

Abstract
Adenosine receptor(s) mediating negative inotropy of paced left atria, isoprenaline-stimulated paced left atria and papillary muscles, and negative chronotropy of spontaneously beating right atria were characterized.
Isometric tension of guinea-pig isolated paced left atria and left ventricular papillary muscles and rate of contraction of spontaneously beating right atria were recorded. Papillary muscles were pre-stimulated with isoprenaline (1×10−8 M). Concentration-response curves (CRCs) for tension or rate reduction by N6-cyclopentyladenosine (CPA), the stereoisomers of N6-(2-phenylisopropyl)adenosine ((+)-PIA and (−)-PIA), 5′-(N-carboxamido)adenosine (NECA), N6-2-(4-aminophenyl)ethyladenosine (APNEA) and N6-(3-iodobenzyl)adenosine-5′-N-methyuromide (IB-MECA) revealed a potency order of CPA=(−)-PIA>NECA in right atria and papillary muscles, which is consistent with involvement of A1-receptors. The potency order in left atria was CPA=NECA>(−)-PIA>(+)-PIA>APNEA, which is not typical of A1 adenosine receptors. Weak activity of APNEA and IB-MECA discounts involvement of A3 receptors.
pA2 values for the antagonism of CPA by 8(p-sulfophenyl)theophylline (8-SPT) were calculated from Schild plots (log concentration-ratio against log 8-SPT concentration), the unity slopes of which indicated competitive antagonism. The pA2 value in the papillary muscles was significantly higher than for atrial preparations, indicating a possible difference in receptor characteristics between atrial and papillary muscle responses.
In left and right atria there was a limit to the displacement of the CPA CRCs at higher concentrations of 8-SPT. The 8-SPT-resistant component of the response is suggested to arise from duality of coupling of a common A1 receptor through either different G proteins or G protein subunits to independent transduction pathways. The results with papillary muscles can be explained by a typical A1 receptor coupled to a single transduction pathway.
Keywords: Adenosine receptors, guinea-pig atria, papillary muscles, 8(p-sulfophenyl)theophylline (8-SPT), N6-cyclo-pentyladenosine (CPA), adenosine agonists, Schild plots
Introduction
The P1-purinoceptor subtype mediating negative inotropy in left atria and negative chronotropy in right atria of the guinea-pig is generally regarded as belonging to the A1-receptor subtype (Brown et al., 1982; Collis, 1983; Böhm et al., 1985; Leung et al., 1986, Collis et al., 1989: Jahnel & Nawrath, 1989; Tawfik-Schlieper et al., 1989; von der Leyen et al., 1989; Wilken et al., 1990). There are few studies on the purinoceptor subtype mediating the indirect anti-adrenergic negative inotropy seen in ventricular muscle preparations of the guinea-pig (von der Leyen et al., 1989).
However, anomalies exist in both the agonist potency orders and antagonist profiles for functional responses in isolated cardiac preparations. For example, 5′-(N-ethylcarboxamido)adenosine (NECA) and (−)-N6-(phenylisopropyl)adenosine ((−)-PIA) show little difference in potency (Ribeiro & Sebastiao, 1986) whereas (−)-PIA would be expected to have greater potency for an A1-receptor-mediated response. Both 8-phenyltheophylline (8-PT) (Urquhart, 1990) and cyclopentyltheophylline (CPT) (Ford & Broadley, 1997) have been shown in this laboratory to exhibit a limit in their antagonism of the functional responses of (−)-PIA and N6-cyclopentyladenosine (CPA), respectively, as the concentration of antagonist is increased in naïve left and right atria. This observation suggests that these responses may be mediated by an atypical A1-receptor peculiar to cardiac tissue or by a heterogeneous population of A1-receptor subtypes. The limit in antagonism seen with both of these adenosine receptor antagonist could also be due to a limit in their solubility.
This study therefore had three aims. The first aim was to clarify the agonist potency order in guinea-pig isolated atrial preparations with the inclusion of the A1/A3 selective agonist N6-2-(4-aminophenyl)ethyladenosine (APNEA) and in left atria the A3-selective agonist N6-(3-iodobenzyl)-5′-N-methyluronamide (IB-MECA) (Gallo-Rodriguez et al., 1994). The second aim was to determine the potency order of adenosine-receptor agonists in guinea-pig papillary muscles stimulated by isoprenaline (ISO). Finally, the inhibition of responses to the selective A1 agonist CPA by the non-selective adenosine receptor antagonist 8(p-sulfophenyl)theophylline (8-SPT) was investigated in a wider range of cardiac tissues (naïve and ISO-stimulated left atria, naïve right atria and ISO-stimulated left papillary muscles) than used in any single previous study. 8-SPT is relatively soluble in water and therefore higher concentrations can be employed in the determination of any limit to antagonism, compared for example with the potent selective antagonist DPCPX (von der Leyen et al., 1989). Also, due to its polar nature 8-SPT penetrates cells to a lesser extent than other P1 receptor antagonists (Bruns et al., 1980; Petrack et al., 1981; Daly, 1982) and this minimizes the possibility of interference from phosphodiesterase inhibitory effects (Ukena et al., 1993) commonly seen with other adenosine receptor antagonists.
Methods
Tissue preparation
Male Dunkin-Hartley guinea-pigs (250–300 g) were killed by a blow to the back of the head followed by exsanguination under running water. The guidelines for the care and use of laboratory animals were followed according to the Animals (Scientific Procedures) Act 1986. The rib cage was removed to expose the heart. The right atrium was removed first as follows.
