, motor agitation and defecation. In both WKY and SHR, thiopilocarpine (31–310 nmoles) induced a dose-dependent increase in MAP which reached its maximal effect between 5–10 min after administration. Like physostigmine, the thiopilocarpine-induced increase in BP remained significant over 15 min and then slowly returned to basal value within 30 min. The magnitude of this increase in BP was significantly greater in SHR than in WKY following thiopilocarpine administration (Student's t-test, P<0.05).
Discussion
The SHR is an animal model of human essential hypertension which exhibits cholinergic abnormalities when compared with its normotensive control, namely the WKY rat. Indeed, several authors reported that SHR develop an exaggerated pressor response following central (or peripheral) administration of cholinergic agonists (see Buccafusco, 1996). Moreover, in this strain, choline acetyltransferase levels were reported to be significantly increased in the locus coeruleus (Helke et al., 1980) and in the rostral ventrolateral medulla (RVLM) (Kubo et al., 1995) while others reported decreased values in the hypothalamus (Helke et al., 1980; Yamada et al., 1984). An increase in central muscarinic binding sites has also been reported in SHR (Hershkowitz et al., 1983; Yamada et al., 1984; Wei et al., 1995; Gattu et al., 1997a,1997b). Finally, Vargas et al. (1988) reported that the chronic reduction of brain acetylcholine with hemicholinium-3 was able to suppress hypertension in SHR.
The present study is, to our best knowledge, the first functional study investigating, in freely moving rats, the involvement of four different central muscarinic cholinoceptor subtypes (M1, M2, M3 and M4) in the central pressor response induced by cholinergic agents. Our results demonstrate that physostigmine (50 μg kg−1), injected by i.v. route, in freely moving WKY and SHR, induced a significant increase in BP. Our data also show that (1) this pressor response was significantly greater in SHR versus WKY rats, (2) this pressor response is mediated in both strains by central muscarinic M2 and M1 cholinoceptors which lead to an increase in sympathetic outflow and vasopressin release, (3) the hyper-responsiveness to cholinomimetic agents in the SHR strain seems to be due to changes in the expression or affinity of the M1 receptor subtypes.
Peripheral mechanisms implicated in the physostigmine-induced blood pressure increase
The present study shows that physostigmine systemic administration induced a significant increase in MAP, which was larger in SH than in WKY rats, with a magnitude comparable to what has already been published with other cholinergic drugs (Hoffman et al., 1978; Buccafusco & Magri, 1990; Lee et al., 1991; Kubo et al., 1997). In both normotensive and hypertensive rats, the pressor response to physostigmine was associated with a slight but significant bradycardia. It is noteworthy that the bradycardia induced by physostigmine was not modified after central pretreatment by atropine or the different selective muscarinic receptor antagonists, confirming the peripheral origin of this phenomenon (Lazartigues et al., 1998). Pretreatment with i.v. atropine induced a slight but significant increase in HR only in WKY rats (+9±4 mmHg). This could be explained by a low vagal tone in freely moving rats. Moreover, some authors suggested a depressed parasympathetic nervous activity in hypertensives explaining the lack of tachycardia following atropine administration in SHR (see Van Zwieten et al., 1995).
The physostigmine-induced pressor response was abolished in both strains by the peripheral administration of both α1-adrenergic receptor and V1A-vasopressin receptor blockers. This has already been reported previously in normotensive rats (Lazartigues et al., 1998). The fact that a pretreatment with both α1-adrenoceptor and vasopressin receptor antagonists also completely suppressed the pressor response to physo-stigmine in SHR eliminates other peripheral mechanisms generating the cholinergic pressor response in this strain. Both adrenergic and vasopressinergic mechanisms appeared to have the same relative contribution in SH as well as in WKY rats. Such a comparable contribution was unexpected because an increase in the SHR sympathetic tone has usually been emphasized in the literature (Arnolda et al., 1997). However, some authors also reported previously indices of excessive vasopressin tone in SHR: urinary excretion, plasma concentrations and pituitary contents of vasopressin being greater in SH than in WKY rat (Crofton et al., 1978). These data agree with the hypothesis that V1A-vasopressin receptors may contribute to the pathogenesis of SHR hypertension, as suggested by the reduced hypertension following treatment with a V1A-vasopressin receptor antagonist in pre-hypertensive SHR (Naitoh et al., 1997). Moreover, it has also been suggested that the potentiated pressor response in SHR may result from an increased vascular responsiveness to vasopressin (Hoffman et al., 1978).
