

Abstract
Volume-Sensitive, Outwardly Rectifying (VSOR) Cl− currents were measured in canine colonic myocytes by whole-cell patch clamp. Decreasing extracellular osmolarity 50 milliosmoles l−1 activated current that was carried by Cl− and 5–7 times greater in the outward direction.
Niflumic acid, an inhibitor of Ca2+-activated Cl− channels, did not inhibit VSOR Cl− current. Glibenclamide, an antagonist of CFTR, and anthracene-9-carboxylate (9-AC) inhibited current less than 25% at 100 μM.
DIDS (4,4-diisothiocyanato-stilbene-2,2′disulphonate) inhibited VSOR Cl− current more potently than SITS (4-acetamido-4′-isothiocyanato-stilbene-2,2′-disulphonate). IC50s were 0.84 and 226 μM, respectively.
VSOR Cl− current was strongly inhibited by tamoxifen ([Z]-1-[p-dimethylaminoethoxy-phenyl]-1,2-diphenyl-1-butene), an anti-oestrogen compound (IC50=0.57 μM).
Gd3+ antagonized VSOR Cl− current more potently than La3+. The IC50 for Gd3+ was 23 μM. In contrast, 100 μM La3+ inhibited current only 35±7%.
Antagonists of VSOR Cl− current had non-specific effects. These compounds blocked voltage-dependent K+ and Ca2+ currents in colonic myocytes. Tamoxifen (10 μM) and DIDS (10 μM) inhibited L-type Ca2+ current 87±7 and 31±5%, respectively. Additionally, in the presence of 300 nM charybdotoxin, tamoxifen (1 μM) and DIDS (10 μM) inhibited delayed rectifier K+ current 38±8 and 10±2%, respectively.
The pharmacology of VSOR Cl− channels overlaps with voltage-dependent cation channels. DIDS and tamoxifen inhibited VSOR Cl− equally. However, because DIDS had much less effect on L-type Ca2+ and delayed rectifier K+ channels than did tamoxifen, it might be useful in experiments to investigate the physiological and pathophysiological role of this conductance in whole tissues.
Keywords: Smooth muscle, tamoxifen, DIDS, volume-sensitive Cl− channels, voltage-gated Ca2+ channels, delayed rectifier K+ channels
Introduction
Distribution of VSOR Cl− channels is widespread in mammalian cells (Nilius et al., 1994), including smooth muscle (Dick et al., 1998; Greenwood & Large, 1998; Xu et al., 1997; Yamazaki et al., 1998). Cl− channels pass currents carried by organic osmolytes and halides and are known to aid in the control of cell volume, pH, and membrane potential (Strange et al., 1996). Swelling-activated Cl− channels are grouped in a gene superfamily known as ClC (Jentsch et al., 1995), with nine known members. There has been debate over the molecular entities that underlie VSOR Cl− currents (Clapham, 1998; Okada, 1997; 1998; Strange, 1998), and different channels may contribute to responses in different cell types. ClC-3, cloned from guinea-pig ventricular myocytes, is thought to be the cardiac VSOR Cl− channel (Duan et al., 1997b). ClC-3 gene transcripts are also present in vascular (Yamazaki et al., 1998) and visceral (Dick et al., 1998) smooth muscle cells. Although the presence of ClC-3 Cl− channels per se has not yet been demonstrated in smooth muscle, the VSOR Cl− conductance present in this cell type could potentially participate in stretch-dependent regulation of excitability and contractility (Nelson, 1998).
Cl− current has been shown to be an important regulator of the electrophysiology of cardiac myocytes (Vandenberg et al., 1997). Recently, a role has been suggested for stretch-sensitive Cl− conductances in the regulation of membrane potential and tone of vascular smooth muscle (Nelson et al., 1997). DIDS and indanyloxyacetic acid hyperpolarized and relaxed isolated arterioles in a concentration-dependent manner, but niflumic acid was without effect. These data suggest that a VSOR Cl− conductance could contribute to the smooth muscle myogenic response (Bayliss, 1902; Bülbring, 1955; Harder, 1984; Meininger & Davis, 1992). Due to the lack of specificity of Cl− channel antagonists, however, the physiological relevance of stretch-sensitive Cl− conductances has been difficult to determine in smooth muscle (Doughty et al., 1998).
There are relatively few studies of Cl− currents in visceral smooth muscles (e.g., Akbarali & Giles, 1993; Sun et al., 1992), and very little is known about the role of VSOR Cl− conductances. The equilibrium potential for Cl− (ECl) in smooth muscle cells is approximately −25 mV (Aickin & Brading, 1982). This is considerably less negative than the resting membrane potential of gastrointestinal muscle and even less negative than the peak of the electrical slow wave (e.g., Smith et al., 1987). Thus, activation of Cl− conductances in gastrointestinal muscles would result in efflux of Cl− (net inward current) and a tendency to depolarize gastrointestinal muscle cells. The link between Cl− efflux and contractility is that depolarization activates voltage-dependent Ca2+ channels and initiates contraction of gastrointestinal muscles (Langton et al., 1989; Ozaki et al., 1991; Ward & Sanders, 1992).
We have recently demonstrated the presence, biophysical properties, and some of the regulatory mechanisms of a VSOR Cl− conductance in smooth muscle cells isolated from the circular layer of the canine colon (Dick et al., 1998). Hypotonic solutions activated an outwardly rectifying Cl− current that demonstrated voltage-dependent inactivation. Tamoxifen, DIDS, and extracellular ATP inhibited the current, and the current was regulated by protein kinase C (Dick et al., 1998). These characteristics mimic the cardiac swelling-activated Cl− current attributed to ClC-3 gene expression in guinea pig ventricular muscle (Duan et al., 1997a,1997b; 1999). ClC-3 gene transcripts were detected in isolated colonic myocytes by the reverse transcriptase polymerase chain reaction (Dick et al., 1998). Thus, it is possible that at least a portion of the VSOR Cl− current in gastrointestinal muscles is due to functional expression of ClC-3. In the present study we have further characterized the VSOR Cl− conductance in canine colonic myocytes by constructing pharmacological profiles of drugs that block the current. We have also tested the effects of VSOR Cl− current blockers on L-type Ca2+ and delayed rectifer K+ channels to determine whether these agents could be used to investigate the role of VSOR Cl− in smooth muscles with assays of electrical or contractile activity. This information is important because it may facilitate the rational design of experiments to investigate the physiological and pathophysiological roles of VSOR Cl− currents in smooth muscles.
