

Abstract
Two in vivo models, in the rat, were used to investigate, in the presence of different substrates, the overall and net intestinal elimination of ciprofloxacin: an open-intestinal perfusion model and an intestinal loop model respectively.
In the presence of quinidine, verapamil and cyclosporin (substrates of the P-glycoprotein (P-gp)), plasma AUCs of ciprofloxacin were 1.5–2 fold increased, while biliary clearance (1.5–2 fold), intestinal overall and net clearances (2–4 fold and 1.5–8 fold respectively) decreased. The weak effect obtained with cyclosporin as compared to verapamil and especially quinidine, suggests, for ciprofloxacin, the existence of transport systems distinct from the P-gp, as the OCT1 transporter which could be inhibited by quinidine.
With cephalexin and azlocillin, two β-lactam antibiotics, plasma AUCs of ciprofloxacin increased and biliary and intestinal overall clearances decreased in a similar fashion (1.3–2 fold), suggesting the involvement of organic anion and/or cation transporters.
In the presence of structural analogues, the effect was dependent on the compound administered: Sparfloxacin had no effect on intestinal clearance of ciprofloxacin. In contrast, with pefloxacin, overall intestinal clearance of ciprofloxacin was decreased and net intestinal clearance increased.
The specificity of ciprofloxacin intestinal transport appears to be different from P-gp as outlined by the lack of competition with sparfloxacin, a P-gp substrate. Ciprofloxacin intestinal elimination seems to be mediated by organic anion and/or cation transporters and a mechanism sensitive to quinidine and verapamil.
Keywords: Ciprofloxacin, intestinal elimination, experimental models, P-glycoprotein, intestinal transporters
Introduction
Ciprofloxacin is mainly eliminated via the kidneys, in man as well as in the rat. However, intestinal secretion of ciprofloxacin in humans represents about 10% of its total elimination (Sörgel et al., 1989. In addition, in the case of chronic renal failure in humans, its intestinal secretion is increased and compensates partially for the reduced renal function (Rohwedder et al., 1990). The importance of intestinal elimination of ciprofloxacin has been confirmed in the rat using an ex vivo intestinal perfusion model (Rubinstein et al., 1994; Metz & Sörgel, 1990) as well as intestinal loops (Dautrey et al., 1995). This compound (Figure 1) has an acidic as well as a basic group and its isoelectric point is at a pH of 7.41 (Ross & Riley, 1994). In the blood, it is mainly found as a zwitterion (roughly 80%) which is one of the electrically neutral forms of the molecule but which also behaves as an anionic and a cationic form. These properties allow ciprofloxacin to cross physiological barriers by passive diffusion but also to use active transport mechanisms such as anion and/or cation transporters, the P-glycoprotein (P-gp) whose substrates are diverse structures, but which are most often hydrophobic, amphiphilic or cationic compounds (Sharom, 1997). The mechanisms implicated in passaging the intestinal barrier for ciprofloxacin in particular and fluoroquinolones in general, have not been completely elucidated. Griffiths et al. (1993; 1994) studied the passage of different fluoroquinolones, including ciprofloxacin, through Caco-2 cells in culture. They showed the presence of a net secretory flux and suggested the existence of active secretory mechanisms common to all fluoroquinolones. Using the same model, Cormet et al. (1995) and Cormet-Boyaka et al. (1998) have demonstrated a net secretory flux of sparfloxacin which was modulated by verapamil, which increased sparfloxacin transport from the apical to the basal side, suggesting the intervention of a multi-drug resistance-like mechanism. The study of intestinal elimination of ofloxacin enantiomers (Rabbaa et al., 1996), carried out in the rat using the ex vivo intestinal perfusion model, showed that intestinal clearance of the R-(+) form is greater than that of the S-(−) form (0.30±0.03 ml min−1 versus 0.23±0.03 ml min−1). Furthermore, the presence of ciprofloxacin, verapamil or quinidine (the latter two substrates block P-gp) decreases the intestinal clearance of the two ofloxacin enantiomers.
Figure 1.