Central muscarinic mechanisms implicated in the physostigmine-induced blood pressure increase
In agreement with previous experiments, the present study showed that the physostigmine-induced pressor response was completely blocked in both strains by a peripheral or a central pretreatment with the non-selective antagonist atropine, confirming the involvement of central muscarinic receptors. Moreover, the ID50 value obtained with atropine was larger in SH (7.33±0.69 nmoles) than WKY rats (3.70±0.52 nmoles) providing further evidence to the cholinergic hyper-responsiveness in this strain, as previously suggested by Buccafusco (1996).
We failed to observe any modification in the baseline BP following pretreatment with atropine, suggesting that muscarinic receptors are not tonically involved in maintaining BP in both strains. According to the site specificity and to the limitations of the i.c.v. injection technique, another explanation for this lack of resting BP modifications could be that atropine injected by i.c.v. route did not reach the areas responsible of the BP maintain. Indeed, experiments performed with bilateral microinjections of atropine, scopolamine or hemicholinium-3 into the RVLM have been shown to lower BP in normotensive rats almost to spinal transection levels (see Buccafusco, 1996; Kubo, 1998). The physostigmine-induced pressor response, following i.v. injection, is known to involve caudal as well as more rostral brain areas (see Brezenoff & Giuliano, 1982). Although the response obtained after i.c.v. injection of muscarinic receptor antagonists represents the contribution of subtypes accessed by the varying distributing amounts of the antagonist, there is no doubt that these subtypes are important in mediating the physostigmine induced pressor response. According to this assumption, in both strains, all ‘selective' muscarinic antagonists (pirenzepine, methoctramine, p-F-HHSiD and tropicamide), when administered centrally, inhibited in a dose-dependent way the physostigmine-induced pressor response. Moreover, for these antagonists, the potency orders were similar (M2>M1>>M3>>M4) in normotensive and hypertensive rats, suggesting the same relative involvement of the four receptor subtypes in the physostigmine-induced pressor response.
The M2 receptor antagonist, methoctramine, had the lowest ID50, being the most potent agent to prevent the increase in BP induced by physostigmine in both strains. These findings suggest that the M2 receptor subtype is the central cholinoceptor subtype which is predominantly involved in the central cholinergic pressor response in both strains. We agree that it is difficult to determine the subtype of receptor which is involved in the cardiovascular effects of physostigmine from only ID50 values of muscarinic receptor antagonists because ID50 of an antagonist is dependent on the magnitude of affinity of the antagonist for receptors. The involvement of a muscarinic M2 receptor subtype in the cardiovascular effects of physostigmine is however confirmed by the linear regression between the Log ID50s calculated from the dose-response curves and the Log Kis published in the literature (Martin, 1992; Lazareno & Birdsall, 1993; Caulfield & Birdsall, 1998). Indeed, although the correlations performed with this method are not very significant, the best linear regressions, in both normotensive and hypertensive rats, were obtained with the muscarinic M2 receptor subtype (r2=0.53 for SHR and 0.34 for WKY) (Figure 3). Methoctramine had the same ID50 in WKY and SHR (4.31±0.55 and 4.63±0.62, respectively). This result suggests that the M2 receptors are not involved in the central mechanisms of the SHR hyper-responsiveness.
The intermediate ID50 values obtained with the M1 receptor antagonist pirenzepine in normotensive and hypertensive rats (10.71±0.87 and 21.60±0.99, respectively) suggest that it is not possible to rule out the mediation of M1 muscarinic cholinoceptors in the central cholinergic pressor response. This has already been suggested in some recent studies performed in normotensive rats (Hori et al., 1995; Lazartigues et al., 1998). However, the dose required to inhibit the pressor response in hypertensive rats was 2 fold higher than in normotensive animals. This increased ID50 value for pirenzepine in the hypertensive strain suggest some putative modifications of the M1 receptors in terms of affinity or expression. On the whole, these functional in vivo data are in agreement with in vitro studies suggesting an increase (a) in cerebral muscarinic receptor binding sites in SHR and in SHR stroke prone (Yamada et al., 1984) and (b) in the mRNA and binding sites of the M1 receptor subtype in various SHR brain areas involved in the physostigmine-induced pressor response (Wei et al., 1994; 1995; Gattu et al., 1997b). The involvement of central M1 receptors in SHR cholinergic hyper-responsiveness is confirmed by the fact that the M1 receptor selective agonist, thiopilocarpine induced a greater increase in BP in hypertensive than in normotensive rats.