Methods
Tissue collection and cell preparation
The care and use of animals followed the recommendations and guidelines of the National Institutes of Health and were approved by the University of Nevada Animal Care and Use Committee. Mongrel dogs of either sex were anaesthetized with 20 mg kg−1 ketamine and 55 mg kg−1 nembutol and the abdomen opened. The proximal colon was removed and placed in Krebs buffer that contained (in mM) NaCl 125, KCl 5.9, CaCl2 2.5, MgCl2 1.2, NaHCO3 15.5, Na2HPO4 1.2, glucose 11.5, pH maintained at 7.4 by bubbling with 95% O2 and 5% CO2. Salts for Krebs and other solutions were purchased from Sigma Chemical Co. (St. Louis, Missouri) and Fisher Scientific (Fairlawn, New Jersey).
Strips of colon were cut and pinned in a dissection dish. The circular tunica muscularis, separated from the longitudinal layer, was dissected free of the surrounding mucosa and connective tissue. Strips of bulk circular muscle were minced with scissors in Ca2+-free Hank's solution. This solution contained (in mM) NaCl 125, KCl 5.36, NaHCO3 15.5, Naz2HPO4 0.336, KH2PO4 0.44, glucose 10, sucrose 2.9, HEPES (N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid]) 11, pH adjusted to 7.4 with NaOH. Minced tissue was then placed in Ca2+-free Hank's that contained 345 units ml−1 collagenase (Worthington Biochemical Corp.; Freehold, New Jersey, U.S.A.), 2 mg ml−1 soybean trypsin inhibitor (Sigma), 2 mg ml−1 fatty acid-free albumin (Sigma), and 0.5 mg ml−1 ATP (Sigma). Tissue pieces were stirred gently for 30 min at 37°C. Enzymes were washed out with Ca2+-free Hank's and then tissue pieces were passed repeatedly through the tip of a fire-polished Pasteur pipette to create cell suspensions. Minimal essential media (Sigma) was added to the cell suspension (50%; v v−1), that was then stored at 4°C and used within 8 h.
Measurement of VSOR Cl− currents
Drops of cell suspension were placed on a glass coverslip that formed the bottom of a recording chamber mounted on the stage of an inverted microscope. Cells were allowed approximately 10 min to adhere to the coverslip and then solutions were suffused over them and aspirated by vacuum. The volume of solution flowing over the cells per min was ten times greater than volume of the chamber. The bath solution contained (in mM) NaCl 125, CsCl 5, MgCl2 1.2, CaCl2 2, HEPES 10, TRIS base (tris[hydroxymethyl]aminomethane) 5, glucose 10; and adjusted to pH to 7.4 with NaOH. Osmolarity of this solution was tested with a freezing point depression osmometer and adjusted to 300 milliosmoles l−1 with mannitol.
Single myocytes were approached with fire-polished patch pipettes fabricated from borosilicate glass with inner and outer diameters of 1.17 and 1.50 mm, respectively (Sutter Instruments Co., Novato, California, U.S.A.). The pipettes had tip resistances of 2–4 MΩ when filled with solution that contained (in mM) TEA (tetraethylammonium) chloride 100, HEPES 10, EGTA (ethylene glycol-bis [β-aminoethyl ether] N,N,N′,N′-tetraacetic acid) 2, Mg-ATP 5, Na2GTP 1, phosphocreatine 2.5; and to pH to 7.1 with TEA hydroxide. The osmolarity of the pipette solution was adjusted to 300 milliosmoles l−1 by adding mannitol. After a ‘giga-seal' was formed, the cell membrane was ruptured and the whole-cell recording technique used. Cells were then exposed to a hypotonic solution having the same composition as the normal bath solution, except that NaCl was reduced to 100 mM and osmolarity was adjusted to 250 milliosmoles l−1 with mannitol.
An AXOPATCH-1D amplifier and CV-4 headstage were used to record Cl− currents (Axon Instruments, Inc., Foster City, California, U.S.A.). The patch clamp amplifier was interfaced with an IBM-compatible 80586 computer by a Lab Master analog-to-digital converter (Scientific Solutions, Inc., Solon, Ohio, U.S.A.). VSOR Cl− currents were sampled at 4 kHz and filtered (Bessel-type, lowpass) at 1 kHz. Data were acquired and analysed with pCLAMP software (Clampex and Clampfit, versions 5.5.1 and 6.0, Axon). Pipette capacitance was nullified by the compensation circuitry of the amplifier in the cell-attached mode (i.e., prior to rupturing the membrane). No other attempt was made to adjust for whole-cell capacitance or series resistance. Cl− currents were measured at room temperature (22–25°C). DMSO was used to dissolve each organic inhibitor except tamoxifen, which was dissolved in ethanol. Solvents did not have any significant inhibitory effect on VSOR Cl− current (−1±4%, n=5 and 9±3%, n=4, for ethanol and DMSO respectively at a dilution of 1 : 1000).
Measurement of L-type Ca2+ and delayed rectifier K+ currents
Techniques similar to those described above were used; however, some important differences require mentioning. An AXOPATCH 200B amplifier and CV203B headstage (Axon) were also used. The amphotericin-perforated patch clamp technique was used to record Ca2+ currents. The bath solution for these experiments contained (in mM) NaCl 125, CaCl2 2, MgCl2 1.2, TEA chloride 10, glucose 10, HEPES 10, TRIS 5; pH adjusted to 7.4 with NaOH. The pipette solution contained (in mM) CsOH 120, aspartate 120, CsCl 20, HEPES 10, EGTA 0.1; and adjusted to pH 7.1 with CsOH. Amphotericin B (250 μg ml−1, Sigma) was added to the pipette solution to perforate the cell membrane and allow electrical access. Cells were held at −60 mV and stepped to −55 mV for 20 ms every 10 s to monitor access development, which took approximately 10–15 min to develop fully. Ca2+ currents were measured at room temperature (22–25°C).
