

Abstract
Human isolated subcutaneous arteries were studied under isometric conditions in a myograph.
Addition of angiotensin II (AII) induced a concentration-dependent increase in tone in isolated arteries. The active metabolite of candesartan (CV 11974), losartan and the active metabolite of losartan, E-3174 antagonized AII-induced tone in a non-competitive manner, but the AT2 selective antagonist, PD123319, was without effect on responses to AII. The effects of candesartan, losartan and E-3174 were analysed using a classical model of non-competitive antagonism and a two-state receptor model.
Mechanical removal of the endothelium; pre-incubation with Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME); pre-incubation with indomethacin, a cyclo-oxygenase inhibitor; or pre-incubation with BQ 485, an endothelin antagonist; had no significant effect on contractions induced by AII.
Our results suggest AII contracts human isolated resistance arteries by an action on AT1 receptors and does not involve release of endothelial factors. Use of a two-state receptor model successfully described the action of the AT1 antagonists without sacrificing assumptions regarding the competitive nature of binding of these antagonists.
Keywords: Angiotensin II, AT1 receptors, endothelium, nitric oxide, endothelin
Introduction
Angiotensin II (AII) plays an important role in regulating blood pressure in the normal physiological state and in hypertension. Mammalian AII receptors have been classified as AT1 and AT2 subtypes on pharmacological grounds and these receptor subtypes may be distinguished by selective non-peptide antagonists; losartan, candesartan (AT1) and PD 123319 (AT2) (De Gasparo et al., 1995). Losartan has been reported to act as a competitive antagonist at AT1 receptors in functional and ligand binding experiments (Chiu et al., 1990a,1990c; Rhaleb et al., 1991; Shibouta et al., 1993). Losartan interacts with amino acids in the transmembrane domains of AT1 receptors different to those binding AII, but this binding is presumed to interfere with binding of AII (Ji et al., 1994). It is likely that closely related drugs such as candesartan cilexetil interact with the AT1 receptor in a manner similar to losartan, although the precise amino acids involved in binding may differ. Both losartan and candesartan cilexetil are metabolized in some species e.g. humans and rats, to a corresponding, more potent acid metabolite in vivo. These metabolites, E-3174 (active metabolite of losartan) and candesartan/CV 11974 (active metabolite of candesartan cilexetil) have been shown to display non-competitive (insurmountable) antagonism of AII-induced contractions in rabbit aortic strips (Shibouta et al., 1993). In contrast, in radioligand binding studies, losartan, candesartan and their metabolites displace AII binding competitively (Shibouta et al., 1993).
In almost all cases direct vasoconstrictor actions of AII are mediated by AT1 receptor activation (Timmermans et al., 1993), though AT2 receptor activation has been reported to be involved in endothelial-dependent responses to AII in rat cerebral arteries (Haberl, 1994). However, vasoconstrictor responses to AII show regional differences as well as variation depending on the preparation studied (Timmermans et al., 1993). This heterogeneity may be due to a variable contribution of the endothelium to AII-induced responses. In some preparations, AII-induced contraction has been reported to be augmented in the presence of endothelium (Lin & Nasjletti, 1991; Manabe et al., 1989), whereas in other preparations AII-induced contraction has been impaired in the presence of endothelium (Cortes et al., 1996; Zhang et al., 1995; Boulanger et al., 1995) or not affected whether endothelium is present or not (Juul et al., 1987).
The endothelium produces a number of vasoactive agents including nitric oxide (NO), eicosanoids and endothelin (ET-1). It has been suggested that endogenous vasodilator nitric oxide (NO) may interact with vasoconstrictor AII responses (Cachofeiro et al., 1995). AII has been reported to induce endothelial release of NO in some isolated preparations; the receptor involved in this effect has been reported to be AT1 in rat carotid artery (Boulanger et al., 1995) and AT2 in rat brain arterioles (Haberl, 1994).
AII has been demonstrated to release endothelin-1 (ET-1) from human endothelial and mesangial cells (Ciafre et al., 1993; Bakris & Re, 1993). The release of endothelin from the endothelium has been reported to mediate responses to AII in both rat tail artery and mesentery, but not in aorta (Chen et al., 1995).
AII has also been reported to stimulate endothelial release of vasoconstrictor cyclo-oxygenase (as well as endothelin) in spontaneously hypertensive rat (SHR) resistance arteries (Dohi et al., 1992; Lang et al., 1995). The endothelial release of contractile cyclo-oxygenase (COX) products has also been observed in Wistar rat mesenteric arteries (Wu et al., 1994). Therefore, it was of interest to study the interaction of AII with the endothelium in human arteries, especially the endothelial release of vasodilator NO and vasoconstrictor ET-1, COX, as these systems may possess opposing effects in vivo.
The aim of this study was to examine the effect of AII and the mode of action of AT1 antagonists on contractile responses in human subcutaneous resistance arteries and examine the possible participation of the endothelium-derived factors in mediating contractile responses to AII in this preparation.
Methods
Experimental protocol
Human subcutaneous resistance arteries (internal diameter 337±27 μm, n=54) were obtained from tissue resected at surgery and mounted as ring segments in an isometric myograph (Mulvany & Halpern, 1977). The myograph contained 10 ml of physiological saline solution (PSS (mM): NaCl, 118; KCl, 4.7; CaCl2.6H2O, 2.5; MgSO4.7H2O, 1.17; NaHCO3, 25.0; NaH2PO4. 2H2O, 1.0; Na2EDTA, 0.03; and glucose, 5.5) maintained at 37°C and aerated with 95% O2 and 5% CO2.