The pericardium was removed and the ventricular apex was clamped with a pair of Spencer-Wells forceps. The right atrium was removed complete with the sino-atrial (SA) node using cottons tied to the superior vena cava which attached the tissue to an isometric force transducer (type UF1, 57 g sensitivity range). Two cotton threads attached to the inferior vena cava and atrioventricular junction were used to secure the atrium to a stainless steel support. The right atrium was then placed in a tissue bath and resting diastolic tension was set at 0.8–1.0 g.
Left atria were then removed from the quiescent heart by means of cotton threads attached to the tip of the left atrial appendage and atrioventricular junction. The latter thread attached the atrium to the electrode tips of a Harvard bipolar platinum electrode and the first thread was attached to an isometric transducer (type UF1, 57 g sensitivity range). A resting diastolic tension of 0.5–1.0 g was then applied.
After removal of the left atria, a discrete papillary muscle from the left ventricle was exposed by cutting along the interventricular septum. A cotton thread was then placed around the chordae tendineae of the papillary muscle and another through its apical end. The papillary muscle was then dissected free of the ventricle. The cotton attached around the chordae tendineae was then attached to an isometric transducer (type UF1, 57 g sensitivity range) with the cotton through its apical end holding it in close contact with bipolar platinum electrodes. A resting tension of 0.4–0.6 g was then applied. All tissues were set up in 50 ml organ baths containing pre-warmed Krebs-bicarbonate buffer gassed with 95% O2 / 5% CO2 and maintained at 37±0.5°C.
The tensions developed by both atria and papillary muscles were measured and displayed on an 8-channel Devices MT8P polygraph. Left atria and papillary muscles were electrically stimulated at 2 Hz using square pulse waves, of 5 msec duration at threshold voltage+50%, delivered by a Harvard 50–72 stimulator. The parameters used for electrical stimulation have previously been shown to drive cardiac tissue without causing significant autonomic transmitter releases, when delivered by electrodes in direct contact with the tissue (Koch-Weser and Blinks 1963). Right atrial preparations were allowed to beat spontaneously and the rate of beating continuously measured electronically, using a Devices (model 4522) rate meter triggered by the signal from the isometric transducer.
Experimental protocol
The Krebs bicarbonate solution was made up in double distilled water and had the following composition in (mM): NaCl 118.0, KCl 4.7, CaCl2 2H2O 2.5, MgSO4 7H2O 1.2, NaHCO3 24.9, KH2PO4.2H2O 1.2, glucose 11.6. All reagents were of Analar standard (Fisons). An equilibration period of 60 min was allowed before commencement of an experimental protocol. During this period, the bathing Krebs solution was replaced with fresh Krebs every 15 min.
Some experiments with left atria and all those with left papillary muscles, involved pre-stimulating the tissue with isoprenaline (ISO). In order to determine a suitable submaximal dose of ISO, cumulative concentration-response curves (CRCs) to ISO were constructed in both tissues (data not shown). The submaximal concentration chosen to pre-stimulate left atria and papillary muscles was 1×10−8 M. This concentration produced an approximate 50% increase in resting developed tension in both preparations. ISO was added 45 min into the equilibration period and the developed tension was allowed to stabilize before construction of CRCs.
After the 60 min equilibration period, cumulative additions of half-logarithmic increases in concentration of agonist, in the case of left and right atria, allowed the construction of CRCs. In the case of left papillary muscles, agonists were added in logarithmic increments of concentration. Enough time was allowed for each response to plateau before a subsequent agonist concentration was added. Wherever possible the volume of individual boluses of agonists added to the bath did not exceed 0.3 ml. Only one concentration-response curve was constructed in an individual tissue.
When the antagonist 8-(p-sulfophenyl)theophylline was used, it was added to the tissue after 30 min of equilibration. The antagonist was then left in contact with the tissue for a further 30 min without changes in the bathing solution. Agonist CRCs were then constructed in the presence of the antagonist as described above.
Data and statistical analysis
Agonist studies
In atrial preparations, responses were determined as % inhibition of resting tension or rate and are reported as mean±s.e.mean (n⩾4). IC50 values (molar concentration producing a 50% fall in basal tension or rate) were calculated from the individual CRCs to each agonist. Since in the papillary muscle, the agonists only exert an indirect negative inotropy against ISO prestimulation, responses were measured as the reduction of tension and expressed as a percentage of the isoprenaline-induced increase in tension. Since not all agonists caused a 50% reduction of the ISO-induced increases in tension, IC35 values (molar concentration producing a 35% fall in the ISO-induced increase in tension) were determined for each agonist. IC50 and IC35 values are expressed as the geometric mean with 95% confidence limits in parenthesis. Orders of potency were determined by firstly performing a one-way analysis of variance. If this test showed statistical differences between population means at a 95% level then it was followed by Duncan's multiple range test (Duncan, 1955; Kramer, 1956) to determine where statistical differences between means lay at a 95% level.
Antagonist studies
The antagonism of N6-cyclopentyladenosine (CPA) by 8(p-sulfophenyl) theophylline (8-SPT) treatment was determined by calculating a pA2 value as follows: A concentration-ratio (CR) was determined for each antagonist concentration used, by dividing the individual IC50 value of the agonist in the presence of the antagonist by the mean value obtained in its absence. In the papillary muscle, all responses were calculated as a percentage of the isoprenaline-induced increase in tension. Also, IC20 values (molar concentration producing 20% of the maximum inhibition of the initial tension or rate) were used to calculated CRs for the antagonism of CPA by 8-SPT rather than IC50 values. This was due to the fact that at high concentrations of 8-SPT full concentration-response curves to CPA could not be constructed (see Figure 3D).
Figure 3.