The ID50 value obtained with the M3 receptor antagonist was also increased in the hypertensive strain, but according to the high doses of p-F-HHSiD necessary to inhibit the physostigmine-induced pressor response in both strains (ID50 for WKY: 60.52±2.38; ID50 for SHR: 139.5±2.82) and to the relative selectivity of this antagonist, it is difficult to suggest that the M3 receptor subtype is indeed involved in the physostigmine-induced pressor response. In spite of conflicting opinions (Brezenoff et al., 1988; Xiao & Brezenoff, 1988; Martin, 1992; Flores et al., 1996), our results are in agreement with previously published reports in the rat (Sundaram et al., 1988; 1989; Özkutlu et al., 1993; Lazartigues et al., 1998) or the cat (Ally et al., 1993; 1995) excluding the involvement of M3 muscarinic cholinoceptors in this pressor response.
To our best knowledge, the involvement of M4 muscarinic cholinoceptors in the pressor response to cholinergic drugs has never been assessed. Our results suggest that these receptors do not mediate the physostigmine-induced pressor effect because of the high ID50 values (WKY: 214.2±2.83 and SHR: 102.7±2.95). This is in agreement with the observation that the M4 receptor, or its corresponding mRNA, was the less abundant subtype in the areas implicated in the physostigmine-induced pressor response like brainstem (see Caulfield, 1993; Levey, 1993).
In conclusion, the present study demonstrates that physostigmine induces a pressor response in freely moving rats mediated by both sympathetic tone and vasopressin release, associated with a bradycardia. This pressor response is mediated by both central M2 and M1 muscarinic receptors. The physostigmine-induced pressor response is greater in the hypertensive strain and both sympathetic and vasopressinergic activities are likely to be increased in SHR. Moreover, to our best knowledge, our study is the first in vivo study showing in the hypertensive rat, a putative modification of the M1 receptor subtypes in terms of affinity or expression. These modifications could be at the origin of the hyper-responsiveness of the SHR to cholinergic drugs and may play a role as a predisposing or initiating factor for the development of hypertension in this hypertensive strain.
Acknowledgments
The authors wish to thank Dr Jean Galitzky for his help in the data analysis and in the conception of the manuscript.
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AVP
arginin-vasopressin
- AVPX
[β-mercapto-β,β-cyclopenta-methylenepropionyl1, O-Me-Tyr2, Arg8]-vasopressin
- BP
blood pressure
- DI50
dose inhibiting 50% of the effect
- HR
heart rate
- MAP
mean arterial pressure
- mRNA
messager ribonucleic acid
- p-F-HHSiD
para-fluoro-hexahydro siladifenidol
- RVLM
rostral ventrolateral medulla
- SHR
spontaneously hypertensive rat
- WKY
Wistar-Kyoto
References
- ALLAIN H., MARUELLE L., BENETON C., ROUAULT G., BELLIARD S. Poussées d'hypertension artérielle lors d'un traitement par tacrine. La Presse Médicale. 1996;25:1388–1389. [PubMed] [Google Scholar]
- ALLY A., HARA Y., MURAYAMA S. Cardiovascular effects of central administration of cholinomimetics in anesthetized cats. Neuropharmacol. 1993;32:185–193. doi: 10.1016/0028-3908(93)90099-o. [DOI] [PubMed] [Google Scholar]
- ALLY A., WILSON L.B., NOBREGA A.C.L., MITCHELL J.H. Cardiovascular effects elicited by central administration of physostigmine via M2 muscarinic receptors in conscious cats. Brain Res. 1995;677:268–276. doi: 10.1016/0006-8993(95)00171-l. [DOI] [PubMed] [Google Scholar]