The vessels were allowed to equilibrate for 1 h and then set at a ‘normalized' internal circumference estimated to be 0.9 times the circumference they would maintain if relaxed and exposed to 100 mmHg transmural pressure (0.9L100). This was calculated for each individual vessel on the basis of the passive length-tension characteristics of the artery and the Laplace relationship (Mulvany & Halpern, 1977). This procedure optimised active force generation by these vessels and the internal diameters referred to were derived from this calculation.
Prior to beginning the studies, vessel viability was assessed by exposing arteries to 118 mM potassium solution (KPSS; PSS with equimolar substitution of 118 mM KCl for NaCl) and noradrenaline (NE) (10 μM). Vessels which failed to reproducibly produce tension equivalent to more than 100 mmHg effective pressure (by Laplace) in response to these stimulants were discarded.
For experiments studying AII receptor antagonists the concentration-response relationships to AII were performed in PSS containing 10.6 mM KCl (PSSK), as increasing extracellular potassium (KCl) had previously been reported to reduce AII-induced tachyphylaxis in other isolated vessels (Juul et al., 1987; Corriu et al., 1995). This was confirmed by us in preliminary studies using human resistance arteries. In our experiments the amount of potassium added to the PSSK (10.6 mM KCl) only produced a small transient contraction which rapidly returned to baseline and there was no sustained active tone. AII concentration-response curves were compared before and after candesartan (1, 10, 100 pM; contact time 30 min), E-3174 (100 pM, 1 nM, 10 nM; contact time 45 min), losartan (1, 10, 100 nM; contact time 45 min), or PD 123319 (100 nM; contact time 60 min). Time-matched controls were performed in the absence of antagonist. The effect of the antagonists on responses to NE (10 μM) in PSS was also investigated. Before each concentration-response curve was constructed the vessels were contracted with KPSS for 2 min to ensure continuing viability.
Endothelial factors
In order to assess the possible influence of release of endothelial factors on AII-induced contractions, responses to AII were compared before and after procedures anticipated to affect endothelial function. These experiments were performed in PSS with a 2 h interval between exposure to AII (and in a balanced random sequence where possible) to minimize tachyphylaxis. The presence or absence of functional endothelium was confirmed by the presence or absence of relaxation in response to substance P (100 nM) when precontracted with NE (10 μM).
The following were investigated on AII responses:
The effect of endothelial removal. A concentration-response relationship to AII (100 pM–100 nM) was constructed in arteries with intact endothelium. Endothelium was then removed by passing a human hair through the lumen of the myograph-mounted artery (Prieto et al., 1995). Abolition of a subsequent relaxation to substance P in vessels preconstricted with 10 μM NE was taken as proof of functional endothelial removal, and the AII concentration-response curve was then repeated.
The effect of inhibition of cyclo-oxygenase on AII responses. Concentration-response data to AII (100 pM–100 nM) were constructed in arteries with intact endothelium in the presence and absence of indomethacin (10 μM, contact time 30 min).
The effect of inhibition of endothelium-derived relaxing factor (EDRF / NO). Concentration-response data to AII (100 pM–100 nM) were constructed in arteries with intact endothelium in the presence and absence of Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME) (10 μM, contact time 30 min).
The effect of inhibition of endothelin-1 (ET-1). Concentration-response data to AII (100 pM–100 nM) were constructed in arteries with intact endothelium in the presence and absence of [(Hexahydro- 1H-azepinyl)carbonyl-Leu-D-Trp-D-Trp-OH.sodium salt] BQ-485 (1 μM, contact time 30 min). Preliminary studies showed that this concentration of BQ-485 abolished contractile responses to ET-1 (100 nM).
Drugs
Candesartan was a gift from Takeda Chemical Industries, Osaka, Japan. Noradrenaline, indomethacin and L-NAME were purchased from Sigma (Poole, Dorset, U.K.). E-3174 and losartan were gifts from Merck & Co. (NJ, U.S.A.). PD 123319 was purchased from Research Biochemicals International (MA, U.S.A.). Amlodipine was a gift from Pfizer (Sandwich, U.K.). Human AII and BQ 485 were purchased from Novabiochem (U.K.).
Statistics and data analysis
Concentration-response data derived from each individual tissue were fitted seperately to a logistic function by non-linear regression and EC50, the concentration of drug producing 50% of the maximal response to the same agent calculated. Non-linear regression was carried out on a PC using commercially available software (Inplot 4.0 and Prism 2.01, GraphPAD Software, CA, U.S.A.). Data are presented as means±s.e.mean calculated from n experiments in separate tissues. Concentration-response data were compared statistically in terms of −log (EC50) and maximum response using a Wilcoxon Paired rank test, a Mann-Whitney U-test or a Kruskal Wallis non-parametric analysis of variance (ANOVA) as appropriate. If ANOVA showed that a significant difference existed between groups it was followed by multiple comparisons of ranks to determine P for individual comparisons (Conover, 1980). A value of P<0.05 was considered significant.
Analysis of non-competitive antagonism and calculation of antagonist affinity (KB) was carried out essentially as described by Kenakin (1993). Accordingly, regression of equieffective concentrations of agonist alone ([A]) and agonist in the presence of antagonist was assumed to give a straight line of slope (1−ρB)−1, where ρB is the fraction of receptors bound by the antagonist. Hence KB=[B]. (slope−1)−1, where [B] is the concentration of antagonist. Analysis was performed on data derived from individual tissues using a custom program written in Excel (Microsoft Corp, Seattle, U.S.A.) and means±s.e.mean of log transformed (−log KB) data calculated from n experiments in separate tissues.