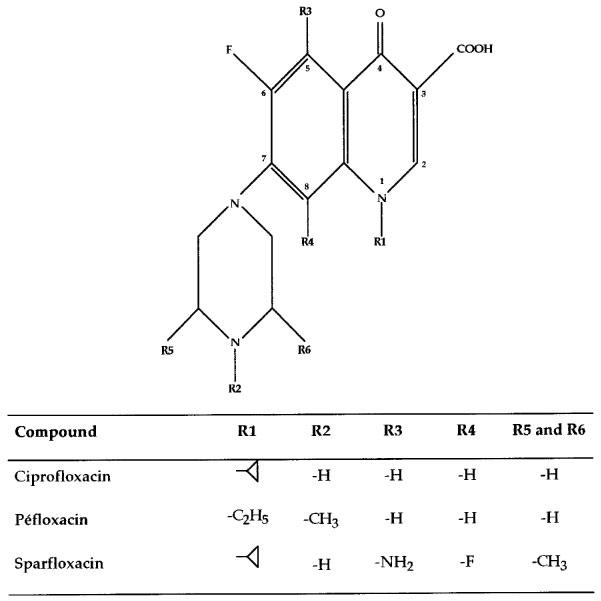
Chemical structures of ciprofloxacin, pefloxacin and sparfloxacin.
The different studies carried out on Caco-2 cells in culture or on the in vivo intestinal perfusion experimental models provide evidence of the existence of intestinal secretory mechanisms common to some fluoroquinolones and implicating P-gp-like transporters.
The aim of the present study was thus to determine the mechanisms implicated in the intestinal secretion of ciprofloxacin using two experimental models in the rat. The first model, using an intestinal loop, allowed us to quantify intestinal secretion taking into account the reabsorption processes. The second model was an open-intestinal perfusion model with a sufficient perfusion flow (0.6 ml min−1) in order to keep the reabsorption phenomenon negligible (Dautrey et al., 1995). Using these models, we were able to calculate respectively net and overall intestinal clearances as well as biliary clearance (bile was collected throughout the experiment). The influence of different competitors of intestinal transport systems (P-glycoprotein, anionic and cationic transporters) on net and overall intestinal elimination of ciprofloxacin were studied first. Then, other fluoroquinolones with different physiochemical properties (Figure 1) were tested in association with ciprofloxacin in order to demonstrate the existence of common mechanisms for intestinal secretion.
Methods
Drugs and chemicals
Ciprofloxacin, pefloxacin and sparfloxacin were generous gifts from Bayer (Puteau, France), Roger Belon (Montrouge, France) and Rhône Poulenc-Rohrer laboratories (Antony, France). Quinidine, cephalexin, azlocillin, verapamil were purchased from Sigma (Saint Quentin Fallavier, France). Cyclosporin was obtained from the injectable form used clinically (Sandimmun® 5 mg ml−1, Novartis laboratories, Rueil Malmaison, France). Monopotassium phosphate, employed in the preparation of solutions used for intestinal perfusion was of analytical grade (Prolabo, Paris, France). Isotonic saline, used for parenteral perfusions and for the preparation of injectable solutions of ciprofloxacin and of the different compounds tested were provided by the ‘Pharmacie Centrale des Hôpitaux' (Paris, France). The reagents used for the chromatographic analyses were of analytical grade: methanol (Carlo-Erba, Milan, Italy), Pic B7® (Waters, Saint Quentin en Yveline, France), triethylamine (Prolabo, Paris, France), formic acid and phosphate monopotassium phosphate (KH2PO4) (Prolabo, Paris, France).
Animals
The animals used in this study were male Sprague-Dawley rats (Charles-River, Saint Aubin les Elbeufs, France), weighing between 250 and 300 g. All the investigations were performed in accordance with the European Regulations for the use of laboratory animals.
Intestinal preparation
The animals were anaesthetized using urethane (ethylcarbamate, Prolabo, Paris, France) at a dose of 1.5 g kg−1 administered i.p. and placed under a heating lamp to ensure constant body temperature. A polyurethane catheter (25×0.9 mm, Insyte® Vialon, Becton-Dickinson Laboratories, Meylan, France) was placed in the carotid artery. A polyethylene tube (internal diameter 0.56 mm) was introduced into the jugular vein.
