

Abstract
The main objective of the present study was to further evaluate the role of nitric oxide (NO) in delayed cardiac protection against ischaemia-reperfusion injury induced by monophosphoryl lipid A (MLA).
For this purpose, rats were administered with either 0.5 or 2.5 mg kg−1 MLA (i.p.). Eight or 24 h later, in vivo NO production in the heart was analysed by electron paramagnetic resonance (EPR) spin trapping technique. In parallel experiments, hearts were removed and perfused according to Langendorff. Functional ventricular parameters and incidence of ventricular fibrillation (VF) were determined after 30 min global ischaemic insult (37°C) followed by 30 min reperfusion. Vascular reactivity of aortic rings was also assessed.
Hearts from rats pretreated with 2.5 mg kg−1 MLA for 24 h (but not those from rats treated with 0.5 mg kg−1 MLA for 8 and 24 h, or with 2.5 mg kg−1 MLA for 8 h) exhibited preservation of ventricular function (LVDP, ±dP/dtmax) and a reduced incidence of VF (25% vs 87.5% in vehicle control) during reperfusion. At the cardioprotective dose of 2.5 mg kg−1 (for 8 or 24 h), MLA did not produce alterations of the contractile response of aortic rings to noradrenaline.
An increased formation of NO was detected in hearts removed from rats pretreated with 2.5 mg kg−1 MLA for 8 h, but not in those from rats treated for 24 h (or with 0.5 mg kg−1 MLA).
Pretreatment of the animals with the inhibitors of inducible NO-synthase, aminoguanidine (2×300 mg kg−1) or L-N6-(1-Iminoethyl)-lysine (L-NIL, 10 mg kg−1) abolished both MLA (2.5 mg kg−1)-induced rise of NO production (observed 8 h after MLA) and cardioprotection (observed 24 h after MLA). However MLA-induced cardioprotection was not attenuated when the hearts were perfused with aminoguanidine (150 μM) for 30 min before the ischaemic insult.
Altogether, the present data suggest that NO acts as a trigger rather then a direct mediator of the delayed cardioprotective effect of MLA in rat heart.
Keywords: Monophosphoryl lipid A, nitric oxide, cardioprotection, inducible NO synthase, spin trapping
Introduction
Monophosphoryl lipid A (MLA) obtained by modification of the lipid A domain of Salmonella minnesota lipopolysaccharide (LPS) is reported to retain some properties of LPS, although it is a thousand times less toxic (Ribi, 1984). Recently, attention has been focused on the ability of MLA to produce delayed cardioprotection against ischaemia-reperfusion injury, pharmacologically mimicking the second window of ischaemic preconditioning (for review Elliott 1998). Administered as a single dose 24 h prior to a cardiac ischaemic insult, MLA reduces reperfusion damage (infarct size, ventricular arrhythmias, myocardial stunning) in dogs (Yao et al., 1993a,1993b, Végh et al., 1996), rabbits (Elliott et al., 1996; Baxter et al., 1996) and rats (Nelson et al., 1991, Song et al., 1998). Due to its long duration and to the low toxicity of MLA, this effect, which is also produced by LPS, could present important benefits in clinical settings.
In rabbit and rat hearts, indirect evidence suggests a role for nitric oxide (NO) in the delayed cardioprotective effects of MLA. Indeed, MLA-induced cardioprotection was abrogated by nitric oxide synthase (NOS) inhibitors (Zhao et al., 1997; Tosaki et al., 1998). In addition it was associated with enhanced NOS activity (assessed by the citrulline assay) in the heart from MLA-treated rabbits (Zhao et al., 1997) and with expression of inducible NOS (iNOS) mRNA in the rat heart (Tosaki et al., 1998). However, to our knowledge no attempt has been made to evaluate nitric oxide production in the hearts of MLA-treated animals in vivo. Furthermore, the temporal relationship between myocardial NO production and cardioprotection has never been evaluated.
Therefore, the main objective of the present study was to further evaluate the role of NO in delayed cardiac protection against ischaemia-reperfusion injury induced by MLA in rat heart. For this purpose, NO production was assessed within cardiac tissue by in vivo NO spin trapping technique and cardioprotection was evaluated in isolated hearts by measuring functional ventricular parameters and the incidence of ventricular fibrillation (VF). The influence of aminoguanidine and L-N6-(1-Iminoethyl)-lysine (L-NIL), two inhibitors of iNOS activity (Misko et al., 1993, Griffiths et al., 1993, Moore et al., 1994) was evaluated in both cases. In addition, as cardioprotective dose of LPS may produce vascular hyporeactivity in rat (Yang et al., 1997), the consequences of MLA-treatment on aortic reactivity to noradrenaline was also examined.