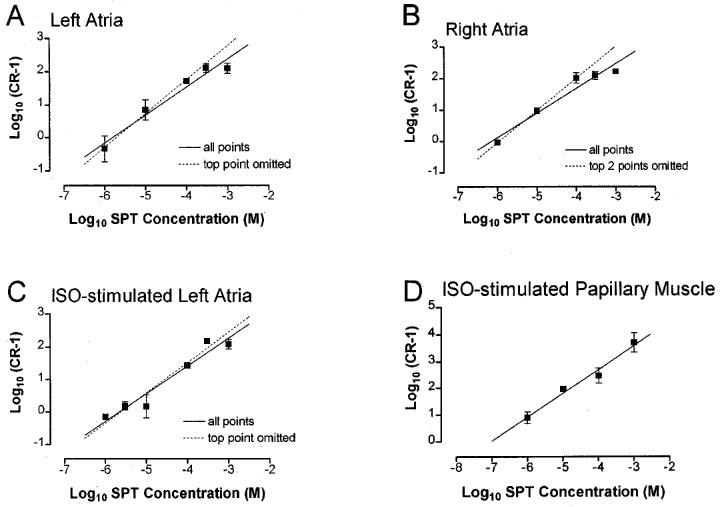
Schild plots for the antagonism of CPA by 8-SPT in (A) left atria, (B) right atria, (C) ISO-stimulated left atria, (D) ISO-stimulated papillary muscle. Mean (n>4) concentration-ratio (CR) for the shift of individual concentration-response curves in the presence of 8-SPT relative to the mean curve in its absence are plotted as log (CR-1) against log concentration (M) of 8-SPT. Vertical bars represent s.e.mean, their absence indicating that they lie within the symbol. Linear regression for all points are indicated by solid lines. Where the higher concentrations appear to deviate from this line, further regression lines are shown (dashed lines).
The mean CR was then plotted as log10 (CR-1) against log antagonist molar concentration according to the method of Arunlakshana & Schild (1959). Lines of best fit, together with their correlation coefficients, were then estimated using linear regression analysis (Bolton, 1990). The pA2 value was estimated together with the 95% confidence interval, by extrapolation using the equation generated by linear regression analysis to find the intercept of the x-axis (log molar concentration of antagonist). The slope of the plot was then tested to determine if it varied significantly from unity (Bolton, 1990).
To compare statistically the pA2 values for the antagonism between 8-SPT and CPA in different tissues, apparent pA2 values were calculated for individual data points according to the method of Mackay (1978). In this method of pA2 estimation, the antagonism is assumed to be competitive and therefore the slope of the Schild plot will be unity. The following equation can then be applied:
The entire set of pA2 values for all tissues were compared by a one-way ANOVA followed by an unpaired Students t-test. The results were taken to be significant if P<0.05.
Drugs and solutions
The drugs used were: N6-2-(4-aminophenyl)ethyladenosine (APNEA) (Semat), N6-cyclopentyladenosine (CPA) (Sigma), N6-(3-iodobenzyl)adenosine-5′-N-methyuromide (IB-MECA) (GlaxoWellcome, Stevenage, U.K.), (−)-isoprenaline acid tartrate (Sigma), 5′-(N-ethylcarboximido)adenosine (NECA) (Sigma), (−)-N6-phenylisopropyladenosine (−)-PIA (Sigma), (+)-N6-phenylisopropyladenosine (+)-PIA (Sigma), 8-(p-sulfophenyl)theophylline (8-SPT) (Semat).
IB-MECA was a kind gift from Dr Julian Reeves, GlaxoWellcome research, Stevenage, U.K. 8-SPT was dissolved and diluted in distilled water. APNEA, CPA, IB-MECA, NECA, (−)-PIA and (+)-PIA were all initially dissolved in polyethylene glycol 400 (PEG, Sigma) : water (50 : 50%). Subsequent dilutions were made in distilled water.
Results
Agonist studies
In guinea-pig left atria all agonists caused a concentration-dependent negative inotropy (Figure 1A). The maximum inhibitions of initial tension together with IC50 values for each agonist are reported in Table 1. The rank order of potency was:
IB-MECA was the least potent agonist but it was not possible to calculate an IC50 value since it had minimal effect on left atrial tension, producing only 22.3±2.7% inhibition at the highest concentration used (1×10−5 M).
Figure 1.
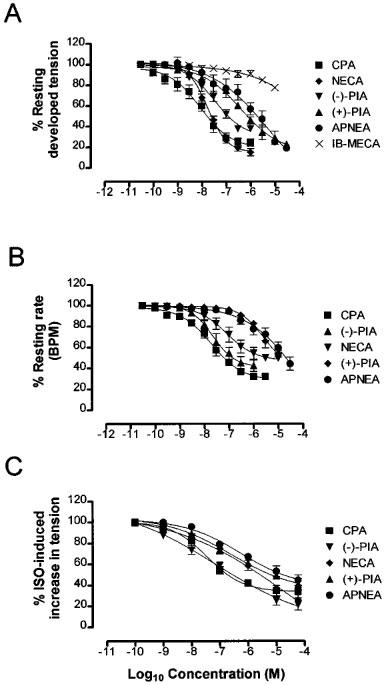
Mean concentration-response curves (n⩾4) for the inhibition of (A) left atrial tension, (B) right atrial rate, (C) ISO-stimulated increase in papillary muscle tension by CPA (▪), (−)-PIA (▾), NECA (⧫), (+)-PIA (▴), APNEA (•) and IB-MECA (X). Each point is the mean reduction of tension or rate. Vertical bars represent the s.e.mean, their absence indicating they lie within the symbol.
Table 1.