The abdominal wall was opened via the linea alba in order to gain access to the small intestine. The common bile duct was isolated and after ligaturing the intestinal extremity, a polyethylene tube (internal diameter 0.3 mm) was inserted for bile collection. A 30 cm length of intestine starting from the Treizt ligament was exposed. A first suture was placed 10 cm from the pylorus, taking care to avoid the mesenteric veins, while a second one was placed 20–30 cm distal to the first one. A polyurethane catheter (25×0.9 mm, Insyte® Vialon, Becton-Dickinson Laboratories, Meylan, France) was introduced, at the level of the first suture, onto which a tube, delivering intestinal perfusion fluid via a peristaltic pump is fixed (Minipuls 2, Gilson, Villiers le Bel, France). The perfusion liquid was maintained at 37°C using a thermostated bath (Polystat 22 S 86602, Bioblock Scientific, Illkirch, France). The liquid left via a polyethylene tube (internal diameter 2.15 mm) place at the end of the isolated intestinal segment. The abdomen was then covered with a compress soaked with physiological saline. The intestinal perfusion liquid was a 0.15 M monopotassium phosphate solution adjusted to a pH of 7.4. Before experiment, the intestinal segment was rinsed with intestinal perfusion liquid at a flow rate of 0.6 ml min−1 during 30 min.
To estimate the overall intestinal elimination, we used an open model based on the continuous perfusion of the isolated intestinal segment, with a constant flow rate maintained at 0.6 ml min−1 throughout the experiment.
The study of net intestinal secretion was carried out on an intestinal loop model. The animals were prepared as described above. The segment was ligatured at both ends and filled with perfusion liquid.
Protocol
Once the animals were prepared, the different compounds tested were administered through the jugular vein. The doses injected depended upon the different compounds and are indicated in Tables 1 and 2. After 15 min, ciprofloxacin was injected at a dose of 12.5 mg kg−1 intra-jugularly in 1 min. A blood sample (250 μl) was taken prior to injection to avoid interference during the measurements, the other samples were taken 2, 5, 10, 15, 30, 45, 60, 75, 90, 105 and 120 min after the end of the injection. Intrajugular perfusion of physiological saline at a flow rate of 3 ml h−1 using an electric syringe provided adequate hydration of the rats. Bile was collected at 15 min intervals. The perfusion liquid for the continuous perfusion model was collected at 15 min intervals. In the case of the model of intestinal loops, contents of the loop were collected at the end of the experiment (after 120 min) and the loop was rinsed with perfusion liquid.
Table 1.
Pharmacokinetic parameters of ciprofloxacin following parenteral administration, in the presence of competitors of intestinal transport systems and structural analogues for the overall intestinal elimination model (n=5 for each group)
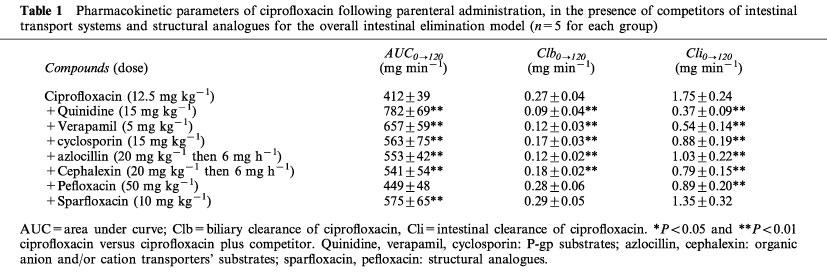
Table 2.
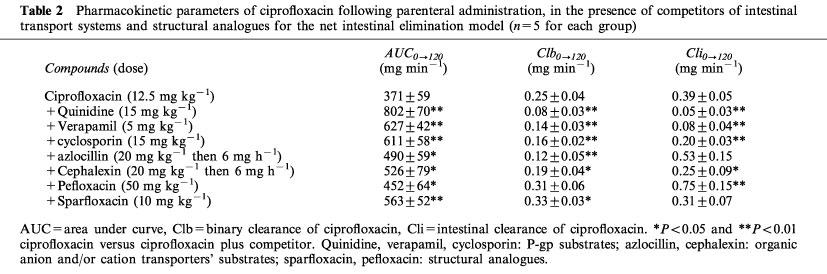
Pharmacokinetic parameters of ciprofloxacin following parenteral administration, in the presence of competitors of intestinal transport systems and structural analogues for the net intestinal elimination model (n=5 for each group)
Validation of the study models of intestinal secretion
The verification of the integrity of the intestinal mucosa was carried out using diethylenetriaminopentaacetate (Sn) 99mTc (TCK-6, Cis bio international, Gif-sur-Yvette, France). This tracer, due to its size, does not cross the intestinal carrier and can be used, in humans, to demonstrate the absence of lesions (Jian, 1983). Two groups of three rats each were studied to validate the models of the net and overall elimination. The animals were prepared as described above. An intrajugular injection of 300 mCi was carried out and the radioactivity of the intestinal content measured after 120 min. In the two groups the activity was less than 0.1% of the injected activity, irrespective of the model of intestinal elimination. The lack of passage of the tracer across the intestinal barrier demonstrated the integrity of the intestinal mucosal lining.