Delayed rectifier K+ currents were measured in the whole-cell dialyzed configuration of the patch clamp technique with the AXOPATCH 1-D and the CV-4 headstage. The following solutions were used. Intracellular solution contained (in mM) KCl 115, K-gluconate 20, Mg-ATP 5, creatine phosphate 2.5, Na2GTP 0.1, HEPES 10, EGTA 0.1; adjusted to pH 7.1 with KOH. The extracellular solution contained (in mM) NaCl 135, KCl 5, MnCl2 2, MgCl2 1.2, glucose 10, HEPES 10, TRIS 5; and adjusted to pH to 7.4 with NaOH. An additional internal solution was used to isolate delayed rectifier current that contained 10 mM BAPTA (1,2-bis[2-aminophenoxy]ethane-N,N,N′,N′-tetraacetic acid) as a Ca2+ chelator.
Statistical analyses and data presentation
Results are shown as individual current traces and/or group data expressed as the mean±standard error of n number of cells. Sigmoid concentration-response curves, cubic splines, and linear regressions were generated with Prism software (version 2.01, GraphPad Software, Inc., San Diego, California, U.S.A.). Statistical analyses were performed with SigmaStat software (version 2.0, Jandel Corp., San Rafael, California, U.S.A.). Unpaired t-tests, paired t-tests, and analysis of variance (with post-hoc analysis) were performed as appropriate and values of P<0.05 were considered significant. Statistical comparisons are explained in the text or figure legends.
Results
VSOR Cl− current in colonic myocytes
The whole-cell dialyzed mode of the patch clamp technique was used to measure VSOR Cl− currents before and after osmotic swelling. An outwardly rectifying current was observed when cells were studied in isotonic solution (Figure 1Aa). While the current was time-independent in the physiological range of membrane potentials, voltage-dependent inactivation was observed at test potentials positive to +40 mV (Jackson & Strange, 1995; Levitan & Garber, 1997; Voets et al., 1997). Exposing cells to hypotonic solution increased current magnitude (Figure 1Ab). Subtracting the current in isotonic solution from that in hypotonic solution yielded a difference current representing the swelling-activated current (Figure 1Ac). This difference current reversed at −14±1 mV (n=5), near the calculated Nernst equilibrium potential for Cl− of −8 mV (inset of Figure 1B). The membrane capacitance of these 5 cells was 76±4 pF; therefore, current density was 7.7±1.5 and 36.8±5.4 pA/pF at +120 mV in isotonic and hypotonic solution, respectively (P<0.005 by paired t-test). Whole-cell conductance in the physiological range of membrane potentials (−80 to −20 mV) was estimated to be 0.9±0.2 and 4.5±0.7 nS in isotonic and hypotonic solution, respectively (n=5; P<0.005 by paired t-test). The whole-cell difference current of these five cells was outwardly rectifying, as dividing the absolute current magnitude at +120 mV by that at −120 mV yielded a ratio of 6.4±0.7. The rectification ratios in isotonic and hypotonic solution were not significantly different (5.8±0.7 vs 6.3±0.5; paired t-test; n=5).
Figure 1.
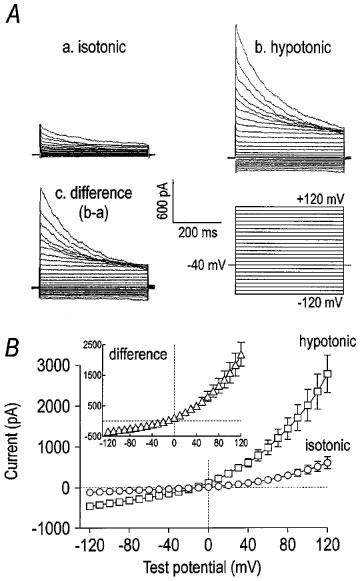
Whole-cell current elicited by hypotonic solution. Panel (A) contains representative traces of whole-cell current under isotonic (a) and hypotonic (b) conditions. The cell was held at −40 mV and stepped for 400 ms to voltages from −120 mV to +120 mV in 10 mV increments with a 5 s pause between steps. The difference current, i.e. current activated by hypotonic solution (b−a), is shown in c. The current activated by hypotonic solution was outwardly rectifying, and it inactivated at potentials positive to +40 mV. The I–V relationships of the whole-cell currents in isotonic and hypotonic solutions are shown in panel (B) (n=5). The inset contains the I–V relationship of the difference currents.
VSOR current was Cl−-dependent, as replacing extracellular Cl− with equimolar gluconate decreased the magnitude of the current (Figure 2A) and shifted the reversal potential to more positive values (Figure 2B). Specifically, gluconate substitution decreased outward current at +120 mV from 2490±170 pA to 594±51 pA. In the same three cells, the reversal potential shifted from −9±3 mV in 111 mM Cl− to 35±11 mV in 36 mM Cl− and 75 mM gluconate. The permeability for gluconate was 38±3% of that for Cl−, as calculated from the Goldman–Hodgkin–Katz equation using the change in reversal potential. Further, the reversal potential varied directly with the log of the extracellular Cl− concentration (Figure 2C), suggesting the current activated by hypotonic solution was carried largely by Cl− under these conditions. Liquid junction potentials (<10 mV) elicited by anion substitution were considered and corrected during data analysis. There was good agreement between the calculated and measured liquid junction potentials using a free-flowing 3 M KCl electrode with the amplifier in current-clamp mode.
Figure 2.