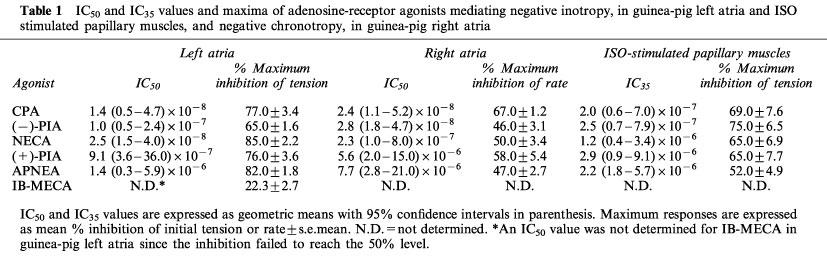
IC50 and IC35 values and maxima of adenosine-receptor agonists mediating negative inotropy, in guinea-pig left atria and ISO stimulated papillary muscles, and negative chronotropy, in guinea-pig right atria
In guinea-pig right atria, all agonists caused a concentration-dependent negative chronotropy (Figure 1B). The maximum inhibitions of initial rate together with IC50 values for each agonist are reported in Table 1. The rank order of potency was:
In ISO-stimulated papillary muscles, all agonists caused a concentration-dependent negative inotropy (Figure 1C). The maximum inhibitions of the ISO-stimulated increases in tension together with IC35 values for each agonist are reported in Table 1. The rank order of potency was:
Antagonist studies
Increasing concentrations of 8-SPT caused concentration-dependent rightward shifts of the CPA concentration-response curves in all preparations (Figure 2). 8-SPT had no significant effect on maximum response to CPA at all concentrations studied in the left atria.
Figure 2.
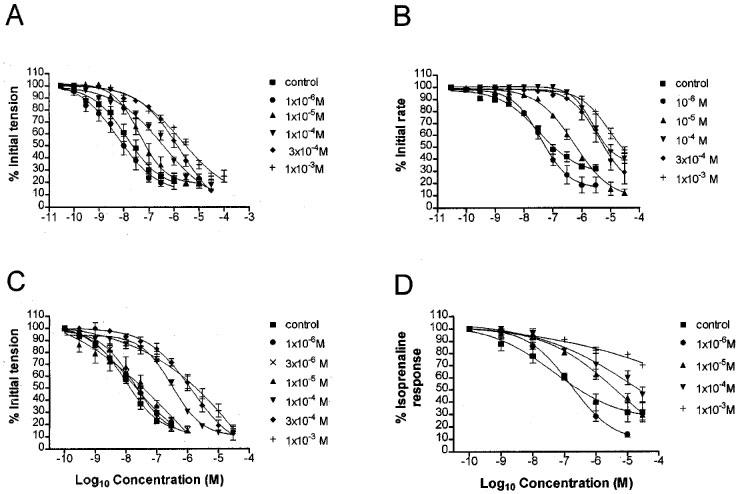
Mean concentration-response curves (n⩾4) for the inhibition of (A) left atrial tension, (B) right atrial rate, (C) ISO-stimulated left atrial tension, (D) ISO-stimulated increase in papillary muscle tension by CPA in the absence (▪) and presence of 8-SPT; 10−6 (•), 3×10−6 M (X), 10−5 M (▴), 10−4 M (▾), 3×10−4 M (⧫), 10−3 M (+). Each point is the mean reduction of tension or rate. Vertical bars represent the s.e.mean, their absence indicating they lie within the symbol.
Schild regression analysis of the interaction between CPA and 8-SPT in all the preparations revealed plots where the data fitted a linear model (Figure 3). None of the regression slopes were significantly different from unity. In the left and right atria, because concentration-ratios did not increase with increase in antagonist concentration, it appeared that a better fit may be obtained by removal of points at higher concentrations of 8-SPT. Further regression analysis is therefore shown in Figure 3A, B and C in which the top two points were omitted. The gradients and r2 values estimated from both regression plots are reported in Table 2 together with pA2 values derived from all points.
Table 2.
Data from Schild plots for the antagonism by 8-SPT of guinea-pig left and right atria and ISO-stimulated papillary muscle responses to CPA
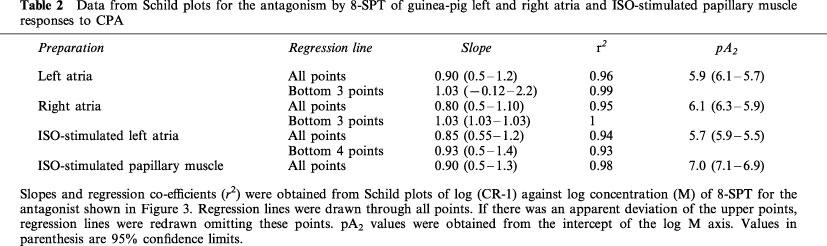
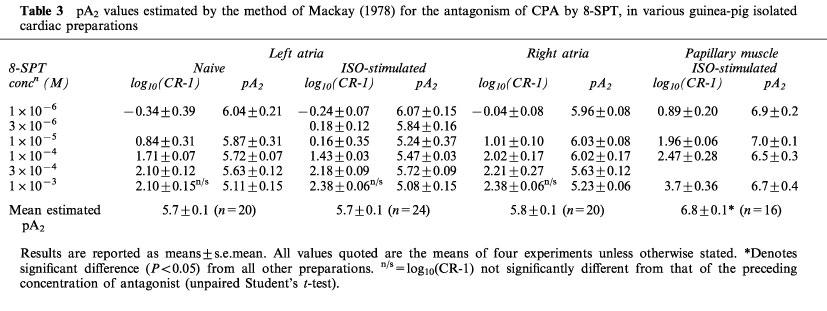
As none of the Schild regression gradients significantly varied from unity, pA2 values were calculated for individual concentrations of 8-SPT according to the method of Mackay (1978). These values and the corresponding concentration-ratios (as log10 (CR-1)) are shown in Table 3. The concentration-ratios increased as concentration of antagonist was increased up to 3×10−4 M, or 1×10−4 M in naïve right atria. However, above this concentration there was no significant increase in the concentration-ratios of left and right atria. As a consequence, the highest concentration of 8-SPT yielded significantly lower pA2 values than with the other concentrations. In the case of the papillary muscle, however, there were progressive increases in the shift of the concentration-response curves and all concentrations of 8-SPT yielded similar pA2 values. Furthermore, the Schild plot was plotted for all points only with the papillary muscle.