Based on these results, the duration of the experiment was fixed at 120 min to avoid eventual alteration in the intestinal mucosa and in the viability of the animals which might occur with longer experimentation times.
Analytical assay
The blood samples were centrifuged and the plasma collected and frozen at −20°C as were the other biological samples. The plasma samples (50 μl) were treated with a methanolic solution containing an internal standard (ofloxacin, Diamant laboratories) in order to precipitate proteins, were again centrifuged and the supernatant collected. Biliary samples (20 μl from each collected sample) were diluted 1/50 in a solution of monopotassium phosphate 0.15 M, pH 7.4, containing the internal standard. Intestinal perfusion liquid (500 μl from each collected sample) was diluted by half in a solution of 0.15 M monopotassium phosphate, pH 7.4 containing the internal standard. Measurements in different biological liquids were carried out by reversed-phase high-performance liquid chromatography coupled with fluorimetric detection (Rubenstein, 1994). The method was linear over the 0.1–40 mg l−1 concentration range. Intra- and inter-day coefficients of variation were less than 10%. The limit of detection was 0.03 mg l−1 while the limit of quantification was 0.1 mg l−1.
Pharmacokinetics and statistical analysis
The amount of ciprofloxacin eliminated intestinally was calculated by cumulating the quantities eliminated during the different time intervals. It is expressed per unit of intestinal area (obtained from the length and diameter of the intestinal segment studied). The quantity eliminated in bile is obtained in a similar manner and is expressed in mg. The area under the curve corresponding to plasma concentrations between 0 and 120 min (AUC0→120) was calculated using the trapezoidal method (Siphar®, SIMED, Créteil, France).
Net and overall intestinal clearances (μl min−1 cm−2) and biliary clearances (ml min−1) of ciprofloxacin were obtained from the ratio of the quantity eliminated between 0 and 120 min for each pathway respectively over AUC0→120.
The results are expressed as means±s.d. and the statistical analysis was performed using the Mann-Whitney non parametric test. The significance level was set at 5%.
Results
Analysis of plasma AUCs0→120 of ciprofloxacin (Tables 1 and 2) showed no significant differences between the two intestinal elimination study models, either in the presence or in the absence of competitor or structural analogues. However, the plasma AUCs0→120 of ciprofloxacin were significantly increased in the presence of different competitors (P<0.05). This was observed in both models studied. Among the structural analogues, only pefloxacin did not significantly increase plasma AUCs0→120 of ciprofloxacin in the net intestinal elimination model.
Biliary clearances of ciprofloxacin (Tables 1 and 2) measured in the presence of competitors in the two models studies reflect the increased plasma AUCs0→120 and the variation in biliary elimination. Thus, biliary clearance of ciprofloxacin was significantly decreased in the presence of the competitors tested, in both models. The concomitant administration of structural analogues gave variable results: in the model of net intestinal elimination pefloxacin did not modify biliary clearance by ciprofloxacin whereas sparfloxacin did.
Net and overall intestinal clearances of ciprofloxacin (Tables 1 and 2) in the presence of the different competitors tested are the reflection of variations in intestinal elimination and of the increase in plasma AUCs0→120. All the competitors as well as the structural analogues with the exception of sparfloxacin significantly decrease overall intestinal clearance of ciprofloxacin. The greatest effect was obtained with quinidine and verapamil which decreased overall intestinal clearance of ciprofloxacin by 79 and 69% respectively. Net intestinal clearance was not modified in the presence of azlocillin and sparfloxacin. Only pefloxacin increased the ciprofloxacin net intestinal clearance, whereas the other compounds decreased it, notably quinidine and verapamil which reduced it by 87% and 79% respectively.
Discussion
The results obtained in plasma, bile or intestine show that substrates of specific transporters or structural analogues of ciprofloxacin are able to modify its pharmacokinetics in vivo. The findings confirm the important role played by transport mechanisms in membrane passages of ciprofloxacin at the biliary or intestinal levels.