Methods
The experiments conform with the National Institutes of Health guidelines for the use of laboratory animals (authorization number 01918 given by the French Government, Department of Agriculture).
Male Wistar rats (10–12-weeks-old, 280–300 g) were pretreated (i.p.) and randomly assigned to one of the following groups (Figure 1): vehicle controls (group I, n=8); 8 h pretreatment with MLA 0.5 mg kg−1 (group II, n=8); 24 h pretreatment with MLA 0.5 mg kg−1 (group III, n=8); 8 h pretreatment with MLA 2.5 mg kg−1 (group IV, n=8); 24 h pretreatment with MLA 2.5 mg kg−1 (group V, n=8). Further experiments were performed in the following groups, pretreated for 24 h with: aminoguanidine (2×300 mg kg−1)+ vehicle control (group VI, n=6); aminoguanidine (2×300 mg kg−1)+MLA (2.5 mg kg−1) (group VII, n=6); L-NIL (10 mg kg−1)+vehicle control (group VIII, n=4); L-NIL (10 mg kg−1)+MLA (2.5 mg kg−1) (group IX, n=4); vehicle control (group X, n=6); MLA (2.5 mg kg−1) (group XI, n=6). In the two latter groups, isolated hearts were perfused during the 30 min period that preceded the onset of ischaemia with aminoguanidine (150 μM). This concentration was described as being effective to inhibit iNOS activity in cells or tissue (Misko et al., 1993; Hasan et al., 1993). In addition, it corresponded to the highest concentration that did not significantly affect ventricular parameters in hearts removed from vehicle-control animals.
Figure 1.

Experimental protocol (*AG=aminoguanidine).
MLA was formulated at 1 mg ml−1 in 40% propylene glycol, 10% ethanol and water for injection. Vehicle control consisted of 40% propylene glycol, 10% ethanol and water for injection. Experiments in rats treated with the vehicle of MLA revealed that there was no significant difference in measured parameters after 8 or 24 h pretreatment (n=4 in each group). Therefore, the control vehicle group (group I) included animals treated with the vehicle of MLA for 8 and 24 h (n=8). Aminoguanidine (300 mg kg−1, dissolved in saline) was given (i.p.) at the same time as MLA (or vehicle) and 6 h later; L-NIL (10 mg kg−1, dissolved in saline) was given at the same time as MLA (or vehicle). This protocol was chosen from preliminary experiments showing the ability of such treatment to abolish the EPR signal for NO observed 8 h after MLA injection.
After 8 or 24 h treatment, the animals were anaesthetized with sodium pentobarbitone (60 mg kg−1 i.p.) and heparinized (500 U kg−1 i.p.).
Heart perfusion
Hearts were excised and placed in ice-cold Krebs-Henseleit buffer. They were then immediately perfused according to Langendorff at 37°C and pH 7.4 with Krebs-Henseleit solution containing (mmol l−1) NaCl 118, NaHCO3 24, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, CaCl2 1.7 and glucose 10, gassed with 95% O2/5% CO2. Perfusion pressure was constant and equivalent to 100 cm H2O. In this condition, the range of coronary flow was about 12–15 ml/min. The heart was placed in a semi-closed, circulating water warmed (37°C) air chamber. A water filled latex balloon connected to a Isotec® pressure transducer (Hugo-Sachs, Germany) was inserted into the left ventricle for measurement of left ventricular pressure. The left ventricular end diastolic pressure was set to 5 mmHg by adjusting the volume of the balloon. This balloon volume was maintained for the duration of the experiment. Cardiac parameters were monitored continuously and included: left ventricular developed pressure (LVDP: difference between left-ventricular peak systolic pressure and end-diastolic pressure), positive and negative first derivatives of LVDP (±dP/dtmax). An epicardial ECG was recorded throughout the experimental period by two electrodes attached directly to the heart. Hearts were equilibrated for 30 min, then subjected to 30 min global, no-flow ischaemia at 37°C, followed by 30 min reperfusion. Haemodynamic parameters were recorded during all experiments.