Table 3.
pA2 values estimated by the method of Mackay (1978) for the antagonism of CPA by 8-SPT, in various guinea-pig isolated cardiac preparations
Discussion
The traditional concept is of cardiac P1 purinoceptors being of the A1-subtype (Collis, 1983; Evans et al., 1982), where an agonist potency order of CPA>(−)-PIA⩾NECA is expected (Collis & Hourani, 1993). In right atria and papillary muscles, the potency orders obtained were CPA=(−)-PIA>NECA, which is consistent with the receptor being of the A1-subtype in these tissues (Dalziel & Westfall, 1994).
The left atrial negative inotropic response displayed a potency order CPA=NECA>(−)-PIA which is not typical of the adenosine A1-receptor. The equipotency of N6-substituted adenosine analogues (CPA and (−)-PIA) and 5′-substituted analogues (NECA) in guinea-pig atria led to the suggestion by Ribeiro & Sebastiao (1986) of a unique adenosine A3 receptor. This receptor was xanthine sensitive, not coupled to adenylyl cyclase and produced effects by modulating calcium entry into cells (Ribeiro & Sebastiao, 1986). Both K+ efflux (Belardinelli & Isenberg, 1983; Cerbai et al., 1988; Visentin et al., 1990; Urquhart et al., 1991; 1993) and closure of L-type calcium channels (DeBiasi et al., 1989; Fassina et al., 1991; Jahnel et al., 1992) have been implicated in the negative inotropic effects of adenosine analogues in the guinea-pig left atria.
A more recently defined A3 adenosine receptor has been cloned from sheep and rat cDNA sequences and expressed in Chinese hamster ovary cells (Zhou et al., 1992; Linden, 1994). This receptor is thought to mediate hypotension in pithed rats (Fozard & Carruthers, 1993) but is distinct from that proposed by Ribeiro & Sebastiao (1986). This receptor displays (−)-PIA and NECA equipotency, is insensitive to most methylxanthine antagonists (Linden, 1994) and the agonist potency order is APNEA>(−)-PIA=NECA>CGS21680 (Collis & Hourani, 1993). In the present study, APNEA was the least potent agonist at mediating any of the responses studied. Furthermore, the selective A3-agonist IB-MECA (Gallo-Rodriguez et al., 1994) was inactive up to a concentration of 3×10−7 M and the responses were susceptible to blockade by 8-SPT. This A3 receptor is therefore unlikely to be responsible for the negative inotropic responses in the left atria.
In ISO-stimulated papillary muscles, CPA, NECA and (−)-PIA were less potent than in left and right atria. These agonists operate as functional antagonists of the isoprenaline response through modulation of cAMP (Dobson & Schrader, 1984). This would require an additional level of efficacy compared with their direct negative inotropy through K+ efflux, which could explain their reduced potency.
Schild analysis of the antagonism of CPA by 8-SPT revealed linear plots with slopes not differing significantly from unity, indicating competitive antagonism. Closer inspection of the data, however, suggests that the antagonism of CPA by 8-SPT may be more complex. In naïve and ISO-stimulated left atria and naïve right atria, there was a plateauing of the Schild plot at higher concentrations of 8-SPT (10−4 M to 10−3 M), which was not observed in the ISO-stimulated papillary muscle. This observation confirms our earlier studies with guinea-pig atria showing a similar limitation in the antagonism by 8-phenyltheophylline (8-PT) (Urquhart, 1990) and cyclopentyltheophylline (CPT) (Ford & Broadley, 1997). Previous pA2 determinations by others in guinea-pig atria have not revealed such limits to the antagonism (von der Leyen et al., 1989), possibly because poor solubility of the antagonist, in this case DPCPX, prevented the use of high concentrations. The primary concern in studies with 8-PT was that the lack of progressive shifts at higher concentrations of the antagonist could have been due to its limited solubility (Bruns & Fergus, 1989). This question of solubility is unlikely to account for the limit in the shift of the CRCs in the present study because 8-SPT is soluble up to a concentration of 2.4×10−2 M in water and secondly, a linear Schild plot was observed in ISO-stimulated papillary muscles up to the highest concentration of 8-SPT used.
Thus, the limit in the antagonism of CPA produced by 8-SPT could have a number of other possible explanations. Firstly, if either 8-SPT or CPA were substrates for an uptake process, this would prevent all the drug molecules from competing at the receptor (Kenakin, 1987). However, no uptake system for 8-SPT or CPA have been reported. A lack of further antagonism at higher concentrations might also suggest that the response is no longer mediated via an 8-SPT-sensitive receptor and that another receptor or subtype is mediating the negative inotropic and chronotropic responses. Indeed, subtypes of the adenosine A1 receptor has been hypothesized (Gustafsson et al., 1989). Identification of a second phase to the Schild plot would require further concentrations of 8-SPT and this would only be seen if the second receptor was sensitive to 8-SPT.
Another possible explanation for the plateauing of the Schild plot is that 8-SPT is acting as an allosteric antagonist, binding to a site distinct from where the agonist exerts its action. Clark & Mitchelson (1976) showed a linear Schild regression for the antagonism of carbachol or acetylcholine by gallamine in guinea-pig left and right atria over a 30 fold concentration range with a limit occurring at high concentrations. This was attributed to gallamine binding entirely to an allosteric site (i.e. no competitive antagonism), which saturates at high concentrations. There are no reports, however, that 8-SPT acts as anything other than a competitive antagonist of P1 adenosine receptors.