The existence of such transport systems has been demonstrated in numerous tissues and notably in organs which have a secretory function, such as the kidney, bile ducts and the intestine. They are localized at the apical membrane of cells like P-gp (Tsui & Tamai, 1996) or at the basolateral membrane like OCT-1 (Zhang et al., 1998). For this reason, in order to study the mechanism(s) involved in its intestinal elimination, ciprofloxacin was administered parenterally at a dose of 12.5 mg kg−1 (a dose which is compatible with a linearity of biliary and intestinal kinetics [Dautrey et al., 1999]) alone, and in the presence of different compounds potentially capable of interacting with it. These compounds were administered parenterally 15 min before ciprofloxacin in order to optimize diffusion into the different organs. They were structural analogues (pefloxacin, sparfloxacin), P-gp substrates (verapamil, quinidine and cyclosporin A) (Tsui & Tamai, 1996) or betalactams: cephalexin whose intestinal absorption depends in the transporter of di- and tripeptides PepT1 (Cormet et al., 1997) and azlocillin which is known to decrease renal and extrarenal clearances of ciprofloxacin in humans (Barrière et al., 1990). The action of the different compounds was studied through plasma AUC, biliary clearance as well as net and overall intestinal clearances of ciprofloxacin. The latter reflects the variations in plasma AUCs and in the quantities of ciprofloxacin eliminated by the intestine. The distinction between overall and net intestinal clearance allows one to determine whether or not the compounds tested had an effect on intestinal reabsorption of ciprofloxacin (Figure 2).
Figure 2.
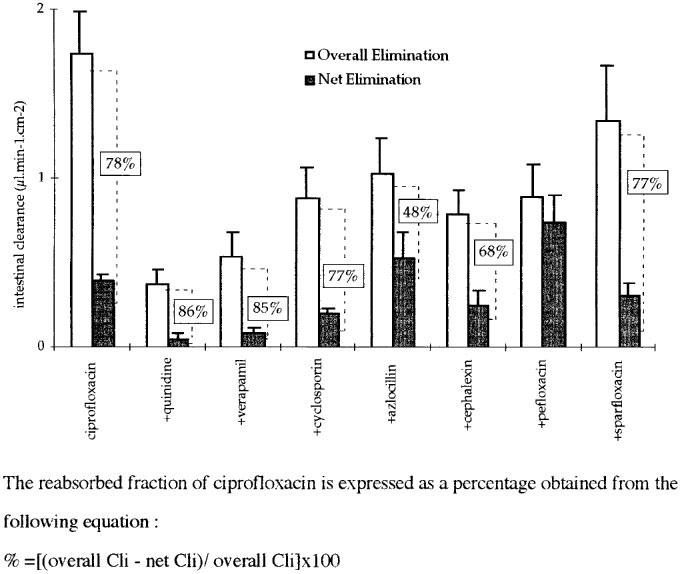
Intestinal clearances (Cli) of ciprofloxacin alone and in the presence of competitors.
In the presence of quinidine, verapamil and cyclosporin, plasma AUCs of ciprofloxacin are increased by a factor of 1.5 to 2, while biliary clearance is decreased in a similar fashion. Overall intestinal clearance is divided by a factor between 2 and 4 depending on the compound administered, whereas for net intestinal clearance the factor lies between 1.5 and 8. The weak effect obtained with cyclosporin, as compared with verapamil and especially quinidine, could result mainly from its vasoconstrictor effect (Mason, 1989) and suggests the existence of other transport systems, distinct from the P-gp for ciprofloxacin. These results support those obtained by Cavet et al. (1997) who showed, using cell culture of the Caco-2 and MDCK lines (epithelial cells of canine renal tubules) that P-gp is not involved in the efflux of ciprofloxacin. Furthermore, a study carried out in our laboratory showed that the accumulation and efflux of ciprofloxacin are identical in wild type LLC-PK1 cells and in the same cells transfected by the human mdr-1 gene (unpublished data). Other arguments are in favour of the existence of other carriers distinct from P-gp: (i) inhibition of P-gp by cyclosporin is greater than that of quinidine and verapamil (Jetté et al., 1995); (ii) quinidine decreases intestinal elimination of cationic substrates by a mechanism distinct from P-gp like inhibition of the OCT1 transporter (Bair et al., 1992; Emi et al., 1998; Nagel et al., 1997) and (iii) a recent study (Cavet et al., 1997) showed that ciprofloxacin is secreted in Caco-2 cells by an anionic transport system which is sensitive to cera and 4-4′diisothiocyanostilbene-2-2′-sulphonic acid (DIDS). This decrease in intestinal secretion of ciprofloxacin under the influence of DIDS was found in a study carried out in parallel on a model of a everted sac in vitro (Dautrey et al., 1998). However, the important action of quinidine on intestinal elimination of ciprofloxacin, observed in vivo, could not be reproduced with this model, the latter being diminished by only 20% suggesting the involvement of specific in vivo factors (Dautrey et al., 1998). The greater effect of verapamil and quinidine on net intestinal elimination with respect to overall intestinal elimination can be explained by an increased reabsorption of ciprofloxacin. Indeed, the inhibition of intestinal elimination may facilitate absorption of a compound by hindering the exit of the cell from the apical side directly following its entry on the same side such as described for substrates of P-gp (Emi et al., 1998).