NO spin trapping and electron paramagnetic resonance (EPR) studies
NO spin trapping was performed in vivo using Fe-diethyldithiocarbamate (DETC) as a trap with following ex vivo EPR spectroscopy of the tissue (Vanin et al., 1984, Mülsch et al., 1995). Vehicle or MLA-pretreated animals were injected with the spin trap components, DETC (500 mg kg−1; i.p.) and FeSO4.7H2O plus Na-citrate (50 and 250 mg kg−1 respectively; s.c.). After 30 min, the animals were decapitated, the hearts were isolated and frozen in liquid nitrogen. EPR spectra were recorded on a Bruker 300E spectrometer with a Dewar flask (Wilmad, U.S.A.) at 77 K. EPR settings were 10 mW microwave power, 0.61 mT amplitude modulation, 9.47 GHz microwave frequency and 100 kHz modulation frequency. NO spin trapping data was quantified by comparison with paramagnetic signal of dinitrosyl iron-thiosulphate complex of known concentration (Mülsch et al., 1995).
Aortic ring preparations and contraction experiments
The thoracic aorta was removed from vehicle or MLA-pretreated animals, cleaned of connective and fat tissues and cut in rings (2–3 mm length). In some rings, endothelium was removed by gently rubbing the intimal surface with curved forceps. Rings were mounted under a passive tension of 2 g in organ bath filled with a Krebs solution (composition in mM: NaCl 119, NaHCO3 25, KCl 4.7, KH2PO4 1.18, MgSO4 1.17, CaCl2 1.25 and glucose 11) continuously kept at 37°C and bubbled with 95% O2/5% CO2. Tension was measured with an isometric force transducer. After 30 min equilibration period, addition of cumulative concentrations of noradrenaline (NA, 0.1 nM to 1 μM) was performed in the absence or in the presence of L-NG-nitroarginine methyl ester (L-NAME, 300 μM, added 15 min before NA). After completion of the concentration-response curve to NA (in rings untreated with L-NAME), acetylcholine (1 μM, to assess the presence or the absence of functional endothelium) or L-arginine (1 mM) was added.
Drugs
MLA was a kind gift of Ribi ImmunoChem Research Inc. (Montana, U.S.A.), pentobarbital was provided by Sanofi Santé Animale (Libourne, France) and L-NIL by Alexis Corporation (Läufelfingen, Switzerland). All the other drugs were obtained from Sigma (Grenoble, France).
Statistical analysis
Ventricular and vascular parameters were expressed as mean±standard error of the mean (s.e.mean). One way analysis of variance was carried out to test for differences between the different groups. If differences were established, the values of the drug-treated groups were compared with those of the corresponding control group by a Dunnett's post hoc test. For comparison of incidence of VF, the Fisher exact probability test was used. P values less than 0.05 were accepted as significant.
Results
Effects of MLA on ventricular functional parameters and arrhythmias
The ventricular functional parameters LVDP, +dP/dtmax and −dP/dtmax were not significantly different between groups at the end of the equilibration period before ischaemia (Table 1). The end-reperfusion values were compared with those obtained at the end of equilibration period (30 min). The recovery of LVDP, +dP/dtmax and −dP/dtmax after ischaemia-reperfusion in the different experimental groups (expressed as a percentage of the values obtained at the end of the equilibration period) is illustrated in Figure 2A, B and C respectively, and in Table 2. The hearts from the control (vehicle) group did not recover their initial values of LVDP, +dP/dtmax, −dP/dtmax after an episode of 30 min no-flow ischaemia, followed by 30 min reperfusion. At the dose of 0.5 mg kg−1 after 8 or 24 h, MLA did not significantly modify the recovery of ventricular contractile function. Eight h after pretreatment with 2.5 mg kg−1 MLA, these parameters presented a slight, albeit not significant recovery. However, 24 h after treatment with the same dose of MLA, recovery of LVDP, +dP/dtmax, −dP/dtmax values were significantly improved. During reperfusion, 87.5% of the vehicle control hearts developed VF (Figure 2D). In groups pretreated with 0.5 mg kg−1 MLA (8 or 24 h) or with 2.5 mg kg−1 MLA (8 h), the incidence of VF was not significantly different from the value obtained in controls. However, it was significantly reduced in the group treated with 2.5 mg kg−1 MLA for 24 h. The influence of aminoguanidine (2×300 mg kg−1) and L-NIL (10 mg kg−1) was studied in animals treated with 2.5 mg kg−1 MLA for 24 h (in which MLA-induced cardioprotection was significant). Both inhibitors prevented the beneficial effects of MLA on LVDP, +dP/dtmax, −dP/dtmax and VF, the parameters reaching values which were not significantly different from those obtained in corresponding controls (Figure 2 and Table 2). However, in heart removed from MLA treated animals (2.5 mg kg−1 for 24 h) and perfused with aminoguanidine (150 μM) 30 min before the onset of ischaemia, the delayed cardioprotection was not attenuated as indicated by the lack of significant recovery of LVDP, ±dP/dtmax and the low incidence of VF (Figure 2 and Table 2).