In addition, data were also analysed on the basis of a two state model as originally proposed by Katz & Thesleff (1957) and further developed by others (Rang & Ritter 1970; Gero 1983; Robertson et al., 1994).
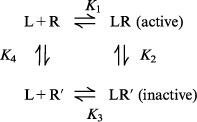 |
This model assumes that the AT1 receptor exists in two interconvertable states: R, a state that is activatable by the agonist and effects a response and R′, an inactive state. It is assumed that a ligand (L), which could be an agonist (A) or antagonist (B), binds to both R and R′ with dissociation equilibrium constants K1 and K3 respectively. Interconversion of R to R′ under basal or drug-bound conditions is governed by the dissociation equilibrium constants K4 and K2 respectively. Consequently, depending on the affinity of a ligand for R and R′, binding can alter the relative proportions of R and R′. For an antagonist this may result in the appearance of competitive or insurmountable antagonism depending on the basal ratio of R : R′ and the relative affinity of the antagonist for the two receptor states.
It is further assumed that binding of L to R (or R′) is more rapid than interconversion of the two receptor states (i.e. a hemi-equilibrium condition). For an agonist this means that responses are the result of an instantaneous interaction with the available pool of R. Even if the agonist has the capacity to alter the ratio between R and R′ this is assumed not to affect production of the response. However when antagonist interactions are simulated, it is assumed that alterations of the ratio of R : R′ do occur as a result of the extended pre-exposure times allowing steady-state to be achieved.
In the model shown interactions between agonist (A), antagonist (B) and receptor (R) are governed by the following equilibrium equations:
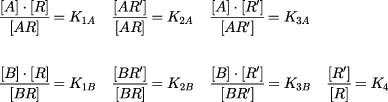 |
where K1–3A and K1–3B are the dissociation equilibrium constants for agonist and antagonists respectively; and K1B and K3B are the affinities of the antagonist for active (R) and inactive (R′) receptor respectively; K4 determines the basal ratio of R and R′. Assuming that the response (y) is equivalent to the fractional occupancy of active receptors by the agonist, then in the presence of an antagonist ([B]):
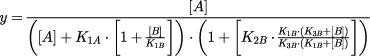 |
For the purposes of the analysis presented here the proportion of receptor in the inactive state at rest was assumed to be 1%. Models where different resting active : inactive proportions were assumed resulted in quantitatively, but not qualitatively different, comparative estimates of state affinity for the various antagonists. Data were fit to models by non-linear regression using macros written in Excel (Microsoft, Seattle, U.S.A.). Estimates of dissociation equilibrium constants derived from the two state model were based on experiments using concentrations of antagonist that depressed maximal responses <50% (Kenakin, 1993).
Results
Action of AII receptor antagonists on AII-induced contraction
Candesartan inhibited AII responses in an insurmountable manner, significantly depressing maximal responses to AII (Figure 1A). Using the model of non-competitive antagonism −log KB for candesartan was calculated as 11.0±0.2 (geometric mean KB=10 pM, n=16). Analysis using the two state model gave estimates of −log KB of 8.9 and 13.3 for active state and inactive state respectively. This can be interpreted as indicating that candesartan has a ∼30,000 fold greater affinity for the inactive compared with the active state of the AT1 receptor.
Figure 1.
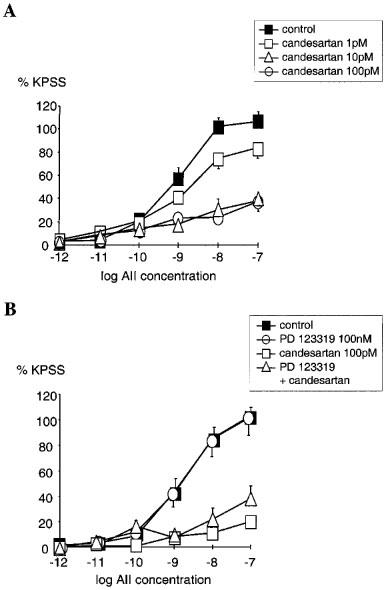
(A) Comparison of the cumulative concentration-response relationships for angiotensin II (AII 1 pM–100 nM) in PSSK (10.6 mM KCl) before (control (▪)) and after exposure to candesartan (1 pM; □), (10 pM; ▵), (100 pM; ○). Points represent mean±s.e.mean of per cent control response to 118 mM potassium-containing saline (KPSS)(n=6 in each case). (B) Comparison of the cumulative concentration-response relationships for angiotensin II (AII 1 pM–100 nM) in PSSK before (control (▪), n=9) and after; PD 123 319 (100 nM (○), n=8), candesartan (100 pM (□), n=7), both PD 123 319 and candesartan (▵), (n=4). Points represent mean±s.e.mean of per cent response to KPSS.
AII responses following PD 123319 (100 nM) were not significantly different from control (Figure 1B). Furthermore, the combination of candesartan (100 pM) and PD 123319 (100 nM) showed no greater inhibition of AII responses than candesartan alone (Figure 1B).