- ARNOLDA L., WANG H., MINSON J., LLEWELLYN-SMITH I., SUZUKI S., PILOWSKY P., CHALMERS J. Central control mechanisms in hypertension. Aust. NZ J. Med. 1997;27:474–478. doi: 10.1111/j.1445-5994.1997.tb02221.x. [DOI] [PubMed] [Google Scholar]
- BIRKENHAGER W., REID J.Experimental and genetic models of hypertension Handbook of Hypertension 1984New York: Elsevier Science Publishers; ed. Jong, W.D [Google Scholar]
- BREZENOFF H.E. Centrally induced pressor responses to intravenous and intraventricular physostigmine evoked via different pathways. Eur. J. Pharmacol. 1973;23:290–292. doi: 10.1016/0014-2999(73)90097-6. [DOI] [PubMed] [Google Scholar]
- BREZENOFF H.E., GIULIANO R. Cardiovascular control by cholinergic mechanisms in the central nervous system. Ann. Rev. Pharmacol. Toxicol. 1982;22:341–381. doi: 10.1146/annurev.pa.22.040182.002013. [DOI] [PubMed] [Google Scholar]
- BREZENOFF H.E., VARGAS H., XIAO Y. Blockade of brain M2 muscarinic receptors lowers blood pressure in spontaneously hypertensive rats. Pharmacology. 1988;36:101–105. doi: 10.1159/000138365. [DOI] [PubMed] [Google Scholar]
- BUCCAFUSCO J.J. The role of central cholinergic neurons in the regulation of blood pressure and in experimental hypertension. Pharmacol. Rev. 1996;48:179–211. [PubMed] [Google Scholar]
- BUCCAFUSCO J.J., BREZENOFF H.E. Pharmacological study of a cholinergic mechanism within the rat posterior hypothalamic nucleus which mediates a hypertensive response. Brain Res. 1979;165:295–310. doi: 10.1016/0006-8993(79)90561-4. [DOI] [PubMed] [Google Scholar]
- BUCCAFUSCO J.J., MAGRI V. The pressor response to spinal cholinergic stimulation in spontaneously hypertensive rats. Brain Res. Bull. 1990;25:69–74. doi: 10.1016/0361-9230(90)90254-w. [DOI] [PubMed] [Google Scholar]
- CAIN J. Hypertension associated with oral administration of physostigmine in a patient with Alzheimer's disease. Am. J. Psychiatry. 1986;143:910–912. doi: 10.1176/ajp.143.7.910. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P. Muscarinic receptors–characterization, coupling and function. Pharmacol. Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P., BIRDSALL N.J.M. International Union of Pharmacology. XVII. Classification of Muscarinic Acetylcholine Receptors. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- CROFTON J., SHARE L., SHADE R., ALLEN C., TARNOWSKI D. Vasopressin in the rat with spontaneous hypertension. Am. J. Physiol. 1978;235:H361–H366. doi: 10.1152/ajpheart.1978.235.4.H361. [DOI] [PubMed] [Google Scholar]
- EGLEN M., WATSON N. Selective muscarinic receptor agonists and antagonists. Pharmacol. Toxicol. 1996;78:59–68. doi: 10.1111/j.1600-0773.1996.tb00181.x. [DOI] [PubMed] [Google Scholar]
- FLORES G., HERNANDEZ S., ROSALES M., SIERRA A., MARTINES-FONG D., FLORES-HERNANDEZ J., ACEVES J. M3 muscarinic receptors mediate cholinergic excitation of the spontaneous activity of subthalamic neurons in the rat. Neurosci. Lett. 1996;203:203–206. doi: 10.1016/0304-3940(95)12297-4. [DOI] [PubMed] [Google Scholar]
- GATTU M., PAULY J., URBANAWIZ S., BUCCAFUSCO J. Autoradiographic comparison of M1 and M2 binding sites in the CNS of spontaneously hypertensive and normotensive rats. Brain Res. 1997b;771:173–183. doi: 10.1016/s0006-8993(97)00691-4. [DOI] [PubMed] [Google Scholar]
- GATTU M., WEI J., PAULY J., URBANAWIZ S., BUCCAFUSCO J. Increased expression of M2 muscarinic receptor mRNA and binding sites in the rostral ventrolateral medulla of spontaneously hypertensive rats. Brain Research. 1997a;756:125–132. doi: 10.1016/s0006-8993(97)00126-1. [DOI] [PubMed] [Google Scholar]