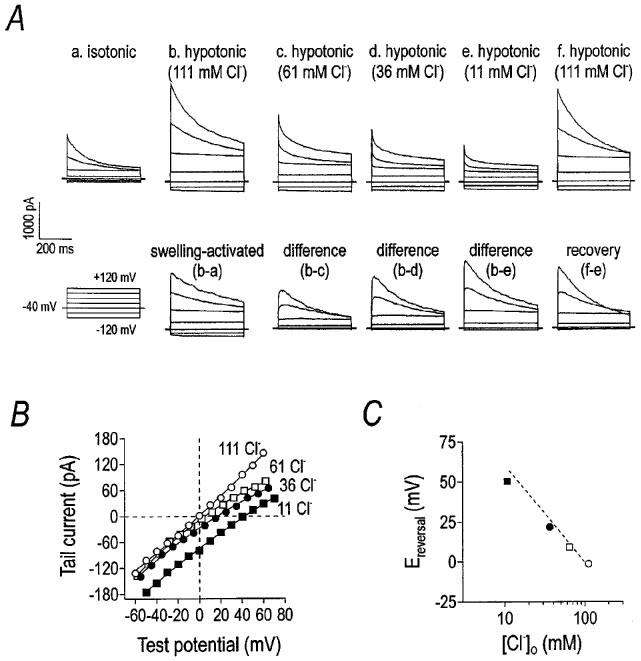
Whole-cell current depends upon the Cl− gradient. Representative current traces demonstrating Cl−-dependence are shown in panel (A). The cell was held at −40 mV and stepped from −120 mV to +120 mV in 40 mV steps in 5 s intervals. Hypotonic solution activated a whole-cell current, the magnitude of which decreased when extracellular NaCl was replaced with equimolar sodium gluconate. Panel (B) demonstrates the shift in the reversal potential and magnitude of the ‘tail' current as a function of Cl− gradient, measured after stepping the membrane to +120 mV for 400 ms and then to the indicated potential. Panel (C) is a plot of ‘tail' current reversal potential versus the extracellular Cl− concentration. The dotted line indicates the relationship predicted by the Nernst equation for Cl−.
Effects of Cl− channel antagonists on VSOR Cl− currents
Many Cl− conductances are sensitive to 9-AC, especially Ca2+-activated Cl− channels (Large & Wang, 1996). 9-AC weakly inhibited the VSOR Cl− current in a concentration-dependent manner (Figure 3A). Maximum inhibition at +120 and −120 mV was 22±8 and 22±9%, respectively (Figure 3C). There was no significant difference in the inhibition caused by 9-AC at positive and negative test potentials and, thus, there was no difference in the rectification ratio (Figure 3D). The effect of 9-AC was significant for the voltage extremes at 10 and 100 μM (P<0.001 by one-way repeated measures ANOVA; difference determined by Tukey post-hoc analysis).
Figure 3.
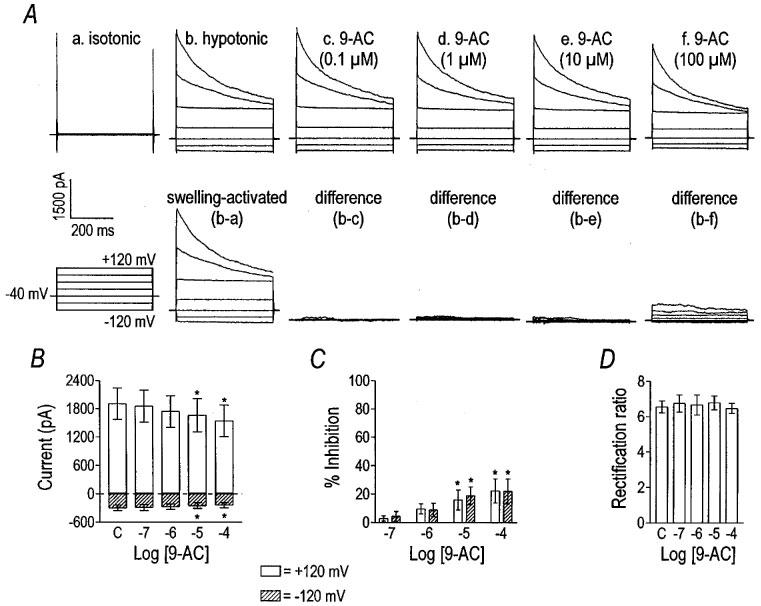
9-AC weakly inhibits VSOR Cl− current. Currents from a representative cell before (panel Ab) and after the application of 9-AC (panel Ac–Af) are shown. Panel (Aa) shows current recorded in isotonic solution, and panel (Ab) shows the activation of VSOR Cl− current in hypotonic solution. The lower traces show difference currents obtained by subtracting responses from the current in panel (Ab). The cell was held at −40 mV and stepped from −120 mV to +120 mV in 40 mV increments. 9-AC weakly inhibited VSOR Cl− current in a concentration-dependent manner (panel B). Asterisks indicate significant differences from control (labelled ‘C' by one-way repeated measures ANOVA and Tukey post-hoc analysis). Inward and outward current were inhibited equally (panel C); therefore, the rectification ratio was unchanged by 9-AC (panel D). Data in panels (B–D) are the mean±s.e.mean from eight cells.
Like 9-AC, niflumic acid is a known antagonist of Ca2+-activated Cl− channels, which are found in many types of smooth muscles (Large & Wang, 1996). Niflumic acid concentrations from 0.l to 100 μM had no significant effect on the VSOR Cl− current (Figure 4A–C). The effect of 100 μM niflumic acid (11±2% reduction; n=3) was not significantly different than the effect of DMSO. VSOR Cl− currents were recorded in the presence of 1 μM nicardipine to block L-type Ca2+ currents that could potentially contaminate recordings of VSOR Cl− currents at 0 mV. Thus, there appears to be little, if any, direct dependence of VSOR Cl− current on Ca2+ influx through L-type channels. These data are consistent with our previous findings in which the VSOR Cl− current was elicited in canine colonic myocytes with 2 mM EGTA in the pipette, with nicardipine (10 μM) in the bath, and in the absence of extracellular Ca2+. Taken together, the data suggest that VSOR Cl− current is not mediated by a Ca2+-activated Cl− conductance (Dick et al., 1998).
Figure 4.
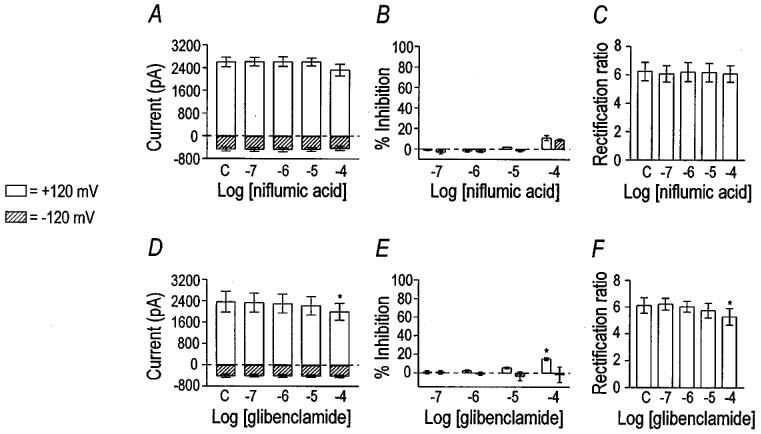
Effect of niflumic acid and glibenclamide on VSOR Cl− current. Niflumic acid was tested on three cells. Niflumic acid had no effect on VSOR Cl− current magnitude (panel A). Per cent inhibition by niflumic acid at the voltage extremes was not significant, even at the highest concentration (panel B). The rectification ratio was unchanged by niflumic acid (panel C). Control values are labelled ‘C'. Glibenclamide had a small inhibitory effect on VSOR Cl− current (n=5), and the rectification ratio was decreased at the highest concentration tested. Asterisks indicate significant differences from control by one-way repeated measures ANOVA with Tukey post-hoc analysis.
