

Abstract
Cardiac pacing, in anaesthetized dogs, protects against ischaemia and reperfusion-induced ventricular arrhythmias when this is initiated 24 h after the pacing stimulus. Now we have examined whether this delayed cardioprotection afforded by cardiac pacing is mediated through nitric oxide.
Twenty-two dogs were paced (4×5 min periods at 220 beats min−1) by way of the right ventricle, 24 h prior to a 25 min period of coronary artery occlusion. Nine of these dogs were given the inhibitor of induced nitric oxide synthase, aminoguanidine (50 mg kg−1 i.v.), 0.5 h prior to coronary artery occlusion. Sham-operated non-paced dogs with and without aminoguanidine treatment served as controls.
Pacing markedly (P<0.05) reduced arrhythmia severity (ventricular fibrillation, VF, during occlusion 15%; survival from the combined ischaemia-reperfusion insult 62%) compared to control, sham-operated, unpaced dogs (VF during occlusion 58%; survival 17%). This protection was attenuated by the administration of aminoguanidine prior to coronary artery occlusion (survival from the combined ischaemia-reperfusion insult 11%, which was significantly (P<0.05) less than in the paced dogs not given aminoguanidine and similar to the controls). Aminoguanidine had no significant effects on coronary artery occlusion when given to dogs that had not been paced. In the dose used aminoguanadine transiently elevated systemic arterial pressure by a mean of 20 mmHg and reduced heart rate by a mean of 22 beats min−1.
These results suggest that nitric oxide, probably derived from induced nitric oxide synthase, contributes significantly to the delayed cardioprotection afforded by cardiac pacing.
Keywords: Cardiac pacing, ischaemic preconditioning, nitric oxide, aminoguanidine, delayed cardioprotection, nitric oxide synthase
Introduction
Rapid cardiac pacing in dogs results in both an immediate (Végh et al., 1991) and delayed (Végh et al., 1994; Kaszala et al., 1996) protection against the early life-threatening arrhythmias that result from acute coronary artery occlusion. It has been argued (Kaszala et al., 1996) that this is a form of ischemic preconditioning. The mechanisms of this delayed protection are unknown but there is some evidence that cardiac pacing (and exercise) may promote the formation of nitric oxide (NO) through nitric oxide synthase gene expression in endothelial cells (Zhao et al., 1997) and in cardiac myocytes (reviewed by Parratt & Végh, 1997). Further, there is recent evidence in rabbits that when the preconditioning stimulus consists of brief periods of coronary artery occlusion there is delayed protection against myocardial stunning and that this is also triggered by the generation of NO (Bolli et al., 1997a, 1997b). Under these conditions there is also enhanced NO formation by the canine myocardium, as demonstrated by elevated nitrite and nitrate levels in coronary sinus blood 24 h after a 10 min preconditioning coronary artery occlusion (Kim et al., 1997).
Although there is a good deal of evidence that, at least in dogs, NO is a key mediator in the antiarrhythmic effects of the early (‘classical') phase of ischaemic preconditioning (Végh et al., 1992a), there have been no studies concerned with the possible role of NO in mediating the delayed antiarrhythmic effects of ischaemic preconditioning, or of cardiac pacing. The purpose of the present study was therefore to examine whether the induced form of nitric oxide synthase (iNOS) is involved in the delayed antiarrhythmic effects of cardiac pacing by examining the effects of aminoguanidine, a reasonably selective inhibitor of iNOS (Corbett et al., 1992; Griffiths et al., 1993; Misko et al., 1993; Kengatharan et al., 1996) in a canine model in which the preconditioning stimulus was right ventricular pacing 24 h prior to coronary artery occlusion (Végh et al., 1994; Kaszala et al., 1996).