Table 1.
Ventricular functional parameters before ischaemia (preischaemic values)
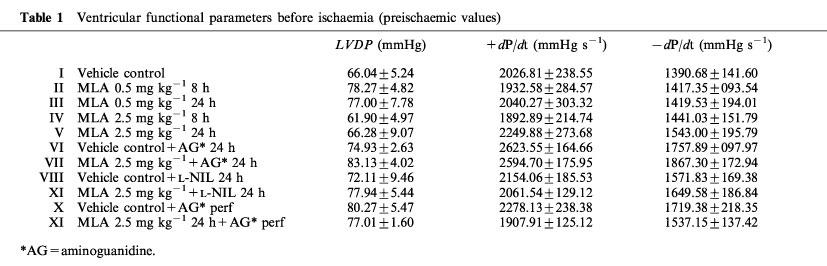
Figure 2.

Postischaemic recovery of the functional ventricular parameters LVDP (A), +dP/dtmax (B), −dP/dtmax (C) and incidence of VF during reperfusion (D) in the different experimental groups (see Methods and Figure 1 for the description of the groups). (*P<0.05 vs control).
Table 2.
Recovery of ventricular functional parameters and the incidence of VF in different control groups

NO spin trapping
No EPR spectra characteristic for NO adducts were found in the hearts of control (vehicle) rats (Figure 3A). In the groups pretreated with 0.5 mg kg−1 MLA for 8 or 24 h, the NO production in the heart was also below detection limit. However, after animals were pretreated with 2.5 mg kg−1 MLA, their hearts exhibited a prominent triplet EPR signal (g=2.035) which is attributed to NO-Fe-DETC complex indicating NO production. The signal was observed only at 8 h (Figure 3B) and disappeared 24 h (Figure 3C) after MLA pretreatment (at 8 h, NO production was 577±73 pmol g−1 wet tissue h−1, n=3). The characteristic EPR signal was not detected in the hearts of animals pretreated for 8 h with MLA and aminoguanidine (Figure 3D) or with MLA and L-NIL (not shown).
Figure 3.

In vivo NO spin trapping in rat heart by Fe-DETC complex. Rats were pretreated with vehicle (A), 2.5 mg kg−1 MLA for 8 h (B), 2.5 mg kg−1 MLA for 24 h (C) or 2.5 mg kg−1 MLA plus 2×300 mg kg−1 aminoguanidine for 8 h (D). EPR spectra were recorded at 77 K. Then spectra of control heart and Cu-DETC complex were substrated. The traces are representative of three experiments. The values of the spectroscopic splitting factor (g⊥ and g‖) and the magnetic field scale are shown at the top and at the bottom, respectively.
Effects of MLA pretreatment on reactivity of aortic rings
At the cardioprotective dose of 2.5 mg kg−1 (for 8 or 24 h), MLA did not significantly affect the contractile effect of NA in aortic rings with or without functional endothelium (Figure 4). The NOS inhibitor L-NAME (300 μM) did not significantly affect the sensitivity and the maximal effect of NA in endothelium-denuded rings removed from vehicle control or MLA pretreated animals, whereas in aortic rings with endothelium from both control and MLA-treated rats, it produced a significant shift of the concentration-response curve of NA to the left (3.4–5.4 fold decrease of the EC50 values of NA). Addition of L-arginine (1 mM), failed to induce a significant relaxing effect (0.4±0.4, 0.6±0.4, 5.0±2.8% relaxation in endothelium-denuded rings removed from control rats, or rats treated with MLA for 8 or 24 h, respectively, and 0.8±0.8, 4.5±4.1, 2.2±1.3% in rings with endothelium from control rats, or those treated with MLA for 8 or 24 h, respectively).
Figure 4.