Hence, the most likely explanation for the plateauing of the Schild plot is the presence of a second methylxanthine resistant mechanism mediating the negative inotropy in naïve and ISO-stimulated left atria and naïve right atria. This may involve a distinct receptor type or subtype or a duality in the coupling of the same receptor to different second messenger systems. The ability of a single receptor type to produce effects through two pathways is referred to as receptor-transducer promiscuity (Kenakin, 1993). With respect to the adenosine-receptors mediating negative inotropy in guinea-pig left atria, it would be expected that a single adenosine receptor (of the A1-subtype) is capable of binding at least two G-proteins. The involvement of a single adenosine-receptor mediating both responses is suggested by the fact that only mRNA for one A1-adenosine receptor has been detected (Linden et al., 1993). There is biochemical evidence that the A1-adenosine receptor can bind with more than one G-protein, since the A1-adenosine receptor from bovine brain co-purifies on an agonist affinity column with three G-proteins, Gi1, Gi2 and Go (Munshi et al., 1991). Alternatively, the single receptor may couple to a common G-protein, which upon receptor activation cleaves into its Gα and Gβγ components. It now appears that it is the Gβγ subunit that directly opens the acetylcholine-gated K+ channel (IKACh) (Clapham & Neer, 1997). In the case of the atrial (but not ventricular) A1 receptor, it is therefore conceivable that the Gα subunit is involved in Ca2+ channel closure possibly via cAMP inhibition, whereas the Gβγ subunit opens the K+ channel.
Different agonists could produce activated receptors with dissimilar ability to activate G-proteins and second messenger pathways. Thus, some agonists may activate both K+ efflux and Ca2+ channel closure, whereas others may only activate one of these second messengers. The ability of different agonists to stimulate different transduction pathways to varying degrees is known as agonist trafficking (Kenakin, 1993). Recently, the traditional model of agonist action has been extended to explain why the same receptor can exhibit different pharmacology when coupled to different effector pathways i.e. promiscuous receptors (Leff et al., 1997; Clarke & Bond, 1998). Two- or three-state receptor models of agonist action may help explain the different agonist potency orders obtained at A1-adenosine receptors in the atria where they appear to be coupled to calcium and potassium channels. The same receptor may exist in different conformational states for which agonists can display different affinities. Only further studies of the molecular nature of the atrial adenosine receptors and their binding of antagonist and agonist would clarify this question.
A second anomoly was that the pA2 values differed between tissue preparations. Although the pA2 values for the atrial preparations did not differ significantly (P>0.05) from each other, that for the ISO-stimulated papillary muscle was significantly (P<0.05) lower than for all the atrial preparations. If the responses of all these preparations were mediated via a single identical receptor, then identical pA2 values would be predicted for all preparations. This evidence and the fact that the antagonism of CPA by 8-SPT produces a linear Schild regression with no plateauing and a unit slope, is indicative of the negative inotropy in papillary muscles being mediated by an adenosine receptor distinct from that in atrial tissue. Binding studies, along with molecular sequencing of the receptor would be required to allow the proposal of a unique class of adenosine receptor (Fredholm et al., 1994).
Acknowledgments
NMG was supported by a Studentship from the Royal Pharmaceutical Society of Great Britain. We are grateful to Dr Julian Reeves of GlaxoWellcome for the generous gift of IB-MECA.
Abbreviations
- APNEA
N6-2-(4-aminophenyl)ethyladenosine
- CPA
N6-cyclopentyladenosine
- CPT
cyclopentyltheophylline
- CR
concentration-ratio, CRCs, concentration-response curves
- IB-MECA
N6-(3-iodobenzyl)adenosine-5′-N-methyuromide
- ISO
isoprenaline
- NECA
5′-(N-carboxamido)adenosine, (+)-PIA, (+)-N6-(2-phenylisopropyl) adenosine
- (−)-PIA
(−)-N6-(2-phenylisopropyl)adenosine
- 8-PT
8-phenyltheophylline
- SA node
sino-atrial node
- 8-SPT
8(p-sulfophenyl)theophylline.