Losartan and E-3174 also inhibited AII responses in an insurmountable manner (Figure 2A and 2B) and using the non-competitive model −log KB for losartan was calculated as 8.1±0.3 (geometric mean KB=7.9 nM, n=21). Analysis using the two state model estimates of 7.3 and 9.1 for −log KB for active and inactive state respectively. So, in this model losartan has a ∼50 fold greater affinity for the inactive compared with the active state of the AT1 receptor.
Figure 2.
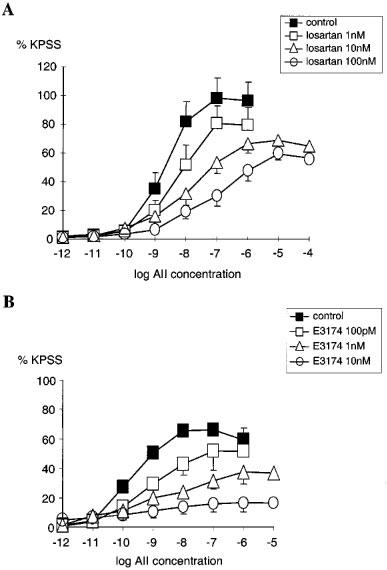
(A) Comparison of the cumulative concentration-response relationships for angiotensin II (AII 1 pM–100 μM) in PSSK (10.6 mM KCl) before (control (▪), n=5) and after exposure to losartan; (1 nM (□), n=7), (10 nM (▵), n=7), (100 nM (○), n=6). (B) Comparison of the cumulative concentration-response relationships for angiotensin II (AII 1 pM–10 μM) in PSSK before (control (▪), n=4) and after exposure to E-3174; (100 pM (□), n=4), (1 nM (▵), n=4), (10 nM (○), n=4). Points represent mean±s.e.mean of per cent response to KPSS.
Using the non-competitive model −log KB for E-3174 was calculated as 9.1±0.4 (KB=0.8 nM, n=20). Analysis using the two state model gave estimates of 8.9 and 11.3 for −log KB for active and inactive states respectively. Therefore the affinity of E-3174 for the inactive is approximately 250 fold greater than for the active state of the AT1 receptor.
Responses to NE (10 μM) and KPSS were unaffected by any of the antagonists at the concentrations studied (data not shown).
Endothelial factors
Removal of endothelium by rubbing had no significant effect on the concentration-response relationship for AII (n=4, NS) (Figure 3A). Pre-incubating the vessels with indomethacin also had no significant effect on the concentration-response relationship for AII when compared with untreated vessels (n=4, NS) (Figure 3B). Pre-incubation with L-NAME had no significant effect on the AII concentration-response relationship when compared with untreated vessels (n=4, NS) (Figure 4A). Similarly, in vessels pre-treated with BQ485 the concentration-response relationship for AII showed no difference compared to control (n=4, NS) (Figure 4B).
Figure 3.
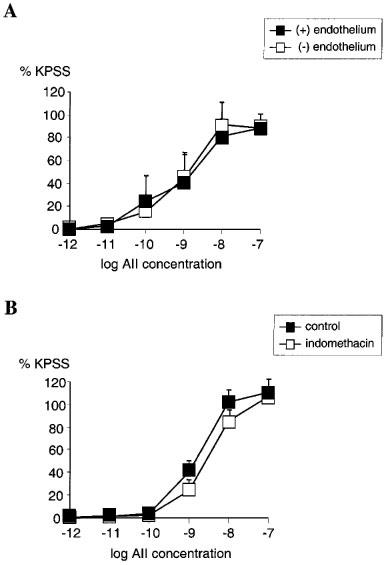
(A) Line graph showing response to increasing concentrations of angiotensin II in vessels with (▪) and without (□) endothelium (n=4). (B) Line graph showing response to increasing concentrations of angiotensin II in vessels with functional endothelium treated with 10 μM indomethacin (□) compared with control (▪) (n=4). Points represent mean±s.e.mean of per cent response to KPSS.
Figure 4.
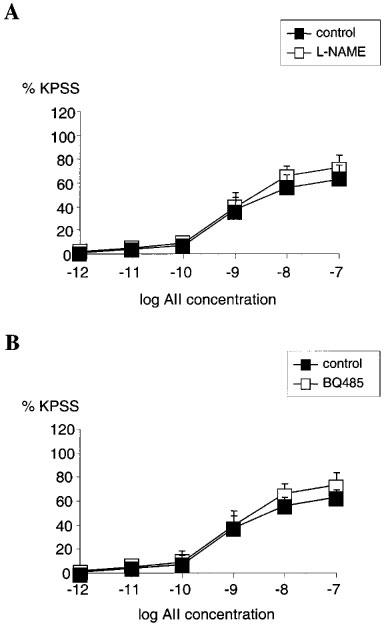
(A) Line graph showing response to increasing concentrations of angiotensin II in vessels with functional endothelium treated with 10 μM L-NAME (□) compared with control (▪) (n=4). (B) Line graph showing response to increasing concentrations of angiotensin II in vessels with functional endothelium treated with 1 μM BQ485 (□) compared with control (▪) (n=4). Points represent mean±s.e.mean of per cent response to KPSS.