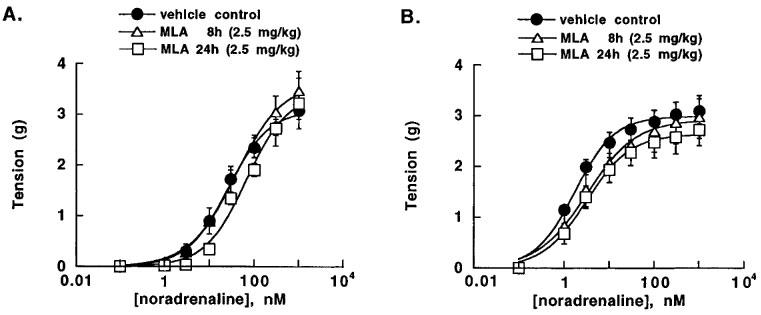
Concentration-response curve of NA in aortic rings with or without functional endothelium, prepared from control vehicle rats and rats treated for 8 or 24 h with 2.5 mg kg−1 MLA. Results are expressed as mean±s.e.mean of five experiments.
Discussion
The main results obtained in the present study are that MLA was able to increase NO production in the rat heart and that there was a temporal dissociation between the NO production and the MLA-induced cardioprotection: the increased NO formation preceeded the myocardial protection and had returned to undetectable levels by the time the protective effect was evident. In addition, both MLA-induced increase in NO production and cardioprotection were abrogated by pretreatment of the rats with iNOS inhibitors.
In the present study, MLA (2.5 mg kg−1) attenuated ventricular contractile dysfunction and decreased the incidence of VF during reperfusion when was injected to rats 24 h before an ischaemic period. These results are in agreement with those obtained by Nelson et al. (1991) and Tosaki et al. (1998) in a similar rat model. The data obtained in the present study also provide some additional information on the dose-response relationship of MLA in rat (no cardioprotection was evident with an i.p. dose of 0.5 mg kg−1) and on the time-course of the protective effect (no cardioprotection with the dose of 2.5 mg kg−1 after 8 h, but after 24 h). In this study, pretreatment of the rats either with aminoguanidine or L-NIL, two selective iNOS inhibitors (Misko et al., 1993, Hasan et al., 1993; Moore et al. 1994, Connor et al., 1995) prevented the protection afforded by MLA on ventricular parameters and on the incidence of VF. Although the ability of aminoguanidine to decrease MLA-induced myocardial NO production was demonstrated in the present study, the contribution of other mechanisms of action of aminoguanidine cannot be excluded on the basis of the data obtained with this drug (Southan & Szabo, 1996). However, as L-NAME (Tosaki et al., 1998) and L-NIL (this study) also abrogated the cardioprotective effect of MLA in rats, inhibition of NOS activity was the most probable mechanism by which these drugs prevented MLA-induced protection. As aminoguanidine and L-NIL are relatively selective for inhibition of iNOS, the present data suggest that the delayed protective effect of MLA was related to the induction of iNOS. This is also supported by the ability of MLA to increase iNOS mRNA expression in rat heart (Tosaki et al., 1998). However it cannot be excluded that the inhibitors used also affected the activity of the constitutive NOS in vivo.
The spin trapping technique, which was used here allows the evaluation of NO availability in tissues in vivo (Vanin et al., 1984, Mülsch et al., 1995). An increased level of NO was detected in non-ischaemic heart 8 h after 2.5 mg kg−1 MLA pretreatment, whereas rise in NO production was not detected 24 h after the same dose of MLA. This time course of NO production after MLA is closely related to that of iNOS mRNA expression, reported previously (Tosaki et al., 1998), consistent with the view that NO which was detected by the EPR method mainly derived from MLA-induced iNOS activity. The involvement of iNOS in MLA-induced enhanced NO production is further supported by the ability of the iNOS inhibitors, aminoguanidine and L-NIL to abolish the EPR signal for NO. However, as mentioned previously an inhibitory effect of aminoguanidine or L-NIL on NO production by constitutive NOS cannot be ruled out.
The difference in the time course of MLA-induced cardioprotection and NO production found here is striking. Indeed 8 h after MLA, NO production was increased in the heart whereas no functional protection was evident. By contrast, 24 h after MLA, there was no EPR detectable NO whereas a cardioprotective effect was observed. At the same time (24 h after MLA) treatment of the isolated hearts with aminoguanidine just before the ischaemic insult did not attenuate the cardioprotection. As the expression of iNOS mRNA was not detected in the rat heart 24 h after MLA (Tosaki et al., 1998), the possibility that an ischaemic insult triggers MLA-induced NO production by a latent form of iNOS as suggested in the rabbit (Zhao et al., 1997), is unlikely in the rat. As previously reported (Elliott, 1998), the mechanism of protection afforded by MLA may differ among species and experimental models. The present data rather suggest that in rat heart elevation of cardiac NO production does not result in simultaneous onset of cardioprotection. However, as pretreatment with iNOS inhibitors abrogated both MLA-induced protection (observed at 24 h) and the rise in NO production (observed at 8 h), it is likely that the NO elevation that occured several hours before the onset of ischaemia is required for MLA-induced cardioprotection.