Glibenclamide, an antagonist of ATP-dependent K+ channels (Noma, 1983), has been reported to inhibit VSOR Cl− currents in a voltage-dependent manner in guinea-pig ventricular myocytes (IC50 ranged from 193–470 μM; Yamazaki & Hume, 1997) and atrial myocytes (IC50 of 60 μM; Sakaguchi et al., 1997). The effect of glibenclamide on the VSOR Cl− channel of smooth muscle is unknown. We found that glibenclamide was largely without effect upon VSOR Cl− current of colonic myocytes (Figure 4D and E). The small inhibitory effect of 100 μM glibenclamide was, however, significantly different than the effect of 0.1% DMSO (P=0.001 by unpaired t-test).
Two stilbene derivatives, DIDS and SITS, were tested for their effects on the VSOR Cl− current (Figures 5 and 6). DIDS inhibited current at +120 mV with an apparent IC50 of 0.84 μM and a Hill coefficient of 0.74 (n=6). Outward currents, elicited positive to 0 mV, were more sensitive to DIDS than inward currents (Figure 5B and C), suggesting voltage-dependent block of the VSOR Cl− current. The IC50 and Hill coefficient for DIDS at −120 mV were 11 μM and 0.34, respectively. The dependence of the block by DIDS on voltage was reflected in the concentration-dependent change in the rectification ratio (Figure 5D). The effect of DIDS was not readily reversible (data not shown). SITS was far less potent as an inhibitor of the VSOR Cl− current than DIDS (Figure 6). The IC50 and Hill coefficient for SITS at +120 mV were 226 and 0.57 μM, respectively (n=5). Outward current was more sensitive to antagonism by SITS than the inward current, as shown by the change in the rectification ratio (Figure 6C). The IC50 and Hill coefficient for SITS at −120 mV were incalculable due to the small inhibitory effect.
Figure 5.
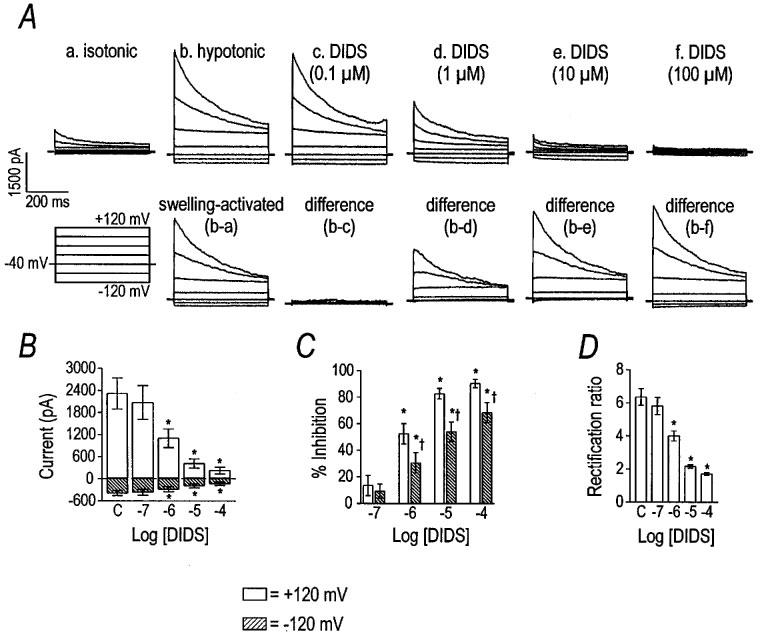
Inhibition of VSOR Cl− current by DIDS. Panel (A) shows representative traces demonstrating the inhibitory effect of DIDS. Current was elicited by holding at −40 mV and stepping from −120 to +120 mV in 40 mV increments. Group data (n=6) for the inhibitory effect of DIDS are shown in panels (B–D). Asterisks indicate differences from control (labelled ‘C') by one-way repeated measures ANOVA with Tukey post-hoc analysis. Per cent inhibition of the current by DIDS at the voltage extremes is plotted in panel (C). Cross symbols indicate differences between the per cent inhibition at +120 and −120 mV by paired t-test. Due to differential inhibition of the current at the voltage extremes, DIDS decreased the rectification ratio in a concentration-dependent manner (panel D).
Figure 6.
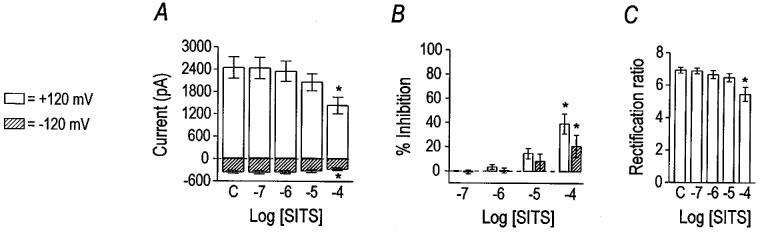
Inhibition of VSOR Cl− current by SITS. Group data (n=5) for the inhibitory effect of SITS are plotted in panels (A), (B), and (C). Current magnitudes at +120 mV and −120 mV are graphed in panel (A), where asterisks indicate differences from control (labelled ‘C') by one-way repeated measures ANOVA with Tukey post-hoc analysis. Per cent inhibition of the current by SITS at the voltage extremes is graphed in panel (B). Asterisks have the same meaning as in panel (A), while cross symbols indicate differences between the two voltages by paired t-test. The effect of SITS on the rectification ratio is shown in panel (C).