Discussion
AII elicited concentration-related contractions of human subcutaneous resistance arteries. The AT1 receptor antagonists, candesartan, losartan and E-3174, reduced the maximal contractile response to AII suggestive of non-competitive antagonism. In the case of losartan the concentration-response curve also appeared to be displaced to higher concentrations giving the overall appearance of mixed competitive and non-competitive antagonism. Analysis of these data using a classical model of non-competitive antagonism (Kenakin, 1993) indicated that candesartan was ∼1000 fold more potent than losartan and ∼100 fold more potent than E-3174 as an antagonist at the AT1 receptor. The finding that losartan behaves as a non-competitive antagonist in human resistance arteries differs from previous observations in rabbit aorta (Chiu et al., 1990c; Wong et al., 1990a), rat aorta and rat mesenteric artery (Corriu et al., 1995) where this drug behaved as a competitive antagonist. In contrast, candesartan (Noda et al., 1993) and E-3174 (Wong et al., 1990b) have previously been reported to act in a non-competitive manner. Nevertheless all three agents generally behave as competitive antagonists in ligand binding assays (Chiu et al., 1990a,1990b; Wong et al., 1990a; Noda et al., 1993; Ojima et al., 1997), although candesartan has been reported to dissociate slowly from the AT1 receptor (Ojima et al., 1997). In an attempt to resolve these observations with our data we modelled the behaviour of the AT1 antagonists using a two-state model as originally outlined by Katz & Thesleff (1957) and developed subsequently (Rang & Ritter, 1970; Gero, 1983; Robertson et al., 1994). The key feature of this model is that ligands may possess different affinities for inactive as opposed to active states of the receptor and consequently can alter the equilibrium between active and inactive states. An overall reduction in active states due to a relatively higher affinity of antagonist for inactive state results in a reduction in maximal response giving the appearance of non-competitive antagonism. In contrast, a drug with equal affinity for active and inactive states behaves as a classical competitive antagonist. Although probably an oversimplification (Kenakin, 1997; Leff et al., 1997), this approach was able to account for our findings and provides an alternative explanation for apparent non-competitive behaviour of non-peptide antagonists without the need to postulate pseudo-irreversible interactions between antagonist and receptor.
Although adopting a slightly different perspective this proposal shares many features with an earlier analysis of peptide antagonists of AT1 receptors using an operational model of agonism (Liu et al., 1993). One interesting consequence of the two-state model is that the apparent behaviour of an antagonist may be influenced by factors governing receptor state transitions at rest, i.e. the proportion of inactive receptors under basal conditions. This could be a mechanism accounting for species- or tissue-based differences in antagonist behaviour.
In our studies the selective AT2 antagonist, PD123319, was not found to alter AII-induced responses in human subcutaneous resistance arteries, either alone or in the presence of candesartan. The majority of cardiovascular effects of AII appear to be mediated via the AT1 receptor and there is limited evidence for participation of AT2 receptors in vasoconstriction in vitro although AT2 binding sites have been characterized in rat cerebral arteries (Tsutsumi & Saavedra, 1991). Nonetheless, a role for the AT2 receptor in blood pressure regulation has been indicated. A study in rats has shown that AII causes an enhanced depressor response when AT1 receptors are blocked, this effect is blocked by the AT2 antagonist, PD123177 (Scheuer et al., 1993). Another study in rats showed that co-infusion of AII and PD123319 results in an enhanced blood pressure response compared to infusion of AII alone (Munzenmaier & Greene, 1996). In addition, in some tissues e.g. chick vasculature, AII has been reported to act via a non-AT1, non-AT2 receptor termed ATx receptor (Le Noble et al., 1993). However, in the absence of a selective antagonist for this receptor its possible functional role in human arteries could not be investigated.
Although AT1 receptors are found on smooth muscle, some studies have implicated the endothelium in modulating responses to AII in blood vessels. The interaction of AII with endothelium-derived relaxing factor (EDRF) / NO has been extensively studied in rat renal vasculature. Vasoconstriction of afferent and efferent arterioles was found to be significantly attenuated by L-NAME in rat kidneys pretreated with losartan (Ye & Healy, 1992). Furthermore, NO has been found to be a physiological antagonist of the glomerular and tubular responses to intra-renal AII (De Nicola et al., 1992). Another study has shown that endothelial release of NO mediated by AT1 receptors decreases contractions in response to AII in rat carotid artery (Boulanger et al., 1995).
AII has also been observed to induce release of endothelin-1 (ET-1) from cultured endothelial cells (Emori et al., 1991). Functional studies found that AII responses were abolished by inclusion of the ETA-receptor antagonist, BQ 123, in rabbit aorta with intact endothelium (Webb et al., 1992) and in rat tail artery (Chen et al., 1995).
Release of eicosanoids has also been proposed to contribute to AII-induced contraction. Indomethacin, a COX antagonist, was found to almost abolish AII responses in the presence of endothelium in spontaneously hypertensive (SHR) and in normotensive Wistar Kyoto (WKY) rats (Cortes et al., 1996). In contrast, indomethacin had no effect on AII-induced contraction in rat renal artery (Zhang et al., 1995) or rat carotid artery (Boulanger et al., 1995).
In our studies, we found no pharmacological evidence of a role for NO, ET-1 or COX products in modulating contractile responses to AII in human resistance arteries in vitro. In addition, the functional removal of vascular endothelium had no effect on contractile responses to AII in these vessels; a finding consistent with other observations in larger human blood vessels (He et al., 1993; Martinez et al., 1994; Jovanovic et al., 1995).
In summary, the present data indicate that the action of AII in human subcutaneous resistance arteries is mediated by AT1 receptors. The functionally important AT1 receptors are probably located on smooth muscle cells and endothelial release of NO, ET-1 and COX products do not contribute to the action of AII in these arteries. In human resistance arteries the non-peptide antagonists, candesartan, losartan and E-3174 act in an insurmountable manner. The apparent non-competitive action of these agents could be explained by competitive binding to two states of the AT1 receptor.