Methods
Animals and pacing procedure
Forty-two mongrel dogs, of either sex and with a mean body weight in excess of 17 kg, were anaesthetized by the intravenous administration of sodium pentobarbitone and were allowed to breathe spontaneously. A Cordat F4 bipolar pacing electrode was inserted, via the right jugular vein, into the right ventricle; the correct placing of this electrode was confirmed by recording the endocardial electrocardiogram. Blood pressure was monitored from the left carotid artery. Thirteen of these dogs were then paced for four 5 min periods, at a rate of 220 beats min−1 with 5 min resting intervals between the pacing stimuli (Végh et al., 1994; Kaszala et al., 1996). The twelve control (sham operated) dogs were those in which the pacing electrode was introduced into the right ventricle, left for the same period of time but these dogs were not paced. The remaining nine dogs, which were also paced as described above, were then given aminoguanidine, as the hemisulphate salt (Sigma; 50 mg kg−1 i.v.) 0.5 h prior to the coronary artery occlusion i.e. 24 h after the pacing stimulus. A further eight dogs were not paced but were given aminoguanidine at the same dose, 0.5 h prior to coronary occlusion. The dose of aminoguanidine has been selected on the basis of those studies performed in dogs which aimed to investigate the effect of aminoguanidine on the inducible nitric oxide synthase (Tárnoky et al., 1996; Numata et al., 1998). The experimental protocol is shown in Figure 1.
Figure 1.
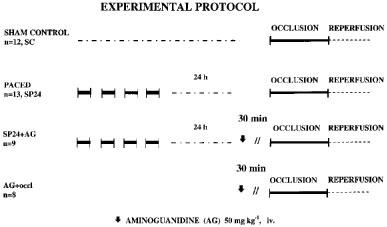
The experimental protocol outlining the procedures observed for the four groups of anaesthetized dogs.
There were no significant differences between the four groups in respect to body weight (sham controls, 24±3 kg; paced dogs, 23±2 kg; paced dogs given aminoguanidine, 27±1 kg; and non-paced dogs given the drug, 25±4 kg).
Haemodynamic measurements
Those dogs subjected to pacing, as well as the sham-operated controls, were allowed to recover from the anaesthetic and 20–24 h later were re-anaesthetized with a mixture of chloralose and urethane (60 and 200 mg kg−1 i.v., respectively), ventilated with room air, thoracotomized and subjected to a 25 min occlusion of the left anterior descending (LAD) coronary artery as previously described (Végh et al., 1992b; Kaszala et al., 1996). Temperature was recorded from the mid-oesphagus and maintained at 37±0.5°C by means of a heating pad. Catheters were inserted into the right femoral artery (for monitoring blood pressure), the left ventricle (for the measurement of left ventricular pressure and dP/dt) and the right femoral vein for drug and anaesthetic administration. Epicardial ST-segment elevation and the degree of inhomogeneity of electrical activation were measured in the area supplied by the LAD coronary artery by means of a composite electrode (Végh et al., 1992b). This electrode gives a summarized recording of R-waves from 30 epicardial measuring points. In the normal, well perfused and oxygenated, myocardium there is a single large spike because all sites are activated almost simultaneously. Following coronary occlusion, however, widening and fractionation of this summarized R-wave occurs since, because of the inhomogeneity of conduction in the ischaemic myocardium, fibres are not simultaneously activated. This inhomogeneity of conduction is expressed in the greatest delay in activation (in ms) within the ischaemic area underlying the composite electrode. All these parameters, together with a standard limb lead electrocardiogram, were recorded on a Graphtec Thermal Array Recorder (Hugo Sachs Electronics, Germany).
Assessment of arrhythmias and area at risk
Ventricular arrhythmias during coronary artery occlusion and following reperfusion were assessed as previously described (Végh et al., 1992b; Kaszala et al., 1996). In brief, the total number of ventricular premature beats (VPBs), the incidence and number of episodes of ventricular tachycardia (VT; defined as a run of four or more ventricular premature beats at a rate faster than the resting heart rate), and the incidence of ventricular fibrillation (VF) were assessed. At the end of the 25 min occlusion period the myocardium was rapidly reperfused. Following reperfusion, ventricular arrhythmias were not assessed in detail but VF occurred in almost all the control, unpaced dogs; those dogs were pronounced as survivors if they were still alive, and predominantly in sinus rhythm, 10 min after reperfusion.