The temporal dissociation between the effect of MLA on cardiac NO production and cardioprotection suggests that NO acts as a trigger of protective mechanisms in hearts from MLA-treated animals. The ability of NO to trigger and/or to mediate the late preconditioning induced by short ischaemic periods was recently suggested by Bolli et al. (1997). There are several potential mechanisms which may be triggered by NO and contribute to protection against ischaemia-reperfusion damages. As most of ischaemia-reperfusion injury is ultimately due to a burst of reactive oxidative species that occurs upon readmission of oxygen (Wang & Zweier 1996, Yasmin et al., 1997), induction of antioxidant enzymes like catalase, superoxide dismutase (SOD) or glutathione peroxidase, likely play an important role in the delayed protection afforded by preconditioning (Richard et al., 1996). It has been demonstrated that NO enhances the expression of Mn SOD (Sano et al., 1996) and that MLA or endotoxin-induced cardioprotection is associated with increased catalase expression and activity (Brown et al., 1989, Nelson et al., 1991). The expression of heat shock proteins (HSP), especially the HSP70 family, is another potential mechanism involved in the delayed protective effect of preconditioning (Richard et al., 1996). Interestingly, HSP expression may also be triggered by NO and MLA in the heart and in cardiac cells, respectively (Malyshev et al., 1996, Nayeem et al., 1997). The contribution of all these potential mechanisms in the NO-triggered delayed cardioprotective effects of MLA remains to be investigated.
Interestingly, the present results also show that although MLA increased NO production in the heart, the ventricular functional parameters were not affected in the experimental conditions. This distinguishes MLA from its parent molecule LPS, which at cardioprotective dose, was reported to decrease basal cardiac contraction by a NO-dependent mechanism (Yang et al., 1997). Another difference between MLA and LPS is their effect on aortic reactivity. In aortic rings removed from rats treated for 8 or 24 h with MLA, the contractile response to NA was not significantly affected both in the absence and in the presence of L-NAME, and the NOS substrate L-arginine failed to induce a significant relaxing effect. These data show no evidence for NO-mediated aortic hyporeactivity or relaxation. Moreover in rings with endothelium, MLA did not affect the potentiating effect of L-NAME on vascular response to NA, showing no modification by MLA of the basal influence of endothelium-derived NO in the aorta. The lack of effect of MLA on vascular contractility and cardiac function (in the absence of ischaemia) is consistent with the observation that in a similar model, MLA did not modify blood pressure or heart rate (Song et al., 1998). By contrast, it was reported that a cardioprotective dose of LPS produced NO-mediated hypo-reactivity to contractile agonists in aortic rings, as well as a decrease of eNOS activity (Yang et al., 1997). The reasons for these differential effects of MLA and LPS, especially in blood vessels, remains to be investigated. In conclusion, the present study shows that MLA increased NO production in rat heart and that the rise of NO production preceeded cardioprotection. Early inhibition of iNOS abolished the rise of cardiac NO, as well as the delayed protective effects of MLA. By contrast inhibition of iNOS just before the ischaemic insult did not attenuate cardioprotection. Taken together, these results suggests that in this model, NO acts as a trigger rather than a direct mediator of the delayed cardioprotective effects of MLA.
Acknowledgments
This work was supported by INTAS (97-0524) and Biomed 2 (PL 950979). A.L. Kleschyov was supported by Fondation de la Recherche Médicale. We are indebted to Dr. G.T. Elliott for the generous gift of MLA and the fruitful discussion of the results. K. György is indebted to Professors J. Gy. Papp and J.R. Parratt for all their input in the realization of this collaborative work between laboratories of Szeged and Strasbourg. The authors also acknowledge C. Untereiner and D. Wagner for technical assistance.