Acknowledgments
This work was supported by an educational grant from Takeda (GmbH). We are grateful to the cardiac surgeons and theatre staff at St. Mary's for supplying us with the tissue used in this study.
Abbreviations
- [A]
concentration of agonist
- AII
angiotensin II
- ANOVA
analysis of variance
- [B]
concentration of antagonist
- BQ-485
(Hexahydro-1H-azepinyl)carbonyl-Leu-D-Trp-D-Trp-OH.sodium salt
- EC50
concentration of drug producing 50% maximum response
- ET-1
endothelin-1
- KB
antagonist affinity
- KPSS
modified physiological saline containing 118 mM KCl
- L100
circumference at 100 mmHg transmural pressure
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- NE
noradrenaline
- PSS
physiological saline
- PSSK
modified physiological saline containing 10.6 mM KCl
References
- BAKRIS G.L., RE R.N. Endothelin modulates angiotensin II-induced mitogenesis of human mesangial cells. Am. J. Physiol. 1993;264:F937–F942. doi: 10.1152/ajprenal.1993.264.6.F937. [DOI] [PubMed] [Google Scholar]
- BOULANGER C.M., CAPUTO L., LEVY B.I. Endothelial AT1-mediated release of nitric oxide decreases angiotensin II contractions in rat carotid artery. Hypertension. 1995;26:752–757. doi: 10.1161/01.hyp.26.5.752. [DOI] [PubMed] [Google Scholar]
- CACHOFEIRO V., MAESO R., RODRIGO E., NAVARRO J., RUILOPE L.M., LAHERA V. Nitric oxide and prostaglandins in the prolonged effects of Losartan and Ramipril in hypertension. Hypertension. 1995;26:236–243. doi: 10.1161/01.hyp.26.2.236. [DOI] [PubMed] [Google Scholar]
- CHEN L., MCNEILL J.R., WILSON T.W., GOPALAKRISHNAN V. Heterogeneity in vascular smooth muscle responsiveness to angiotensin II (role of endothelin) Hypertension. 1995;26:83–88. doi: 10.1161/01.hyp.26.1.83. [DOI] [PubMed] [Google Scholar]
- CHIU A.T., MCCALL D.E., ALDRICH P.E., TIMMERMANS P.B. [3H]DUP 753, a highly potent and specific radioligand for the angiotensin II-1 receptor subtype. Biochem. Biophys. Res. Commun. 1990a;172:1195–202. doi: 10.1016/0006-291x(90)91575-d. [DOI] [PubMed] [Google Scholar]
- CHIU A.T., MCCALL D.E., ARDECKY R.J., DUNCIA J.V., NGUYEN T.T., TIMMERMANS P.B. Angiotensin II receptor subtypes and their selective nonpeptide ligands. Receptor. 1990b;1:33–40. [PubMed] [Google Scholar]
- CHIU A.T., MCCALL D.E., PRICE W.A., WONG P.C., CARINI D.J., DUNCIA J.V., WEXLER R.R., YOO S.E., JOHNSON A.L., TIMMERMANS P.B. Nonpeptide angiotensin II receptor antagonists. VII. Cellular and biochemical pharmacology of DuP 753, an orally active antihypertensive agent. J. Pharmacol. Exp. Ther. 1990c;252:711–718. [PubMed] [Google Scholar]
- CIAFRE S.A., D'ARMIENTO F.P., DI GREGORIO F., COLASANTI P., DI BENEDETTO A., LANGELLA A., DI IESO N., LIGUORI A., COLASANTI R., NAPOLI C. Angiotensin II stimulates endothelin-1 release from human endothelial cells. Recent Prog. Med. 1993;84:248–253. [PubMed] [Google Scholar]
- CONOVER W.J.Practical Nonparametric statistics 1980New York: John Wiley and sons; 2nd edn [Google Scholar]
- CORRIU C., BERNARD S., SCHOTT C., STOCLET J.C. Effects of losartan on contractile responses of conductance and resistance arteries from rats. J. Cardiovasc. Pharmacol. 1995;26:688–692. doi: 10.1097/00005344-199511000-00003. [DOI] [PubMed] [Google Scholar]
- CORTES S.F., ANDRIANTSITOHAINA R., STOCLET J.C. Alterations of cyclo-oxygenase and NO in responses to angiotensin II of resistance arteries from the spontaneously hypertensive rat. Br. J. Pharmacol. 1996;119:1635–1641. doi: 10.1111/j.1476-5381.1996.tb16083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE GASPARO M., HUSAIN A., ALEXANDER W., CATT K.J., CHIU A.T., DREW M., GOODFRIEND T., HARDING J.W., INAGANI T., TIMMERMANS P.B.M.W.M. Proposed update of angiotensin receptor nomenclature. Hypertension. 1995;25:924–927. doi: 10.1161/01.hyp.25.5.924. [DOI] [PubMed] [Google Scholar]
- DE NICOLA L., BLANTZ R.C., GABBAI F.B. Nitric oxide and angiotensin II. J. Clin. Invest. 1992;89:1248–1256. doi: 10.1172/JCI115709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOHI Y., HAHN A.W.A., BOULANGER C.M., BUHLER F.R., LUSCHER T.F. Endothelin stimulated by angiotensin II augments contractility of spontaneously hypertensive rat resistance arteries. Hypertension. 1992;19:131–137. doi: 10.1161/01.hyp.19.2.131. [DOI] [PubMed] [Google Scholar]