The risk area following coronary artery occlusion was assessed in each dog at the end of the experiment by injecting patent blue V dye into the re-occluded coronary artery, and was expressed as a percentage of the left ventricular wall together with the septum (Kaszala et al., 1996).
Statistical evaluation
All data are expressed as means±s.e.mean and the differences between means were compared by analysis of variance (ANOVA for repeated measures) or the Student's t-test as appropriate. A one-way ANOVA was undertaken to determine whether or not there were significant haemodynamic differences between the groups. Ventricular premature beats were compared by using the Mann-Whitney Rank Sum test, and the incidence of arrhythmias was compared using the Fisher Exact test. Differences between groups were considered significant when P<0.05.
Results
Haemodynamic effects of aminoguanidine
The administration of aminoguanidine, when given 0.5 h prior to coronary artery occlusion, resulted in transient increases in arterial pressure (maximal at 5 min) and a reduction in heart rate (Table 1). Both blood pressure and heart rate, however, had started to return to the initial levels (i.e. to those before injection) by the time the coronary artery was occluded.
Table 1.
Haemodynamic effects of aminoguanidine (50 mg kg−1 i.v.) in dogs paced 24 h previously and in unpaced dogs

Effects of coronary artery occlusion
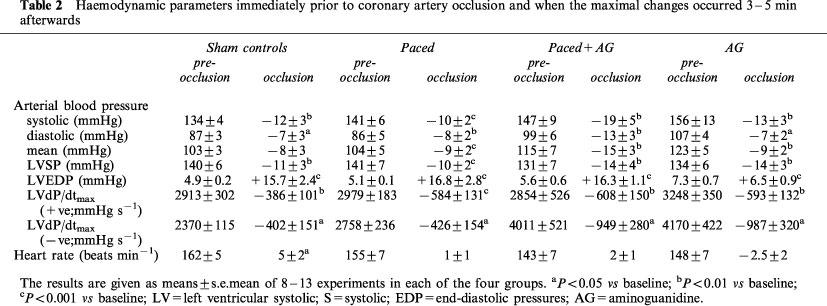
The haemodynamic effects of coronary artery occlusion were similar in control (sham operated) and paced dogs and are summarized in Table 2. In each of the groups there were significant reductions in arterial pressure and in LVdP/dt and marked increases in LVEDP. These changes are similar to those previously reported for both preconditioned (paced) and control dogs (e.g. Kaszala et al., 1996) and there was no marked difference between the haemodynamic effects of coronary artery occlusion in the controls, in the paced dogs and in the dogs given aminoguanidine 0.5 h prior to coronary occlusion (Table 2).
Table 2.
Haemodynamic parameters immediately prior to coronary artery occlusion and when the maximal changes occurred 3–5 min afterwards
Ventricular arrhythmias during coronary artery occlusion
In control (sham-operated) dogs coronary artery occlusion led to marked ventricular ectopic activity; the distribution of these ventricular premature beats is shown in Figure 2. There were 330±110 ventricular premature beats over the 25 min occlusion period, and a mean of 6.4±3.6 episodes of VT per dog (Figure 3). All but three of the 12 control dogs exhibited VT at sometime during the occlusion period and seven of the 12 dogs fibrillated during occlusion (Figure 2). Three of the remaining five dogs fibrillated almost immediately on reperfusion. There were thus two survivors from the combined ischaemia-reperfusion insult in these 12 sham-operated, control dogs (i.e. a survival of 17%).
Figure 2.