Globally, these results point towards a preferential transport of ciprofloxacin by anionic and cationic carriers, which is confirmed by the inhibitory effects of cephalexin and of azlocillin on biliary and overall intestinal clearances of ciprofloxacin. Indeed, in the presence of cephalexin and azlocillin, plasma AUC of ciprofloxacin are increased by a 1.3 to 1.5 factor. By contrast biliary and overall intestinal clearances are decreased by a 1.3 to 2 and 1.7 to 2.2 factor respectively. Cephalexin, like ciprofloxacin, is an amphoteric molecule which at physiological pH is in the form of a zwitterion and an anion (Inui et al., 1985). It is secreted at the level of the renal tubules by anionic and cationic transport systems (Inui et al., 1985). It could thus compete with ciprofloxacin at both biliary and intestinal levels. Azlocillin is a ureidopenicillin found in anionic form at physiological pH which reduces urinary elimination of ciprofloxacin probably by competing at the level of tubular secretion (Barrière et al., 1990). The decrease in biliary and overall intestinal clearances of ciprofloxacin observed in the presence of these two betalactams could thus be explained by competition at the level of organic anion and/or cation transport systems.
Plasma AUC of ciprofloxacin in the presence of pefloxacin are little or not modified. By contrast, sparfloxacin increases them by about 40%. Biliary clearance of ciprofloxacin is unchanged in the presence of structural analogues with the exception of the model studying net intestinal elimination with sparfloxacin. In the intestine, sparfloxacin, which is demonstrated to be a substrate of the P-gp (Cormet-Boyaka et al., 1998) has no effect and pefloxacin decreases, by a factor of 2, overall intestinal elimination, and increases net intestinal elimination of ciprofloxacin by 90%. The results with pefloxacin confirm those of Griffiths et al. (1994). With respect to net intestinal clearance, its increase in the presence of pefloxacin suggests a competition at the level of ciprofloxacin reabsorption, which supports the decrease of the intracellular accumulation of ciprofloxacin in the presence of pefloxacin observed by Griffiths et al. (1994). The difference observed between the two fluoroquinolones tested may be explained by their different structures (Figure 1). Pefloxacin is methylated in position N4, which leads to a decreased pI of the molecule which becomes lower than 7 (Sörgel & Kinzig, 1993a). By contrast, sparfloxacin is not N-methylated and its pI is close to 7.4, as for ciprofloxacin (Sörgel & Kinzig, 1993a).
In conclusion, the results of this study demonstrate that the pharmacokinetics of ciprofloxacin are not mainly driven by passive diffusion. The diversity of the compounds studied and the differences in their effects in the pharmacokinetics of ciprofloxacin suggest the involvement of one or many active transport mechanisms at the intestinal, as well as at the biliary levels, in the rat. The pharmacokinetics of ciprofloxacin are more sensitive to the administration of substrates which compete with intestinal and biliary anion and cation transport systems than to specific inhibitors of P-gp such as cyclosporin. The use of anionic and/or cationic transporters has already been proposed with respect to renal secretion (Sörgel & Kinzig, 1993b). However, the important effect of quinidine could suggest the existence of a multi-drug resistant like transport system, with a high affinity for cationic substances as the OCT1 transporter. The active elimination mechanisms of ciprofloxacin are not completely elucidated. However their existence is confirmed by the modifications of ciprofloxacin pharmacokinetics by compounds such as quinidine or betalactams.