- EMORI T., HIRATA Y., OHTA K., KANNO K., EGUCHII S., IMAI T., SCHICHIRI M., MARUMO F. Cellular mechanism of endothelin-1 release by angiotensin and vasopressin. Hypertension. 1991;18:165–170. doi: 10.1161/01.hyp.18.2.165. [DOI] [PubMed] [Google Scholar]
- GERO A. Desensitization, two-state receptors and pharmacological parameters. J. Theor. Biol. 1983;103:137–161. doi: 10.1016/0022-5193(83)90204-7. [DOI] [PubMed] [Google Scholar]
- HABERL R.L. Role of angiotensin receptor subtypes in the response of rabbit brain arterioles to angiotensin. Stroke. 1994;25:1476–1479. doi: 10.1161/01.str.25.7.1476. [DOI] [PubMed] [Google Scholar]
- HE G.W., SHAW J., HUGHES C.F., YANG C.Q., THOMSON D.S., MCCAUGHAN B., HENDLE P.N., BAIRD D.K. Predominant alpha 1-adrenoceptor-mediated contraction in the human internal mammary artery. J. Cardiovasc. Pharmacol. 1993;21:256–263. [PubMed] [Google Scholar]
- JI H., LEUNG M., ZHANG Y., CATT K.J., SANDBERG K. Differential structural requirements for specific binding of nonpeptide and peptide antagonists to the AT1 angiotensin receptor: identification of amino acid residues that determine binding of the antihypertensive drug losartan. J. Biol. Chem. 1994;269:16533–16536. [PubMed] [Google Scholar]
- JOVANOVIC A., GRBOVIC L., ZIKIC I., TULIC I. Characterization of arginine vasopressin actions in human uterine artery: lack of role of the vascular endothelium. Br. J. Pharmacol. 1995;115:1295–1301. doi: 10.1111/j.1476-5381.1995.tb15039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUUL B., AALKJAER C., MULVANY M.J. Responses of femoral resistance vessels to angiotensin in vitro. Eur. J. Pharmacol. 1987;135:61–68. doi: 10.1016/0014-2999(87)90757-6. [DOI] [PubMed] [Google Scholar]
- KATZ B., THESLEFF S. A study of the ‘desensitization' produced by acetylcholine at the motor endplate. J. Physiol. (Lond.) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENAKIN T.P. Pharmacologic analysis of drug-receptor interaction 1993New York: Raven; 2nd edn [Google Scholar]
- KENAKIN T. Agonist-specific receptor conformations. Trends Pharmacol. Sci. 1997;18:416–417. doi: 10.1016/s0165-6147(97)01127-9. [DOI] [PubMed] [Google Scholar]
- LANG M.G., NOLL G., LUSCHER T.F. Effect of aging and hypertension on contractility of resistance arteries: modulation by endothelial factors. Am. J. Physiol. 1995;269:H837–H844. doi: 10.1152/ajpheart.1995.269.3.H837. [DOI] [PubMed] [Google Scholar]
- LE NOBLE F.A., SCHREURS N.H., VANSTRAATEN H.W., SLAAF D.W., SMITS J.F., ROGG H., STRUIJKER-BOUDIER H.A. Evidence for a novel angiotensin II receptor involved in angiogenesis in chick embryo chorioallantoic membrane. Am. J. Physiol. 1993;264:R460–R465. doi: 10.1152/ajpregu.1993.264.2.R460. [DOI] [PubMed] [Google Scholar]
- LEFF P., SCARAMELLINI C., LAW C., MCKECHNIE K. A three-state receptor model of agonist action. Trends Pharmacol. Sci. 1997;18:355–362. doi: 10.1016/s0165-6147(97)01105-x. [DOI] [PubMed] [Google Scholar]
- LIN L., NASJLETTI A. Role of endothelium-derived prostanoid in angiotensin II-induced vasoconstriction. Hypertension. 1991;18:158–164. doi: 10.1161/01.hyp.18.2.158. [DOI] [PubMed] [Google Scholar]
- LIU Y.J. Antagonist effect of losartan on angiotensin II induced contraction in five isolated smooth muscle assays. Eur. J. Pharmacol. 1993;240:147–154. doi: 10.1016/0014-2999(93)90892-l. [DOI] [PubMed] [Google Scholar]
- MANABE K., SHIRAHASE H., USUI H., KURAHASHI K., FUJIWARA M. Endothelium-dependent contractions induced by angiotensin I and angiotensin II in canine cerebral artery. J. Pharmacol. Exp. Ther. 1989;251:317–320. [PubMed] [Google Scholar]
- MARTINEZ M.C., VILA J.M., ALDASORO M., MEDINA P., FLOR B., LLUCH S. Relaxation of human isolated mesenteric arteries by vasopressin and desmopressin. Br. J. Pharmacol. 1994;113:419–424. doi: 10.1111/j.1476-5381.1994.tb17005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- MUNZENMAIER D.H., GREEN A.S. Opposing actions of angiotensin II on microvascular growth and arterial blood pressure. Hypertension. 1996;27:760–765. doi: 10.1161/01.hyp.27.3.760. [DOI] [PubMed] [Google Scholar]