Tamoxifen, an oestrogen receptor antagonist, also blocked the VSOR Cl− current (Figure 7). Like DIDS and SITS, tamoxifen altered the rectification ratio, indicating voltage-dependent block of VSOR Cl− current (Figure 7D). Tamoxifen, at +120 mV, inhibited VSOR Cl− with an IC50 and Hill coefficient of 0.57 μM and 1.18, respectively (n=5). The IC50 and Hill coefficient for tamoxifen at −120 mV were 0.90 μM and 1.18, respectively. In a minority of cells, the inhibitory effect of tamoxifen was very slowly reversible (data not shown).
Figure 7.
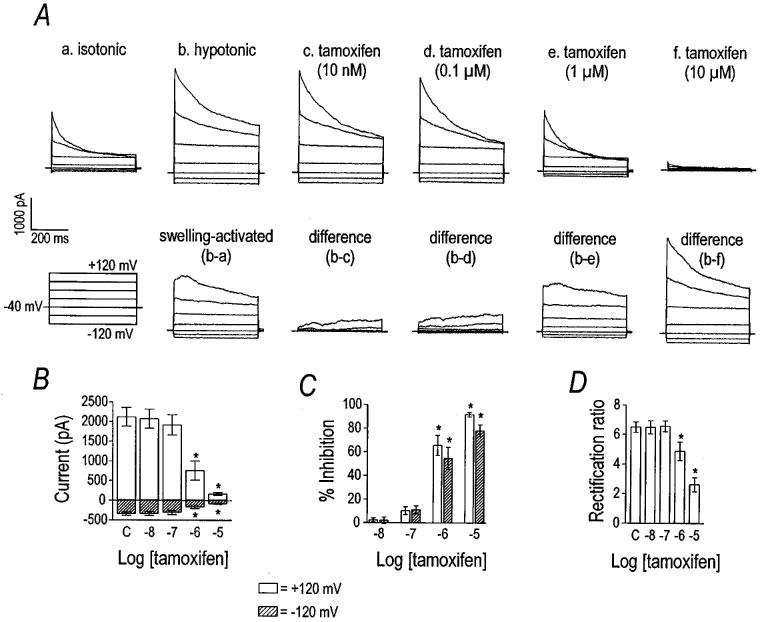
Inhibition of VSOR Cl− current by tamoxifen. Representative current traces in panel (A) illustrate the inhibitory effect of tamoxifen on the VSOR Cl− current. The cell was held at −40 mV and stepped from −120 to +120 mV in 40 mV increments. Current was inhibited by tamoxifen in a concentration-dependent manner. Group data (n=5) for the inhibitory effect of tamoxifen are plotted in panels (B), (C), and (D). Current magnitudes at +120 mV and −120 mV are graphed in panel (B), where asterisks indicate differences from control (labelled ‘C') by one-way repeated measures ANOVA with Tukey post-hoc analysis. Per cent inhibition of the current by tamoxifen at those voltages is shown in panel (C). Open and hatched bars represent data for +120 mV and −120 mV, respectively. Asterisks have the same meaning as in panel (B). The effect of tamoxifen on the rectification ratio is shown in panel (D) and asterisks indicate significant differences from control.
Effects of multivalent cations on VSOR Cl− currents
Multivalent cations have been demonstrated previously to inhibit the hyperpolarization- and Ca2+-activated Cl− currents of Xenopus oocytes (Tokimasa & North, 1996); therefore, we tested the effect of Gd3+ and La3+ on VSOR Cl− current in canine colonic myocytes (Figures 8 and 9). Gd3+ inhibited VSOR Cl− current with an estimated IC50 of 23 μM and Hill coefficient of 0.64 (n=8). The block by Gd3+ appeared largely independent of voltage, as the rectification ratio was decreased only at 100 μM Gd3+ (P<0.001 by one-way repeated measures ANOVA with the difference indicated by Tukey post-hoc analysis). Thus, outward current was inhibited more potently than inward current at the highest concentration. As reported for the Cl− channels of Xenopus oocytes (Tokimasa & North, 1996), we also observed that the inhibitory effect of Gd3+ was only partially reversible. After application of 100 μM Gd3+ to three cells, the current at +120 mV recovered only 52±8% after washing out the cation for 20 min. La3+ also inhibited VSOR Cl− current concentration-dependently, but less potently (Figure 9). The inhibitory effect of 100 μM Gd3+ was significantly greater than equimolar La3+ (P<0.01 by unpaired Student's t-test). La3+, in contrast to Gd3+, had no significant effect upon the rectification ratio. Thus, La3+, at the concentrations tested, inhibited inward and outward current equally.
Figure 8.
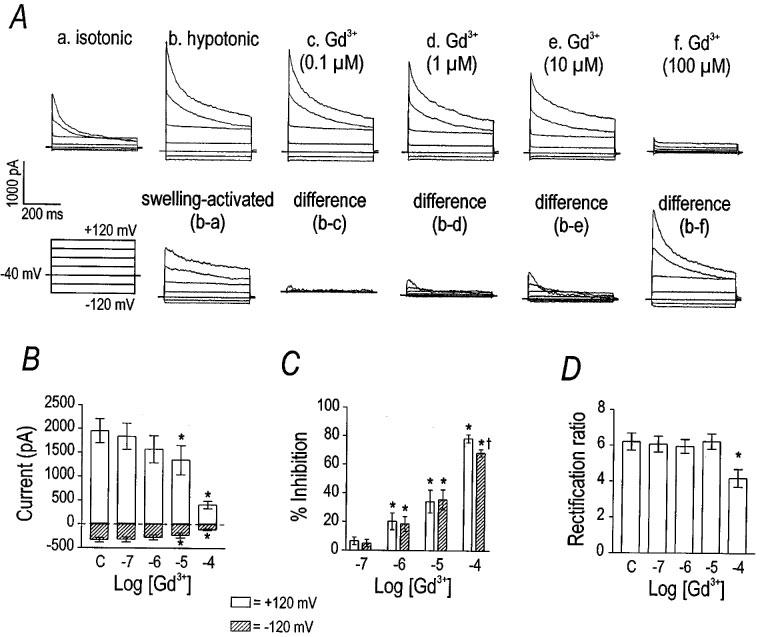
Inhibition of VSOR Cl− current by Gd3+. Panel (A) shows representative current traces demonstrating the inhibitory effect of Gd3+ on VSOR Cl− current. The cell was stepped from −120 to +120 mV in 40 mV increments. The currents in isotonic and hypotonic solutions with increasing concentrations of Gd3+ are shown. Gd3+ inhibited VSOR Cl− current in a concentration-dependent manner. Group data (n=8) for the effect of Gd3+ on current at +120 and −120 mV are plotted in panel (B). Asterisks indicate differences from control (labelled ‘C') by one-way repeated measures ANOVA with Tukey post-hoc analysis. Panel (C) expresses the data in panel (B) as per cent inhibition (asterisks have the same meaning). Cross symbol in panel (B) indicates that Gd3+ inhibited outward current more than inward current; therefore, explaining the change in the rectification properties (panel C).