Abbreviations
- AUC
area under the curve
- Clb
biliary clearance
- Cli
intestinal clearance
- DIDS
4-4′diisothiocyanostilbene-2-2′sulphonic acid
- P-gp
P-glycoprotein
References
- BAIR C.-H., TANG M.-J., HUANG J.-D. Concentration-dependent exsorption of quinidine in the rat intestine. J. Pharm. Pharmacol. 1992;44:659–662. doi: 10.1111/j.2042-7158.1992.tb05489.x. [DOI] [PubMed] [Google Scholar]
- BARRIÈRE S.L., CATLIN D.H., ORLANDO P., NOE A., WAYNE FROST R. Alteration in the pharmacokinetic disposition of ciprofloxacin by simultaneous administration of azlocillin. Antimicrob. Agents Chemother. 1990;34:823–826. doi: 10.1128/aac.34.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAVET M.E., WEST M., SIMMONS N.L. Fluoroquinolone (ciprofloxacin) secretion by human intestinal epithelial (Caco-2) cells. Br. J. Pharmacol. 1997;121:1567–1578. doi: 10.1038/sj.bjp.0701302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORMET E., BARLIER A.M., HUNEAU J.F., RUBINSTEIN E., CARBON C., FARINOTTI R., TOMÉ D. Sparfloxacin secretion across Caco-2 cells involves a multidrug resistance-like mechanism. Drugs. 1995;29 suppl. 2:307–309. doi: 10.2165/00003495-199500492-00080. [DOI] [PubMed] [Google Scholar]
- CORMET E., HUNEAU J.F., BOURAS M., CARBON C., RUBENSTEIN E., TOMÉ D. Evidence for a passive diffusion mechanism for sparfloxacin uptake at the brush-border membrane of the human intestinal cell-line Caco-2. J. Pharm. Sci. 1997;86:33–36. doi: 10.1021/js960262s. [DOI] [PubMed] [Google Scholar]
- CORMET-BOYAKA E., HUNEAU J.-F., MORDRELLE A., BOYAKA P., CARBON C., RUBENSTEIN E., TOMÉ D. Secretion of sparfloxacin from the human intestinal Caco-2 cell-line is altered by P-glycoprotein inhibitors. Antimicrob. Agents Chemother. 1998;42:2207–2211. doi: 10.1128/aac.42.10.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAUTREY S., BERLIOZ F., VEAU C., ROZE C., LACOUR B., CARBON C., FARINOTTI R.Ciprofloxacin (CIP) is eliminated by rat intestine via a DIDS-sensitive, PGP-like transporter 1998San Diego, California, USA; 38th ICAAC, September 24–271998 [Google Scholar]
- DAUTREY S., RABBAA L., LAOUARI D., LACOUR B., CARBON C., FARINOTTI R. Influence of renal failure on the intestinal clearance of ciprofloxacin in rat. Antimicrob. Agents Chemother. 1999;43:678–680. doi: 10.1128/aac.43.3.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAUTREY S., RABBAA L., PETIET A., CARBON C., FARINOTTI R. Comparison of models for studying the intestinal elimination of ciprofloxacin in the rat. Drugs. 1995;49 suppl. 2:310–311. doi: 10.2165/00003495-199500492-00081. [DOI] [PubMed] [Google Scholar]
- EMI Y., TSUNASHIMA D., OGAWARA K.-I., HIGAKI K., KIMURA T. Role of P-glycoprotein as a secretory mechanism in quinidine absorption from rat small intestine. J. Pharm. Sci. 1998;87:295–299. doi: 10.1021/js970294v. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS N.M., HIRST B.H., SIMMONS N.L. Active secretion of the fluoroquinolone ciprofloxacin by human intestinal epithelial Caco-2 cell layers. Br. J. Pharmacol. 1993;108:575–576. doi: 10.1111/j.1476-5381.1993.tb12844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIFFITHS N.M., HIRST B.H., SIMMONS N.L. Active intestinal secretion of the fluoroquinolone antimicrobials ciprofloxacin, norofloxacin and pefloxacin; a common secretory pathway. J. Pharmacol. Exp. Ther. 1994;269:496–502. [PubMed] [Google Scholar]