- HELKE C., MUTH E., JACOBOWITZ D. Changes in central cholinergic neurons in the spontaneously hypertensive rat. Brain Res. 1980;188:425–436. doi: 10.1016/0006-8993(80)90042-6. [DOI] [PubMed] [Google Scholar]
- HERSHKOWITZ M., ELIASH S., COHEN S. The muscarinic cholinergic receptors in the posterior hypothalamus of hypertensive and normotensive rats. Eur. J. Pharmacol. 1983;86:229–236. doi: 10.1016/0014-2999(82)90320-x. [DOI] [PubMed] [Google Scholar]
- HOFFMAN W., SCHMID P., PHILLIPS M. Central cholinergic and noradrenergic stimulation in spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 1978;206:644–651. [PubMed] [Google Scholar]
- HORI H., HARUTA K., NANKI M., SAKAMOTO N., UEMURA K., MATSUBARA T., ITOH K., IGUCHI A. Pressor response induced by the hippocampal administration of neostigmine is suppressed by M1 muscarinic antagonist. Life Sci. 1995;57:1853–1859. doi: 10.1016/0024-3205(95)02165-f. [DOI] [PubMed] [Google Scholar]
- KRSTIC M.K., DJURKOVIC D. Cardiovascular response to intracerebroventricular administration of acetylcholine in rats. Neuropharmacol. 1978;17:341–347. doi: 10.1016/0028-3908(78)90004-7. [DOI] [PubMed] [Google Scholar]
- KUBO T. Cholinergic mechanism and blood pressure regulation in the central nervous system. Brain Res. Bull. 1998;46:475–481. doi: 10.1016/s0361-9230(98)00041-0. [DOI] [PubMed] [Google Scholar]
- KUBO T., ISHIZUKA T., FUKUMORI R., ASARI T., HAGIWARA Y. Enhanced release of acetylcholine in the rostral ventrolateral medulla of spontaneously hypertensive rats. Brain Res. 1995;686:1–9. doi: 10.1016/0006-8993(95)00433-q. [DOI] [PubMed] [Google Scholar]
- KUBO T., TAGUCHI K., SAWAI N., OZAKI S., HAGIWARA Y. Cholinergic mechanisms responsible for blood pressure regulation on sympathoexcitatory neurons in the rostral ventrolateral medulla of the rat. Brain Res. Bull. 1997;42:199–204. doi: 10.1016/s0361-9230(96)00256-0. [DOI] [PubMed] [Google Scholar]
- LAZARENO S., BIRDSALL N.J.M. Pharmacological characterization of acetylcholine-stimulated [35S]-GTPγS binding mediated by human muscarinic m1–m4 receptors: antagonist studies. Br. J. Pharmacol. 1993;109:1120–1127. doi: 10.1111/j.1476-5381.1993.tb13738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAZARTIGUES E., FRESLON J.L., TELLIOGLU T., BREFEL-COURBON C., PELAT M., TRAN M.A., MONTASTRUC J.L., RASCOL O. Pressor and bradycardic effects of tacrine and other acetylcholinesterase inhibitors in the rat. Eur. J. Pharmacol. 1998;361:61–71. doi: 10.1016/s0014-2999(98)00717-1. [DOI] [PubMed] [Google Scholar]
- LEE S.B., KIM S.Y., SUNG K.W. Cardiovascular regulation by cholinergic mechanisms in rostral ventrolateral medulla of spontaneously hypertensive rats. Eur. J. Pharmacol. 1991;205:117–123. doi: 10.1016/0014-2999(91)90809-5. [DOI] [PubMed] [Google Scholar]
- LEVEY A. Immunological localization of m1-m5 muscarinic acetylcholine receptors in peripheral tissues and brain. Life Sci. 1993;52:441–448. doi: 10.1016/0024-3205(93)90300-r. [DOI] [PubMed] [Google Scholar]
- MARTIN J.R. Pressor response to posterior hypothalamic administration of carbachol is mediated by muscarinic M3 receptor. Eur. J. Pharmacol. 1992;215:83–91. doi: 10.1016/0014-2999(92)90612-8. [DOI] [PubMed] [Google Scholar]
- NAITOH M., BURRELL L., RISVANIS J., ALDRED K., ROCKELL M., JOHNSTON C., PHILIPPS P. Modulation of genetic hypertension by short-term AVP V1A or V2 receptor antagonism in young SHR. Am. J. Physiol. 1997;272:F229–F234. doi: 10.1152/ajprenal.1997.272.2.F229. [DOI] [PubMed] [Google Scholar]