Figure 9.
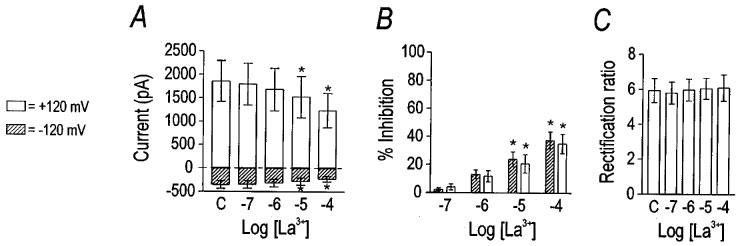
Inhibition of VSOR Cl− current by La3+. Group data (n=7) are plotted to illlustrate the inhibitory effect of La3+. La3+ significantly inhibited current at 100 μM (panel A). Per cent inhibition was equivalent at positive and negative potentials (panel B); therefore, the rectification ratio was unchanged (panel C). Asterisks indicate differences from control (labelled ‘C'), as detected by one-way repeated measures ANOVA with Tukey post-hoc analysis.
Antagonists of VSOR Cl− currents also block Ca2+ and K+ currents
In order to use blockers of VSOR Cl− currents for tests of the role of this conductance in intact muscles, it is important to determine non-specific effects of these compounds on other conductances. We tested the specificity of tamoxifen, Gd3+, and DIDS by examining the effects of these agents on L-type Ca2+ and delayed rectifier K+ current because some Cl− channel antagonists inhibit voltage-dependent conductances in vascular smooth muscles (Doughty et al., 1998). In colonic myocytes the voltage-dependent Ca2+ channels are ‘L-type' based on molecular identification (Rich et al., 1993) and pharmacology (Langton et al., 1989; Rich et al., 1993). These channels are more permeable to Ba2+ than Ca2+ and blocked by nifedipine. We used the amphotericin-perforated patch clamp technique to prevent ‘run-down' of the Ca2+ current. Cells were held at −80 mV and stepped to 0 mV every 15 s. Tamoxifen inhibited peak Ca2+ current in a concentration-dependent manner, as shown in Figure 10A–C. Gd3+ (10 μM) inhibited Ca2+ current 93±8% (n=3; Figure 10D). DIDS was the least non-specific of the VSOR Cl− channel antagonists tested (Figure 10E and F), as the inhibition of Ca2+ current elicited by 10 μM DIDS was significantly less than that caused by 10 μM tamoxifen (P<0.01 by unpaired t-test). We did not test the effects of niflumic acid, 9-AC, SITS, glibenclamide, or La3+ because these agents were poor inhibitors of the VSOR Cl− current.
Figure 10.
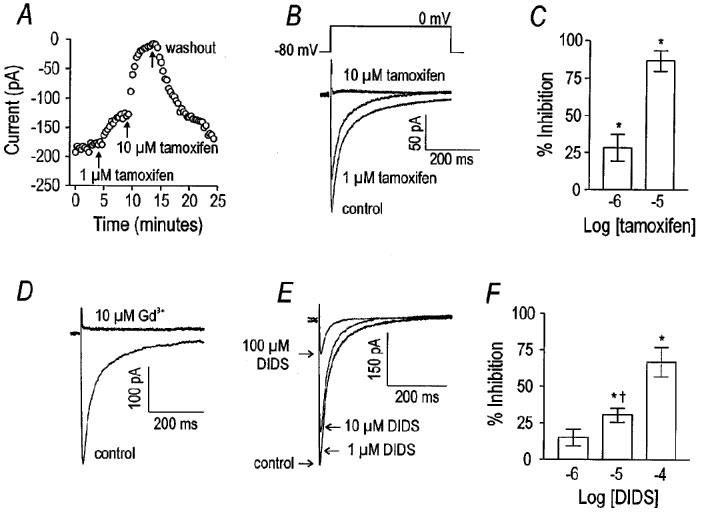
Tamoxifen and DIDS antagonize L-type Ca2+ current. The effect of tamoxifen to antagonize L-type Ca2+ current is shown in panel (A) where current is plotted versus time. The myocyte was held at −80 mV and stepped to 0 mV (peak of the I–V relationship, data not shown). Tamoxifen inhibited Ca2+ current in a concentration-dependent and reversible manner. Representative current tracings demonstrate the inhibitory effect of tamoxifen on L-type Ca2+ current (panel B). Group data (n=5) for the inhibitory effect of tamoxifen on Ca2+ current are displayed in panel (C). Tamoxifen significantly inhibited L-type Ca2+ current at both 1 and 10 μM (by one-way repeated measures ANOVA with Tukey post-hoc analysis). An example of the inhibitory effect of 10 μM Gd3+ on L-type Ca2+ current is shown in panel (D), where the same voltage step protocol shown in (B) was used (see text for group data). DIDS inhibited L-type Ca2+ current less potently than either tamoxifen or Gd3+, as the example in panel (E) demonstrates. Group data (n=4) are plotted in panel (F). DIDS significantly inhibited L-type Ca2+ current at 10 and 100 μM (asterisks indicate difference from control by one-way repeated measures ANOVA). The inhibitory effect of 10 μM DIDS was significantly less than an equal concentration of tamoxifen or Gd3+ (indicated by cross; tested by one-way ANOVA).