- INUI K.-I., TAKANO M., OKANO T., HORI R. H+ gradient-dependent transport of aminocephalosporins in rat renal brush border membrane vesicles: role of H+/organic cation antiport system. J. Pharmacol. Exp. Ther. 1985;233:181–185. [PubMed] [Google Scholar]
- JETTÉ L., MURPHY G., LECLERC J.-M., BOLIVEAU R. Interaction of drugs with P-glycoprotein in brain capillaries. Biochem. Pharmacol. 1995;50:1701–1709. doi: 10.1016/0006-2952(95)02073-x. [DOI] [PubMed] [Google Scholar]
- JIAN R. Mise au point. Intérêt clinique de la scintigraphie dans l'exploration du tube digestif. Gastroenterol. Clin. Biol. 1983;7:693–701. [PubMed] [Google Scholar]
- MASON J. Pharmacology of cyclosporin (Sandimmune) VII. Pathophysiology and toxicology of cyclosporin in humans and in animals. Pharmacol. Rev. 1989;42:423–434. [PubMed] [Google Scholar]
- METZ R., SÖRGEL F.The gastrointestinal secretion of quinolones – Preliminary evaluation of in vivo animal model 1990Vancouver, Canada; 3rd International Symposium of New Quinolones, July 12–14 [Google Scholar]
- NAGEL G., VOLK C., FRIEDRICH T., ULZHEIMER J.C., BAMBERG E., KOEPSELL H. A reevaluation of substrate specificity of the rat cation transporter rOCT1. J. Biol. Chem. 1997;272:31953–31956. doi: 10.1074/jbc.272.51.31953. [DOI] [PubMed] [Google Scholar]
- RABBAA L., DAUTREY S., COLAS-LINHART N., CARBON C., FARINOTTI R. Intestinal elimination of ofloxacin enantiomers in the rat: evidence of a carrier-mediated process. Antimcrob. Agents Chemother. 1996;40:2126–2130. doi: 10.1128/aac.40.9.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROHWEDDER R., BERGAN T., THORSTEINSSON S.B., SCHOLL H. Transintestinal elimination of ciprofloxacin. Chemotherapy. 1990;36:77–84. doi: 10.1159/000238751. [DOI] [PubMed] [Google Scholar]
- ROSS D.L., RILEY C.M. Dissociation and complexation of the fluoroquinolone antimicrobials – an update. J. Pharm. Biomed. Anal. 1994;12:1325–1331. doi: 10.1016/0731-7085(94)00081-6. [DOI] [PubMed] [Google Scholar]
- RUBINSTEIN E., ST JULIEN L., RAMON J., DAUTREY S., FARINOTTI R., HUNEAU J.-F., CARBON C. The intestinal elimination of ciprofloxacin in the rat. J. Infect. Dis. 1994;169:218–221. doi: 10.1093/infdis/169.1.218. [DOI] [PubMed] [Google Scholar]
- SHAROM F.J. The P-glycoprotein efflux pump: how does it transport drugs. J. Membrane Biol. 1997;160:161–175. doi: 10.1007/s002329900305. [DOI] [PubMed] [Google Scholar]
- SÖRGEL F., KINZIG M. Pharmacokinetics of gyrase inhibitors, Part 1: Basic chemistry and gastronintestinal disposition. Am. J. Med. 1993a;94 suppl. 3A:44S–55S. [PubMed] [Google Scholar]
- SÖRGEL F., KINZIG M. Pharmacokinetics of gyrase inhibitors, Part 2: Renal and hepatic elimination pathways and drug interactions. Am. J. Med. 1993b;94 suppl. 3A:56S–69S. [PubMed] [Google Scholar]
- SÖRGEL F., NABER G., JAEDHE U., REITER A., SEELMANN R., SIGL G. Brief report: Gastrointestinal secretion of ciprofloxacin. Am. J. Med. 1989;87 suppl. 5A:S62–S65. doi: 10.1016/0002-9343(89)90025-9. [DOI] [PubMed] [Google Scholar]
- TSUI A., TAMAI I. Carrier-mediated intestinal transport of drugs. Pharm. Res. 1996;13:963–977. doi: 10.1023/a:1016086003070. [DOI] [PubMed] [Google Scholar]
- ZHANG L., BRETT C.L., GIACOMINI K.M. Role of organic cation transporters in drug absorption and elimination. Annu. Rev. Pharmacol. Toxicol. 1998;38:431–460. doi: 10.1146/annurev.pharmtox.38.1.431. [DOI] [PubMed] [Google Scholar]