Abbreviations
- DETC
diethyldithiocarbamate
- EPR
electron paramagnetic resonance
- iNOS
inducible nitric oxide synthase
- L-NAME
L-NG-nitroarginine methyl ester
- L-NIL
L-N6-(1-Iminoethyl)-lysine
- LPS
lipopolysaccharide
- LVDP
left ventricular developed pressure
- MLA
monophosphoryl lipid A
- NA
noradrenaline
- NO
nitric oxide
- NOS
nitric oxide synthase
- SOD
superoxide dismutase
- VF
ventricular fibrillation
References
- BAXTER G.F., GOODWIN R.W., WRIGHT M.J., KERAC M., HEADS R.J., YELLON D.M. Myocardial protection after monophosphoryl lipid A: studies of delayed anti-ischaemic properties in rabbit heart. Br. J. Pharmacol. 1996;117:1685–1692. doi: 10.1111/j.1476-5381.1996.tb15340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLLI R., MANCHIKALAPUDI S., TANG X-L., TAKANO H., QIU Y., GUO Y., ZHANG Q., JADOON A.K. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Circ. Res. 1997;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- BROWN J.M., GROSSO M.A., TERADA L.S., WHITMAN G.J.R., BANERJEE A., WHITE C.W., HARKEN A.H., REPINE J.E. Endotoxin pretreatment increases endogenous myocardial catalase activity and decreases ischaemia-reperfusion injury of isolated rat hearts. Proc. Natl. Acad. Sci. 1989;86:2516–2520. doi: 10.1073/pnas.86.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOR J.R., MANNING P.T., SETTLE S.L., MOORE W.M., JEROME G.M., WEBBER R.K., TJOENG F.S., CURRIE M.G. Suppression of adjuvant-induced arthritis by selective inhibition of inducible nitric oxide synthase. Eur. J. Pharmacol. 1995;273:15–24. doi: 10.1016/0014-2999(94)00672-t. [DOI] [PubMed] [Google Scholar]
- ELLIOTT G.T. Monophosphoryl lipid A induces delayed preconditioning against cardiac ischaemia-reperfusion injury. J. Mol. Cell. Cardiol. 1998;30:3–17. doi: 10.1006/jmcc.1997.0586. [DOI] [PubMed] [Google Scholar]
- ELLIOTT G.T., COMERFORD M.L., SMITH J.R., ZHAO L. Myocardial ischaemia/reperfusion protection using monophosphoryl lipid A is abrogated by the ATP-sensitive potassium channel blocker, glibenclamide. Cardiovasc. Res. 1996;32:1071–1080. doi: 10.1016/s0008-6363(96)00154-x. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS M.J.D., MESSENT M., MACALLISTER R.J., EVANS T.W. Aminoguanidine selectively inhibits inducible nitric oxide synthase. Br. J. Pharmacol. 1993;11:963–968. doi: 10.1111/j.1476-5381.1993.tb13907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASAN K., HEESEN B.-J., CORBETT J.A., MCDANIEL M.L., CHANG K., ALLISON W., WOLFFENBUTTEL B.H.R., WILLIAMSON J.R., TILTON R.G. Inhibition of nitric oxide formation by guanidines. Eur. J. Pharmacol. 1993;249:101–106. doi: 10.1016/0014-2999(93)90667-7. [DOI] [PubMed] [Google Scholar]
- MALYSHEV I.Y., MALUGIN A.V., MANUKHINA E.B., LARIONOV N.P., MALENYUK E.B., MALYSHEVA E.V., MIKOYAN V.D., VANIN A.F. Is HSP70 involved in nitric oxide-induced protection of the heart. Physiol. Res. 1996;45:267–272. [PubMed] [Google Scholar]
- MISKO T.P., MOORE W.M., KASTEN T.P., NICKOLS G.A., CORBETT J.A., TILTON R.G., MCDANIEL M.L., WILLIAMSON J.R., CURRIE M.G. Selective inhibition of inducible nitric oxide synthase by aminoguanidine. Eur. J. Pharmacol. 1993;233:119–125. doi: 10.1016/0014-2999(93)90357-n. [DOI] [PubMed] [Google Scholar]
- MOORE W.M., WEBBER R.K., JEROME G.M., TJOENG F.S., MISKO T.P., CURRIE M.G. L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J. Med. Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- MÜLSCH A., MORDVINTCEV P., BASSENGE E., LUNG F., CLEMENT B., BUSSE R. In vivo spin trapping of glycerol trinitrate-derived nitric oxide in rabbit blood vessels and organs. Circulation. 1995;92:1876–1882. doi: 10.1161/01.cir.92.7.1876. [DOI] [PubMed] [Google Scholar]