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative use of drug antagonists. Br. J. Pharmacol. Chemother. 1959;257:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELARDINELLI L., ISENBERG G. Isolated atrial myocytes: adenosine and acetylcholine increase potassium conductance. Am. J. Physiol. 1983;224:H734–H737. doi: 10.1152/ajpheart.1983.244.5.H734. [DOI] [PubMed] [Google Scholar]
- BÖHM M., BRUCKNER R., MEYER W., NOSE M., SCHMITZ W., SCHOLZ H., STARBATTY J. Evidence for adenosine receptor-mediated isoprenaline-antagonistic effects of the adenosine analogs PIA and NECA on force of contraction in guinea-pig atrial and ventricular cardiac preparations. Naunyn-Schmiedeberg's Arch. Phamacol. 1985;331:131–139. doi: 10.1007/BF00634229. [DOI] [PubMed] [Google Scholar]
- BOLTON S. Pharmaceutical Statistics 199044New York: Marcel Dekker INC; 2nd ed [Google Scholar]
- BROWN C.M., BURNSTOCK G., CUSACK N.J., MEGHJI P., MOODY C.J. Evidence for the stereospecificity of the P1-purinoreceptor. Br. J. Pharmacol. 1982;78:110–117. doi: 10.1111/j.1476-5381.1982.tb08762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUNS R.F., DALY J.W., SNYDER S.H. Adenosine receptors in brain membranes: binding of N6-cyclohexyl [3H] adenosine and 1,3-diethyl-8-[3H] phenylxanthine. Proc. Natl. Acad. Sci. USA. 1980;77:5547–5551. doi: 10.1073/pnas.77.9.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUNS R.F., FERGUS J.H. Solubilities of adenosine antagonists determined by radioreceptor assay. J. Pharm. Pharmacol. 1989;41:590–594. doi: 10.1111/j.2042-7158.1989.tb06537.x. [DOI] [PubMed] [Google Scholar]
- CERBAI E., KLOCKNER U., ISENBERG G. Ca-antagonistic effects of adenosine in guinea-pig atrial cells. Am. J. Physiol. 1988;254:H872–H878. doi: 10.1152/ajpheart.1988.255.4.H872. [DOI] [PubMed] [Google Scholar]
- CLAPHAM D.E., NEER E.J. G protein βγ subunits. Annu. Rev. Pharmacol. Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- CLARK A.L., MITCHELSON F. The inhibitory effect of gallamine on muscarinic receptors. Br. J. Pharmacol. 1976;58:323–331. doi: 10.1111/j.1476-5381.1976.tb07708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARKE W.P., BOND R.A. The elusive nature of intrinsic efficacy. Trends Pharmacol. Sci. 1998;19:270–276. doi: 10.1016/s0165-6147(97)01138-3. [DOI] [PubMed] [Google Scholar]
- COLLIS M.G. Evidence for A1-adenosine receptors in the guinea-pig atria. Br. J. Pharmacol. 1983;78:207–212. doi: 10.1111/j.1476-5381.1983.tb09381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLLIS M.G., HOURANI S.M.O. Adenosine-receptor subtypes. Trends Pharmacol. Sci. 1993;14:360–366. doi: 10.1016/0165-6147(93)90094-z. [DOI] [PubMed] [Google Scholar]
- COLLIS M.G., STOGGALL S.M., MARTIN F.M. Apparent affinity of 1,3-dipropyl-8-cyclopentylxanthine for adenosine A1 and A2 receptors in isolated tissues from guinea-pigs. Br. J. Pharmacol. 1989;97:1274–1278. doi: 10.1111/j.1476-5381.1989.tb12589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALY J.W. Adenosine receptors: Targets for future drugs. J. Med. Chem. 1982;25:197–207. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]
- DALZIEL H.H., WESTFALL D.P. Receptors for adenine nucleotides and nucleosides: subclassification, distribution, and molecular characterisation. Pharmacol. Rev. 1994;46:449–466. [PubMed] [Google Scholar]
- DE BIASI M., FROLDI G., RAGAZZI E., PANDOLFO L., CAPARROTTA L., FASSINA G. Potassium channel blockers differentially affect carbachol and (−)N6-phenylisopropyladenosine on guinea-pig atria. Br. J. Pharmacol. 1989;97:866–872. doi: 10.1111/j.1476-5381.1989.tb12026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOBSON J.G. , JR, SCHRADER J. Role of extracellular and intracellular adenosine in attenuation of catecholamine evoked responses in guinea-pig heart. J. Mol. Cell. Cardiol. 1984;16:813–822. doi: 10.1016/s0022-2828(84)80005-x. [DOI] [PubMed] [Google Scholar]
- DUNCAN D.B. Multiple range and multiple-F tests. Biometrics. 1955;11:1–42. [Google Scholar]
- EVANS D.B., SCHENDEN J.A., BRISTOL J.A. Adenosine receptors mediating cardiac depression. Life Sci. 1982;31:2425–2432. doi: 10.1016/0024-3205(82)90746-9. [DOI] [PubMed] [Google Scholar]
- FASSINA G., DEBIASI M., RAGAZZI E., CAPARROTTA L. Adenosine: a natural modulator of L-type calcium channels in atrial myocardium. Pharmacol. Res. 1991;23:319–326. doi: 10.1016/1043-6618(91)90047-2. [DOI] [PubMed] [Google Scholar]
- FORD W., BROADLEY K.J. Functional classification of P1-purinoreceptors in guinea-pig left and right atria: anomalous characteristics of antagonism by cyclopentyltheophylline. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;355:759–766. doi: 10.1007/pl00005010. [DOI] [PubMed] [Google Scholar]
- FOZARD J.R., CARRUTHERS A.M. Adenosine A3 receptors mediate hypotension in the angiotensin II-supported circulation of the pithed rat. Br. J. Pharmacol. 1993;109:3–5. doi: 10.1111/j.1476-5381.1993.tb13522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREDHOLM B.B., ABBRACCHIO P., BURNSTOCK G., DALY J.W., HARDEN K., JACOBSON K.A., LEFF P., WILLIAMS M. Nomenclature and classification of purinoreceptors. Pharmacol. Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- GALLO-RODRIGUEZ C., JI X.D., MELMAN N., SIEGMAN B.D., SANDERS L.H., ORLINA J., FISCHER B., PU Q., OLAH M.E., VAN GALEN K.J., STILES G.L., JACOBSON K.A. Structure-activity relationships of N6-benzyl-5′-uronamides as A3 selective agonists. J. Med. Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUSTAFSSON L.E., WIKLUND C.U., WIKLUND N.P., STELIUS L.Identification of subclasses of adenosine A1 receptors. Suggestion of A1a and A2a subclasses and evidence from antagonist data Adenosine Receptors in the Nervous System 1989London: Taylor & Francis; ed. Ribeiro, J.A. p 194 [Google Scholar]