- ÖZKUTLU U., ONAT F., ASLAN A.N., OKTAY S. Central muscarinic M2 cholinoceptors involved in cholinergic hypertension. Eur. J. Pharmacol. 1993;250:349–354. doi: 10.1016/0014-2999(93)90020-i. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. Sydney: Academic Press; 1982. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- RASCOL O., MONTASTRUC J.L., GAUQUELIN G., TRAN M.A., GEELEN G., GHARIB C., MONTASTRUC P. Cardiovascular effects of central injection of acetylcholine in anaesthetized dogs: a role for vasopressin release. Br. J. Pharmacol. 1990;100:471–476. doi: 10.1111/j.1476-5381.1990.tb15831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWARTZ R. Autoradiographic distribution of high affinity muscarinic and nicotinic cholinergic receptors labelled with [3H] acetylcholine in rat brain. Life Sci. 1986;38:2111–2119. doi: 10.1016/0024-3205(86)90210-9. [DOI] [PubMed] [Google Scholar]
- SUNDARAM K., KRIEGER A., SAPRU H. M2 muscarinic receptors mediate pressor responses to cholinergic agonists in the ventrolateral medullary pressor area. Brain Res. 1988;449:141–149. doi: 10.1016/0006-8993(88)91032-3. [DOI] [PubMed] [Google Scholar]
- SUNDARAM K., MURUGAIAN J., WATSON M., SAPRU H. M2 muscarinic receptor agonists produce hypotension and bradycardia when injected into the nucleus tractus solitarii. Brain Res. 1989;477:358–362. doi: 10.1016/0006-8993(89)91427-3. [DOI] [PubMed] [Google Scholar]
- VAN ZWIETEN P.A., HENDRIKS M.G.C., PFAFFENDORF M., BRUNING T.A., CHANG P.C. The parasympathetic system and its muscarinic receptors in hypertensive disease. J. Hypertens. 1995;13:1079–1090. doi: 10.1097/00004872-199510000-00002. [DOI] [PubMed] [Google Scholar]
- VARGAS H., BREZENOFF H.E. Suppression of hypertension during chronic reduction of brain acetylcholine in spontaneously hypertensive rats. J. Hypertens. 1988;6:739–745. doi: 10.1097/00004872-198809000-00008. [DOI] [PubMed] [Google Scholar]
- WAINER B., LEVEY A., MUFSON E., MESULAM M.M. Cholinergic systems in mammalian brain identified with antibodies against choline acetyltransferase. Neurochem. Int. 1984;6:163–182. doi: 10.1016/0197-0186(84)90089-5. [DOI] [PubMed] [Google Scholar]
- WATSON M., ROESKE W., VICKROY T., SMITH T., AKIYAMA K., GULYA K., DUCKLES S., SERRA M., ADEM A., NORDBERG A., GEHLERT D., WAMSLEY J., YAMAMURA H. Biochemical and functional basis of putative muscarinic receptor subtypes and its implications. Trends Pharmacol. Sci. 1986. pp. 46–55.
- WEI J., MILICI A., BUCCAFUSCO J. Alterations in the expression of the genes encoding specific muscarinic receptor subtypes in the hypothalamus of spontaneously hypertensive rats. Circ. Res. 1995;76:142–147. doi: 10.1161/01.res.76.1.142. [DOI] [PubMed] [Google Scholar]
- WEI J., WALTON E., MILICI A., BUCCAFUSCO J. m1-m5 muscarinic receptor distribution in rat CNS by RT–PCR and HPLC. J. Neurochem. 1994;63:815–821. doi: 10.1046/j.1471-4159.1994.63030815.x. [DOI] [PubMed] [Google Scholar]
- XIAO Y.F., BREZENOFF H.E. The role of M2 muscarinic receptors in the posterior hypothalamus in the pressor response to intracerebroventricularly-injected neostigmine. Neuropharmacol. 1988;27:1061–1065. doi: 10.1016/0028-3908(88)90068-8. [DOI] [PubMed] [Google Scholar]
- YAMADA S., ISHIMA T., HAYASHI M., TOMITA T., HAYASHI E. Muscarinic cholinoceptors and choline acetyltransferase activity in the hypothalamus of spontaneously hypertensive rats. Life Sci. 1984;34:2151–2158. doi: 10.1016/0024-3205(84)90314-x. [DOI] [PubMed] [Google Scholar]