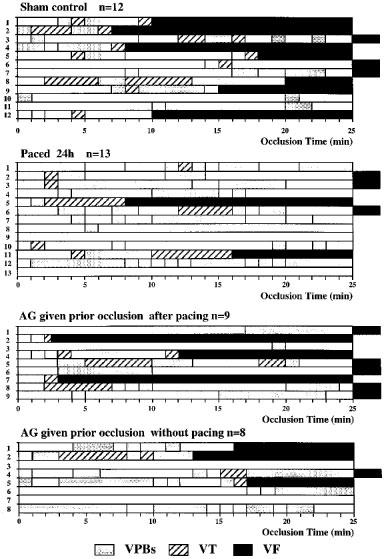
The distribution of ventricular arrhythmias; ventricular premature beats (VPBs), ventricular tachycardia (VT) and ventricular fibrillation (VF) during a 25 min occlusion of the left anterior descending coronary artery in anaesthetized dogs; this was followed by rapid reperfusion. Seven of the 12 sham-operated controls fibrillated during the occlusion and all but three had periods of ventricular tachycardia. Only two of these controls survived the combined ischaemia-reperfusion insult. These arrhythmias were markedly suppressed in the paced 24 h group; thus only two dogs fibrillated during the occlusion period and there was a 62% survival from the ischaemia-reperfusion insult. In the paced dogs given aminoguanidine (AG) only one of the nine dogs (i.e. 11%) survived ischaemia and reperfusion. There was no evidence that AG given prior to occlusion in unpaced dogs was pro-arrhythmic.
Figure 3.

The total number of ventricular premature beats (VPBs) and the number of episodes of ventricular tachycardia (VT) during coronary artery occlusion in sham control dogs (SC), in dogs paced 24 h previously (SP24), in paced dogs given aminoguanidine (AG) prior to occlusion (SP24+AG) and in unpaced dogs also given AG (AG+occl.). *P<0.05 compared to sham controls.
The pacing stimulus markedly reduced ventricular ectopic activity when the coronary artery was occluded 24 h later (Figure 3). There was a mean of only 70±28 ventricular premature beats during the occlusion period with only seven of the 13 dogs exhibiting VT (with a mean of 2.2±1.9 episodes per dog). Only two dogs fibrillated during occlusion (15%; P<0.05 compared to controls) and eight of the remaining dogs also survived reperfusion (Figure 2), giving a survival rate of 62% (P<0.05 compared to controls).
In the paced dogs that were given aminoguanidine prior to coronary artery occlusion there were 285±120 ventricular ectopic beats and there were 5.0±3.2 episodes of VT (Figure 3). The incidences of VT (56%) and of VF (33%) during the occlusion period were not significantly different to the sham controls (75 and 58%, respectively) and somewhat higher than in the paced dogs that had not been given aminoguanidine (incidences of VT and VF 46 and 15%, respectively). Ventricular fibrillation resulting from ischaemia and reperfusion was 89% in the aminoguanidine-treated dogs (Figure 4); this was significantly higher than in the paced dogs (38%; P<0.05) and not different to that in the controls (83%). Thus, 1/9 of the paced dogs given aminoguanidine survived the occlusion-reperfusion insult in contrast to 8/13 of the paced dogs and 2/12 of the sham controls (Figure 4). To determine whether aminoguanidine itself modified arrhythmia severity, eight sham-operated dogs, that had not been paced, were given the drug 0.5 h prior to the coronary artery occlusion. The number of ventricular premature beats (139±68) was somewhat less than in the controls and 3/8 of these dogs fibrillated during occlusion (38%). Four of the remaining five dogs survived reperfusion, giving a survival rate of 50% from the combined occlusion-reperfusion insult (Figure 4).
Figure 4.
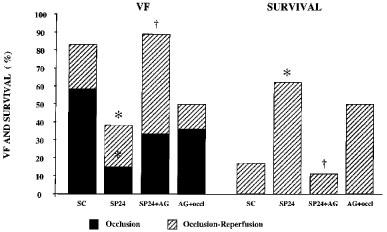
The incidence of ventricular fibrillation during coronary artery occlusion and following reperfusion at the end of the 25 min occlusion in control dogs (SC), in dogs subjected to right ventricular pacing 24 h prior to the occlusion (SP24) and in paced (SP24+AG) and non-paced (AG+occl.) dogs given aminoguanidine (AG) 0.5 h prior to coronary artery occlusion. Also shown in the right hand panels is survival from the combined ischaemia-reperfusion insult. The marked protection against ventricular fibrillation during both occlusion and reperfusion which results from cardiac pacing is markedly attenuated by the prior administration of aminoguanidine. *P<0.05 compared to sham controls; †P<0.05 compared to paced dogs given aminoguanidine.