- JAHNEL U., NAWRATH H. Characterisation of adenosine receptors in guinea-pig left atria. Br. J. Pharmacol. 1989;97:1182–1190. doi: 10.1111/j.1476-5381.1989.tb12577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAHNEL U., NAWRATH H., OCHI R. Adrenoceptor-mediated effects on calcium channel currents are antagonised by 5′-(N-ethyl)-carboxamido-adenosine in guinea-pig atrial cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;345:564–569. doi: 10.1007/BF00168950. [DOI] [PubMed] [Google Scholar]
- KENAKIN T.P. Pharmacological Analysis of Drug Receptor Interactions. New York: Raven Press; 1987. [Google Scholar]
- KENAKIN T.P. Pharmacologic Analysis of Drug-Receptor Interactions 1993New York: Raven Press; 2nd Edn [Google Scholar]
- KOCH-WESER J., BLINKS J.R. The influence of the interval between beats on myocardial contractility. Pharmacol. Rev. 1963;15:601–652. [PubMed] [Google Scholar]
- KRAMER C.Y. Extension of the multiple range test to group means with unequal numbers of replication. Biometrics. 1956;12:307–310. [Google Scholar]
- LEFF P., SCARAMELLINI C., LAW C., MCKECHNIE K. A three-state receptor model of agonist action. Trends Pharmacol. Sci. 1997;18:355–362. doi: 10.1016/s0165-6147(97)01105-x. [DOI] [PubMed] [Google Scholar]
- LEUNG E., JOHNSTON C.I., WOODCOCK E.A. A comparison between the adenosine receptors mediating adenylate cyclase inhibition and cardiac depression in guinea-pig heart. J. Cardiovasc. Pharmacol. 1986;8:1003–1008. doi: 10.1097/00005344-198609000-00018. [DOI] [PubMed] [Google Scholar]
- LINDEN J. Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 1994;15:298–306. doi: 10.1016/0165-6147(94)90011-6. [DOI] [PubMed] [Google Scholar]
- LINDEN J., TUCKER A.L., ROBEVA A.S., GRABER S.G., MUNSHI R. Properties of recombinant adenosine receptors. Drug Dev. Res. 1993;28:232–236. [Google Scholar]
- MACKAY D. How should values of pA2 and affinity constants for pharmacological competitive antagonists be estimated. J. Pharm. Pharmacol. 1978;30:312–313. doi: 10.1111/j.2042-7158.1978.tb13237.x. [DOI] [PubMed] [Google Scholar]
- MUNSHI R., PANG I-H., STERWEIS P.C., LINDEN J. A1-adenosine receptors of the bovine brain couple to guanine nucleotide-binding proteins Gi1, Gi2 and G0. J. Biol. Chem. 1991;266:22285–22289. [PubMed] [Google Scholar]
- PETRACK B., CZERNIK A., ANSELL J., CASSIDY J. Potentiation of arginine-induced glucagon secretion by adenosine. Life Sci. 1981;28:2611–2615. doi: 10.1016/0024-3205(81)90718-9. [DOI] [PubMed] [Google Scholar]
- RIBEIRO J.A., SEBASTIAO A. Adenosine receptors and calcium: basis for proposing a third (A3) receptor. Progr. Neurobiol. 1986;26:179–210. doi: 10.1016/0301-0082(86)90015-8. [DOI] [PubMed] [Google Scholar]
- TAWFIK-SCHLIEPER H., KLOTZ K-N., KREYE V.A.W., SCHWABE U. Characterisation of the K+-channel-coupled adenosine receptor in guinea-pig atria. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989;340:648–688. doi: 10.1007/BF00717745. [DOI] [PubMed] [Google Scholar]
- UKENA D., SCHUDT C., SYBRECHT G.W. Adenosine receptor blocking xanthines as inhibitors of phosphodiesterase isozymes. Biochem. Pharmacol. 1993;45:847–851. doi: 10.1016/0006-2952(93)90168-v. [DOI] [PubMed] [Google Scholar]
- URQUHART R.A.The pharmacology of adenosine receptor agonists in cardiac and vascular smooth muscle 1990University of Wales, Cardiff; PhD Thesis [Google Scholar]
- URQUHART R.A., FORD W.R., BROADLEY K.J. Potassium channel blockade of atrial negative inotropic responses to P1-purinoceptor and muscarinic receptor agonists and to cromakalim. J. Cardiovasc. Pharmacol. 1993;21:279–288. doi: 10.1097/00005344-199302000-00014. [DOI] [PubMed] [Google Scholar]
- URQUHART R.A., ROTHAUL A.L., BROADLEY K.J. 86Rubidium efflux and negative inotropy induced by P1- and muscarinic agonists in guinea-pig left atria. Biochem. Pharmacol. 1991;42:655–662. doi: 10.1016/0006-2952(91)90329-4. [DOI] [PubMed] [Google Scholar]
- VISENTIN S., WU S.N., BELARDINELLI L. Adenosine-induced changes in atrial potential: contribution of Ca and K currents. Am. J. Physiol. 1990;258:H1070–H1078. doi: 10.1152/ajpheart.1990.258.4.H1070. [DOI] [PubMed] [Google Scholar]
- VON DER LEYEN H., SCHMITZ H., SCHOLZ J., LOHSE M.J., SCHWABE U. Effects of 1,3-dipropyl-8-cyclopentylxanthine (DCPCX), a highly selective adenosine receptor antagonist, on force of contraction in guinea-pig atrial and ventricular cardiac preparations. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989;340:204–209. doi: 10.1007/BF00168970. [DOI] [PubMed] [Google Scholar]
- WILKEN A., TAWFIK-SCHLIEPER H., KLOTZ H.N., SCHWABE U. Pharmacological characterisation of the adenylate cyclase-coupled adenosine receptor in isolated guinea-pig atrial myocytes. Mol. Pharmacol. 1990;37:916–920. [PubMed] [Google Scholar]
- ZHOU Q.Y., LI C., OLAH M.E., JOHNSON R.A., STILES G.L., CIVELLI O. Molecular cloning and characterisation of an adenosine receptor: the A3-adenosine receptor. Proc. Natl. Acad. Sci. USA. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]