- NODA M., SHIBOUTA Y., INADA Y., OJIMA M., WADA T., SANADA T., KUBO K., KOHARA Y., NAKA T., NISHIKAWA K. Inhibition of rabbit aortic angiotensin II (AII) receptor by CV-11974, a new nonpeptide AII antagonist. Biochem. Pharmacol. 1993;46:311–318. doi: 10.1016/0006-2952(93)90420-2. [DOI] [PubMed] [Google Scholar]
- OJIMA M., INADA Y., SHIBOUTA Y., WADA T., SANADA T., KUBO K., NISHIKAWA K. Candesartan (CV-11974) dissociates slowly from the angiotensin AT1 receptor. Eur. J. Pharmacol. 1997;319:137–146. doi: 10.1016/s0014-2999(96)00837-0. [DOI] [PubMed] [Google Scholar]
- PRIETO D., SIMONSEN U., NYBORG N.C. Regional involvement of an endothelium-derived contractile factor in the vasoactive actions of neuropeptide Y in bovine isolated retinal arteries. Br. J. Pharmacol. 1995;116:2729–2737. doi: 10.1111/j.1476-5381.1995.tb17234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANG H.P., RITTER J.M. On the mechanism of desensitization at cholinergic receptors. Mol. Pharmacol. 1970;6:357–382. [PubMed] [Google Scholar]
- RHALEB N.E., ROUISSI N., NANTEL F., D'ORLEANS-JUSTE P., REGOLI D. DuP 753 is a specific antagonist for the angiotensin receptor. Hypertension. 1991;17:480–484. doi: 10.1161/01.hyp.17.4.480. [DOI] [PubMed] [Google Scholar]
- ROBERTSON M.J., DOUGALL I.G., HARPER D., MCKECHNIE K.C.W., LEFF P. Agonist-antagonist interactions at angiotensin receptors: application of a two state receptor model. Trends Pharmacol. Sci. 1994;15:364–369. doi: 10.1016/0165-6147(94)90156-2. [DOI] [PubMed] [Google Scholar]
- SCHEUER D.A., PERRONE M.H. Angiotensin type 2 receptors mediate depressor phase of biphasic pressure response to angiotensin. Am J. Physiol. 1993;33:R917–R923. doi: 10.1152/ajpregu.1993.264.5.R917. [DOI] [PubMed] [Google Scholar]
- SHIBOUTA Y., INADA Y., OJIMA M., WADA T., NODA M., SANADA T., KUBO K., KOHARA Y., NAKA T., NISHIKAWA K. Pharmacological profile of a highly potent and long-acting angiotensin II receptor antagonist, CV-11974 and its prodrug, TCV-116. J. Pharmacol. Exp. Ther. 1993;266:114–120. [PubMed] [Google Scholar]
- TIMMERMANS P.B.M.W.M., WONG P.C., CHIU A.T., HERBLIN W.F., BENFIELD P., CARINI D.J., LEE R.J., WEXLER R.R., SAYE J.A.M., SMITH R.D. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- TSUTSUMI K., SAAVEDRA J.M. Characterization of AT2 angiotensin II receptors in rat anterior cerebral arteries. Am. J. Physiol. 1991;261:H667–H670. doi: 10.1152/ajpheart.1991.261.3.H667. [DOI] [PubMed] [Google Scholar]
- WEBB M.L., DICKINSON K.E.J., DELANEY C.L., LIU E.C.K., SERAFINO R., COHEN R.B., MONSHIZADEGAN H., MORELAND S. The endothelin receptor antagonist BQ 123 inhibits angiotensin II-induced contractions in rabbit aorta. Biochem. Biophys. Res. Commun. 1992;185:887–892. doi: 10.1016/0006-291x(92)91710-8. [DOI] [PubMed] [Google Scholar]
- WONG P.C., HART S.D., ZASPEL A.M., CHIU A.T., ARDECKY R.J., SMITH R.D., TIMMERMANS P.B. Functional studies of nonpeptide angiotensin II receptor subtype-specific ligands: DuP 753 (AII-1) and PD123177 (AII-2) J. Pharmacol. Exp. Ther. 1990a;255:584–592. [PubMed] [Google Scholar]
- WONG P.C., PRICE W.A. , JR, CHIU A.T., DUNCIA J.V., CARINI D.J., WEXLER R.R., JOHNSON A.L., TIMMERMANS P.B. Nonpeptide angiotensin II receptor antagonists. XI. Pharmacology of EXP3174: an active metabolite of DuP 753, an orally active antihypertensive agent. J. Pharmacol. Exp. Ther. 1990b;255:211–217. [PubMed] [Google Scholar]
- WU X.C., JOHNS E., MICHAEL J., RICHARDS N.T. Interdependence of contractile rsponses of rat small mesenteric arteries on nitric oxide and cyclo-oxygenase and lipoxygenase products of arachidonic acid. Br. J. Pharmacol. 1994;112:360–368. doi: 10.1111/j.1476-5381.1994.tb13080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE M.Q., HEALY D.P. Characterization of an angiotensin type 1 receptor partial cDNA from rat kidney- evidence for a novel AT1B receptor subtype. Biochem. Biophys. Res. Commun. 1992;185:204–210. doi: 10.1016/s0006-291x(05)80976-3. [DOI] [PubMed] [Google Scholar]
- ZHANG J., PFAFFENDORF M., ZHANG J.S., VAN ZWEITEN P.A. Influence of the vascular endothelium on angiotensin II-induced contractions in rabbit renal artery. Fundam. Clin. Pharmacol. 1995;9:25–29. doi: 10.1111/j.1472-8206.1995.tb00261.x. [DOI] [PubMed] [Google Scholar]