There was no significant difference in the area at risk between the paced dogs given aminoguanidine (37±1%), the sham controls (37±3%), the paced dogs without aminoguanidine (43±1%) and the non-paced dogs given aminoguanidine (37±3%).
Changes in ischaemia severity following coronary artery occlusion
This was assessed in two ways; by epicardial ST-segment mapping and by changes in the degree of electrical inhomogeneity within the ischaemic area. The changes following coronary artery occlusion are illustrated in Figure 5 (for ST-segment changes) and Figure 6 (for changes in the degree of electrical inhomogeneity). They demonstrate that in the paced dogs ST-segment changes were less than in the controls and that this was largely reversed in those paced dogs given aminoguanidine prior to occlusion. Changes in the degree of electrical inhomogeneity were also less in paced dogs compared to controls and, again, this was reversed by aminoguanidine.
Figure 5.
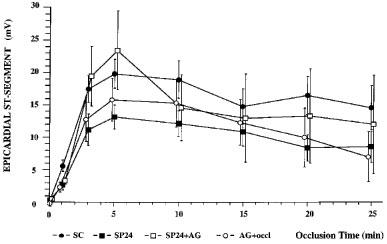
Changes in ST-segment elevation recorded from epicardial electrodes during a 25 min occlusion of the left anterior descending coronary artery in control dogs, dogs paced 24 h previously, and in dogs given aminoguanidine (50 mg kg−1) prior to the coronary artery occlusion with or without pacing. Pacing decreased ST-segment elevation recorded over the ischaemic area; this was markedly attenuated by the prior administration of aminoguanidine.
Figure 6.
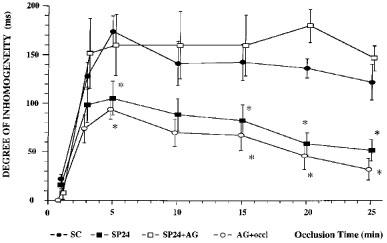
Changes in the inhomogeneity of electrical activation within the ischaemic area in control dogs, dogs paced 24 h previously, and in dogs given aminoguanidine (50 mg kg−1) prior to the coronary artery occlusion with or without pacing. Cardiac pacing reduced the severity of the changes in inhomogeneity; this was not seen in those paced dogs given aminoguanidine. *P<0.05 compared to sham controls.
Discussion
These studies confirm our previous findings (Végh et al., 1994; Kaszala et al., 1996) that right ventricular pacing markedly reduces the severity of arrhythmias that occur when a major coronary artery is occluded 24 h later. For example, only two of the paced dogs succumbed in ventricular fibrillation during the occlusion period; this incidence of VF during occlusion (15%) is similar to that observed by Kaszala et al. (1996) in a similar group of paced dogs. Other indices of ischaemia severity were also reduced by cardiac pacing, e.g. epicardial ST-segment changes mapped over the ischaemic area and delayed conduction, as assessed by changes in the degree of electrical inhomogeneity within the ischaemic area. The time course of this protection (absent 6 h after pacing, present 24 h after pacing but lost after 48 and 72 h; Kaszala et al., 1996) suggests protein induction, and the fact that the protection at 24 h is not seen in dogs treated with dexamethasone (Végh et al., 1994) suggests the involvement of induced nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), or both.
The present experiments suggest that NO is involved in this protection, since the administration of aminoguanidine attenuated the protection afforded by cardiac pacing. There were more ventricular premature beats and episodes of VT following coronary artery occlusion, and a higher incidence of VF and a significantly lower rate of survival following reperfusion at the end of the occlusion period. Further, the reduction in other indices of ischaemia severity (epicardial ST-segment elevation; changes in the degree of inhomogeneity of electrical activation within the ischaemic area), which are markedly reduced by cardiac pacing, were not seen in those paced dogs given aminoguanidine prior to coronary artery occlusion.