- NAYEEM M.A., ELLIOTT G.T., SHAH M.R., HASTILLO-HESS S.L., KUKREJA R.C. Monophosphoryl lipid A protects adult rat cardiomyocytes with specific induction of the 72-KD heat shock protein. J. Mol. Cell. Cardiol. 1997;29:2303–2310. doi: 10.1006/jmcc.1997.0452. [DOI] [PubMed] [Google Scholar]
- NELSON D.W., BROWN J.M., BANERJEE A., BENSARD D.D., ROGERS K.B., LOCKE-WINTER C.R., ANDERSON B.O., HARKEN A.H. Pretreatment with a nontoxic derivative of endotoxin induces functional protection against cardiac ischemia/reperfusion injury. Surgery. 1991;110:365–369. [PubMed] [Google Scholar]
- RIBI E. Beneficial modification of the endotoxin molecule. J. Biol. Response Modif. 1984;3:1–9. [PubMed] [Google Scholar]
- RICHARD V., KAEFFER N., THUILLEZ C. Delayed protection of the ischaemic heart–from pathophysiology to therapeutic applications. Fundam. Clin. Pharmacol. 1996;10:409–415. doi: 10.1111/j.1472-8206.1996.tb00595.x. [DOI] [PubMed] [Google Scholar]
- SANO H., HIRAI M., SAITO H., NAKASHIMA I., ISOBE K. Nitric oxide releasing reagent, S-nitroso-N-acetylpenicillamine, enhances the expression of manganese superoxide dismutase mRNA in rat vascular smooth muscle cells. J. Cell. Biochem. 1996;62:50–55. doi: 10.1002/(sici)1097-4644(199607)62:1<50::aid-jcb6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- SONG W., FURMAN B.L., PARRATT J.R. Monophosphoryl lipid A reduces both arrhythmia severity and infarct size in a rat model of ischaemia. Eur. J. Pharmacol. 1998;345:285–287. doi: 10.1016/s0014-2999(98)00126-5. [DOI] [PubMed] [Google Scholar]
- SOUTHAN G.J., SZABO C. Selective pharmacological inhibition of distinct nitric oxide synthases with variable isoforms selectivity. Biochem. Pharmacol. 1996;51:383–394. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- TOSAKI A., MAULIK N., ELLIOTT G.T., BLASIG I.E., ENGELMAN R.M., DAS D.K. Preconditioning of rat heart with monophosphoryl lipid A: a role for nitric oxide. J. Pharmacol. Exp. Ther. 1998;285:1274–1279. [PubMed] [Google Scholar]
- VANIN A.F., MORDVINTCEV P.I., KLESCHYOV A.L. Appearance of nitrogen oxide in animal tissues in vivo. Stud. Biophys. 1984;102:135–143. [Google Scholar]
- VÉGH A., PAPP J.GY., ELLIOTT G.T., PARRATT J.R.Pretreatment with monophosphoryl lipid A (MPL-C) reduces ischaemia reperfusion-induced arrhythmias in dogs J. Mol. Cell. Cardiol. 199628A56(abstract) [DOI] [PubMed] [Google Scholar]
- WANG P., ZWEIER J.L. Measurement of nitric oxide and peroxynitrite generation in the postischaemic heart: evidence for peroxynitrite-mediated reperfusion injury. J. Biol. Chem. 1996;271:29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- YANG B.C., CHEN L.Y., SALDEEN T.G.P., MEHTA J.L. Reperfusion injury in the endotoxin-treated rat heart: reevaluation of the role of nitric oxide. Br. J. Pharmacol. 1997;120:305–311. doi: 10.1038/sj.bjp.0700891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAO Z., AUCHAMPACH J.A., PIEPER G.M., GROSS G.J. Cardioprotective effects of monophosphoryl lipid A, a novel endotoxin analog, in the dog. Cardiovasc. Res. 1993a;27:832–838. doi: 10.1093/cvr/27.5.832. [DOI] [PubMed] [Google Scholar]
- YAO Z., RASMUSSEN J.L., HIRT J.L., MEI D.A., PIEPER G.M., GROSS G.J. Effects of monophosphoryl lipid A on myocardial ischaemia/reperfusion injury in dogs. J. Cardiovasc. Pharmacol. 1993b;22:653–663. doi: 10.1097/00005344-199310000-00021. [DOI] [PubMed] [Google Scholar]
- YASMIN W., STRYNADKA K.D., SCHULZ R. Generation of peroxynitrite contributes to ischaemia-reperfusion injury in isolated rat hearts. Cardiovasc. Res. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- ZHAO L., WEBER P.A., SMITH J.R., COMERFORD M.L., ELLIOTT G.T. Role of inducible nitric oxide synthase in pharmacological ‘preconditioning' with monophosphoryl lipid A. J. Mol. Cell. Cardiol. 1997;29:1567–1576. doi: 10.1006/jmcc.1997.0390. [DOI] [PubMed] [Google Scholar]