The attenuation of the protective effects of pacing by aminoguanidine could conceivably be due to a pro-arrhythmic effect of the drug, especially as this drug has some effect on cNOS, as evidenced by the studies of LÁSZLÓ et al. (1995) in rats. In fact, aminoguanidine did not modify significantly arrhythmia severity during coronary artery occlusion and indeed tended to increase, rather than decrease, survival from the combined ischaemia-reperfusion insult (Figure 4). This was perhaps as a result of the less severe ischaemic changes following coronary artery occlusion as suggested by the less marked changes in epicardial ST-segment (Figure 5) and inhomogeneity of electrical activation (Figure 6).
Most workers (Corbett et al., 1992; Griffiths et al., 1993; Misko et al., 1993; Kengatharan et al., 1996) have provided evidence that aminoguanidine is a rather selective inhibitor of iNOS. Aminoguanidine, although in larger doses than those used in the present study, can inhibit other enzymes, such as catalase (Ou & Wolff, 1993) and histaminase (Kusche et al., 1977). In the present study aminoguanidine administration resulted in a short-lasting increase in systemic arterial blood pressure and a reduction in heart rate, presumably reflex in origin. This suggests that nitric oxide derived from the constitutive synthase (cNOS) might also have been inhibited. However, the blood pressure changes were not nearly as marked as those following administration of unselective inhibitors of the L-arginine nitric oxide pathway, such as NG-nitro-L-arginine methyl ester. When such inhibitors are given under similar conditions to those described in the present study there are marked and sustained increases in systemic arterial blood pressure (Végh et al., 1992a). It is not possible, therefore, to state categorically that it is NO derived from the induced enzyme which is responsible for the delayed protection; it could be derived from the enhanced synthesis of NO through cNOS in coronary vascular endothelium. In a recent study, although in a quite different model of ischaemic preconditioning (conscious rabbits; recovery of contractile function following a period of ischaemia and reperfusion), Bolli and his colleagues (1997b) found that three different NOS inhibitors (aminoguanidine and S-methylisothiourea sulphate, both of which are relatively selective for iNOS, and Nω-nitro-L-arginine which is non-selective) all exacerbated stunning in the preconditioned but not in the normal myocardium. The authors suggest that nitric oxide has a dual role in late preconditioning acting both as a trigger and as a mediator. We had earlier suggested a similar hypothesis to account for the antiarrhythmic effects of preconditioning, induced by brief periods of coronary artery occlusion, during the first (classical) window of protection (Végh et al., 1992a). This concept has been recently reviewed (Parratt & Végh, 1996). Our concept (Végh et al., 1994; Végh & Parratt, 1998), that NO plays an important role in the delayed phase of protection has been confirmed by two, more recent studies, from other laboratories. Takano et al. (1998) and Imagawa et al. (1999) have found that in conscious rabbits, the induction of iNOS, following preconditioning by brief coronary artery occlusions, and the resultant formation of NO, play a role in the delayed cardioprotective (infarct size limiting) effect of preconditioning, since the protection was abolished by aminoguanidine.
These studies do not eliminate the possibility that other endogenous myocardial protective substances are also involved in the delayed protective effects of cardiac pacing and of ischaemic preconditioning induced by brief periods of coronary artery occlusion. This possibility is suggested by the fact that whereas aminoguanidine only attenuated the protective delayed effects of pacing, dexamethasone abolished this protection (Végh et al., 1994). It remains a possibility that protective prostanoids, such as prostacyclin, which are also profoundly antiarrhythmic (Parratt, 1989), are also involved in the delayed antiarrhythmic effects of pacing-induced preconditioning. This possibility is at present under investigation.
Acknowledgments
This work was supported by a collaborative grant from the British Council, and the Hungarian Ministry of Culture and Education, by the European Union (BIOMED II, Grant No. BMH4-CT96-0979), by a Scientific Network Grant from the European Union (CIPA CT92 4009) and by the Hungarian State Government (OTKA).
Abbreviations
- AG
aminoguanidine
- NO
nitric oxide
- cNOS
constitutive nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- LAD
left anterior descending coronary artery
- VPBs
ventricular premature beats
- VT
ventricular tachycardia
- VF
ventricular fibrillation
References
- BOLLI R., BHATTI Z.A., TANG X.-L., QIU Y., ZHANG Q., GUO Y., JADOON A.K. Evidence that late preconditioning against myocardial stunning in conscious rabbits is triggered by the generation of nitric oxide. Circ. Res. 1997a;81:42–52. doi: 10.1161/01.res.81.1.42. [DOI] [PubMed] [Google Scholar]
- BOLLI R., MANCHIKALAPUDI S., TANG X.-L., TAKANO H., QIU Y., GUO Y., ZHANG Q., JADOON A.K. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Circ. Res. 1997b;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- CORBETT J.A., TILTON R.G., CHANG K., HASAN K.S., IDO Y., WANG J.L., SWEETLAND M.A., MANCASTER J.R., WILLIAMSON J.R., MCDANIEL M.L. Aminoguanidine, a novel inhibitor of nitric oxide formation, prevents diabetic vascular dysfunction. Diabetes. 1992;41:552–556. doi: 10.2337/diab.41.4.552. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS M.J.D., MESSENT M., MACALLISTER R.J., EVANS T.W. Aminoguanidine selectively inhibits inducible nitric oxide synthase. Br. J. Pharmacol. 1993;110:963–968. doi: 10.1111/j.1476-5381.1993.tb13907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMAGAWA J., YELLON D.M., BAXTER G.F. Pharmacological evidence that inducible nitric oxide synthase is a mediator of delayed preconditioning. Br. J. Pharmacol. 1999;126:701–708. doi: 10.1038/sj.bjp.0702368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KASZALA K., VÉGH Á., PAPP J. GY. &, PARRATT J.R. Time course of the protection against ischaemia and reperfusion-induced ventricular arrhythmias resulting from brief periods of cardiac pacing. J. Mol. Cell. Cardiol. 1996;28:2085–2095. doi: 10.1006/jmcc.1996.0201. [DOI] [PubMed] [Google Scholar]
- KENGATHARAN K.M., DE KIMPE S.J., THIEMERMANN C. Role of nitric oxide in the circulatory failure and organ injury in a rodent model of Gram-positive shock. Br. J. Pharmacol. 1996;119:1411–1421. doi: 10.1111/j.1476-5381.1996.tb16053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM S.-J., GHALEH B., JUDEJ R.K., HUANG C.-H., HINTZE T.H., VATNER S.F. Delayed enhanced nitric oxide-mediated coronary vasodilation following brief ischaemia and prolonged reperfusion in conscious dogs. Circ. Res. 1997;81:53–59. doi: 10.1161/01.res.81.1.53. [DOI] [PubMed] [Google Scholar]
- KUSCHE J., STAHLKNECHT C.D., LORENZ W., REICHERT G., RICHTER H. Diamine oxidase activity and histamine release in dogs following acute mesenteric artery occlusion. Agents Actions. 1977;7:81–84. doi: 10.1007/BF01964885. [DOI] [PubMed] [Google Scholar]
- LÁSZLÓ F., EVANS S.M., WHITTLE B.J.R. Aminoguanidine inhibits both constitutional and inducible nitric oxide synthase isoforms in rat intestinal microvasculature in vivo. Eur. J. Pharmacol. 1995;272:169–175. doi: 10.1016/0014-2999(94)00637-m. [DOI] [PubMed] [Google Scholar]
- MISKO T.P., MOORE W.M., KASTEN T.P., NICKOLS G.A., CORBETT J.A., TILTON R.G., MCDANIEL M.L., WILLIAMSON J.R., CURRIE M.G. Selective inhibition of the inducible nitric oxide by aminoguanidine. Eur. J. Pharmacol. 1993;233:119–125. doi: 10.1016/0014-2999(93)90357-n. [DOI] [PubMed] [Google Scholar]
- NUMATA M., SUZUKI S., MIYAZAWA N., MIYASHITA A., NAGASHIMA Y., INOUE S., KANEKO T., OKUBO T. Inhibition of inducible nitric oxide synthase prevents L-S-induced acute lung injury in dogs. J. Immunol. 1998;160:3031–3037. [PubMed] [Google Scholar]
- OU P., WOLFF P. Aminoguanidine: a drug proposed for prophylaxis in diabetes inhibits catalase and generates hydrogene peroxide in vitro. Biochem. Pharmacol. 1993;48:1139–1144. doi: 10.1016/0006-2952(93)90461-5. [DOI] [PubMed] [Google Scholar]
- PARRATT J.R.Eicosanoids and arrhythmogenesis 1989Springer, Berlin; 569–589.In: Vaughan Williams, E.M. (Ed.). Antiarrhythmic Drugs(Handbook of Experimental Pharmacology volume 89) [Google Scholar]
- PARRATT J.R., VÉGH Á. Endothelial cells, nitric oxide and ischaemic preconditioning. Basic Res. Cardiol. 1996;91:27–30. doi: 10.1007/BF00788857. [DOI] [PubMed] [Google Scholar]
- PARRATT J.R., VÉGH Á. Delayed protection against ventricular arrhythmias by cardiac pacing. Heart. 1997;78:423–425. doi: 10.1136/hrt.78.5.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKANO H., MANCHIKALAPUDI S., TANG X.-L., QIU Y., RIZVI A., JADOON A.K., ZHANG Q., BOLLI R. Nitric oxide synthase is the mediator of the late preconditioning against myocardial infarction in conscious rabbits. Circulation. 1998;98:441–449. doi: 10.1161/01.cir.98.5.441. [DOI] [PubMed] [Google Scholar]
- TÁRNOKY K., KASZAKI J., SZALAY L., BOROS M., NAGY S. Comparative study of the circulatory effects of aminoguanidine and N-nitro-L-arginine in hyperdynamic endotoxemia. Acta. Physiol. Hung. 1996;84:157–170. [PubMed] [Google Scholar]
- VÉGH Á, , KOMORI S., SZEKERES L., PARRATT J.R. Antiarrhythmic effects of preconditioning in anaesthetised dogs and rats. Cardiovasc. Res. 1992b;26:487–495. doi: 10.1093/cvr/26.5.487. [DOI] [PubMed] [Google Scholar]
- VÉGH Á, , PAPP J. GY. &, PARRATT J.R. Prevention by dexamethasone of the marked antiarrhythmic effects of preconditioning induced 20 h after rapid cardiac pacing. Br. J. Pharmacol. 1994;113:1081–1082. doi: 10.1111/j.1476-5381.1994.tb17104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VÉGH Á. &, PARRATT J.R.Delayed protection against ventricular arrhythmias Delayed Preconditioning and Adaptive Cardioprotection 1998Kluwer Academic Publishers, London; 63–91.Baxter, G.F. & Yellon, D.M. (Eds.) [Google Scholar]
- VÉGH Á., SZEKERES L., PARRATT J.R. Transient ischaemia induced by rapid cardiac pacing results in myocardial preconditioning. Cardiovasc. Res. 1991;25:1051–1053. doi: 10.1093/cvr/25.12.1051. [DOI] [PubMed] [Google Scholar]
- VÉGH Á., SZEKERES L., PARRATT J.R. Preconditioning of the ischaemic myocardium: involvement of the L-arginine nitric oxide pathway. Br. J. Pharmacol. 1992a;107:648–652. doi: 10.1111/j.1476-5381.1992.tb14501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO G., ZHANG X., XU X., OCHOA M., HINTZE T.H. Short-term exercise training enhances reflex cholinergic nitric oxide-dependent coronary vasodilation in conscious dogs. Circ. Res. 1997;80:868–876. doi: 10.1161/01.res.80.6.868. [DOI] [PubMed] [Google Scholar]