To test the effects of VSOR Cl− current antagonists on delayed rectifier K+ current, myocytes were dialyzed with 135 mM K+ and 0.1 mM EGTA (buffering intracellular Ca2+ to approximately 100 nM), and bathed in HEPES-buffered solution in which equimolar Mn2+ replaced Ca2+. These conditions moderately reduce Ca2+-activated K+ channel current and emphasize delayed rectifier K+ current in these cells (Carl, 1995). Cells were held at −80 mV and stepped to 0 mV or ramped from −80 mV to +80 mV. Ramp protocols confirmed that the outward current is composed of delayed rectifier and Ca2+-activated K+ currents as previously reported (Cole et al., 1989). Responses to ramp protocols revealed an outward ‘hump' near 0 mV, that was previously shown to be due to delayed rectifier K+ current, and Ca2+-activated K+ currents, which were activated at more positive potentials (Cole et al., 1989). In voltage step protocols, delayed rectifier current appeared as a smooth, slowly-developing outward current at potentials up to 0 mV. Tamoxifen (1 μM) inhibited current at 0 mV by 72±5% (n=7), but a 10 fold lower concentration was without significant effect (−5±7%; n=7). In contrast, current at +80 mV was potentiated 151±27% by 1 μM tamoxifen (n=5). Unlike tamoxifen, 1 and 10 μM DIDS inhibited current at 0 mV by only 9±3 and 20±8%, respectively. The inhibitory effects of DIDS (1 μM) on delayed rectifier K+ current were significantly less than the effects of tamoxifen at the same concentration (P<0.001 by unpaired t-test). DIDS was approximately one order of magnitude less potent in inhibiting delayed rectifier current than tamoxifen.
The identity of the tamoxifen- and DIDS-sensitive outward current at 0 mV was confirmed using 300 nM charybdotoxin. To more carefully isolate delayed rectifier K+ current, cells were dialyzed with 10 mM BAPTA and superfused with Ca2+-free PSS to more strongly reduce intracellular Ca2+ and thus the contribution of Ca2+-activated K+ channels. Cells were also treated with 300 nM charybdotoxin to block Ca2+-activated K+ current. With these steps taken to isolate delayed rectifier K+ current, 1 μM tamoxifen inhibited current at 0 mV by 38±8% (n=3). Similarly, under the recording conditions designed to isolate delayed rectifier current, DIDS inhibited current at 0 mV by 10±2% (n=3). Again, when delayed rectifier K+ current was more carefully isolated, tamoxifen was still shown to be a more potent inhibitor of this current than DIDS.
Discussion
We have constructed a partial pharmacological profile for the VSOR Cl− current in canine colonic myocytes, a current that may be due to expression of ClC-3 (Dick et al., 1998). It is possible that the pharmacological profile outlined here could be tissue specific; however, it is a point of reference from which to start investigating physiological/pathophysiological roles of VSOR Cl− currents by techniques that require the use of intact smooth muscle preparations. Exposing colonic smooth muscle cells to hypotonic solution activated a VSOR Cl− current. The VSOR Cl− current was largely insensitive to 9-AC and niflumic acid (inhibitors of Ca2+-activated Cl− channels) and glibenclamide (an inhibitor of KATP and CFTR). DIDS, SITS, tamoxifen, Gd3+, and La3+ blocked the VSOR Cl− current, with DIDS, tamoxifen, and Gd3+ being the most potent. The rank order potency at +120 mV was tamoxifen⩾DIDS>4Gd3+ and tamoxifen>DIDS>Gd3+ at −120 mV. Inhibition of the VSOR Cl− current by DIDS was more voltage-dependent than that of tamoxifen or Gd2+. Neither tamoxifen nor DIDS was highly specific for the VSOR Cl− current. Both drugs inhibited L-type Ca2+ and delayed rectifier K+ currents, as does Gd3+. It is important to note, however, that DIDS was considerably less potent than tamoxifen in inhibiting L-type Ca2+ and delayed rectifier K+ current. This pharmacological agent might be useful in testing the role of VSOR Cl− currents in intact smooth muscles (e.g., with microelectrode or isometric tension measurements). It might be difficult to determine the role of the swelling-activated (or volume- or stretch-sensitive) Cl− conductance on mechanical activity with the channel antagonists currently available, because of the confounding effects of the inhibitors on other voltage-gated conductances. Electrophysiologically, however, it may be possible to study the role of the VSOR Cl− conductance on resting potentials, where the open probabilities of L-type Ca2+ and delayed rectifier K+ channels are low (Ward & Sanders, 1992).
Niflumic acid and 9-AC, antagonists of Ca2+-activated Cl− channels, had little effect on the VSOR Cl− current in canine colonic smooth muscle cells. Previously, we have shown that the VSOR Cl− current activated by hypotonicity in colonic myocytes can be recorded in the absence of extracellular Ca2+, in the presence of L-type Ca2+ channel antagonists, and when the intracellular solution is buffered with 2 mM EGTA (Dick et al., 1998). These data suggest that little or none of the VSOR Cl− current is due to the direct activation of a Ca2+-dependent current. Glibenclamide, a well-known blocker of KATP (Noma, 1983), has also been shown to block CFTR Cl− channels (Sheppard & Welsh, 1992). High concentrations of glibenclamide have also been shown to reversibly inhibit the swelling-activated Cl− conductance of guinea-pig cardiac myocytes (Sakaguchi et al., 1997; Yamazaki et al., 1997). In that preparation, maximal (100%) inhibition of atrial swelling-activated Cl− current was achieved with >500 μM glibenclamide, with an IC50 in the 100 μM range. In the present study we found that glibenclamide had little effect upon the VSOR Cl− current of canine colonic myocytes in the concentration range of 0.1 to 100 μM.
The stilbene derivatives (DIDS and SITS) and tamoxifen, inhibited the VSOR Cl− but the efficacy of these drugs differed approximately four orders of magnitude. SITS was by far the least potent of these three antagonists. DIDS and SITS were more effective at inhibiting the outward portion of the current than the inward current, suggesting the block by these agents depends upon the voltage. With respect to the outward Cl− current, tamoxifen and DIDS had nearly equivalent potency, but the block by tamoxifen was less voltage-dependent than that of DIDS, resulting in more inhibition of inward current. Tamoxifen was more potent than DIDS for inhibiting inward current and, it is, of course, the inward current (outward movement of Cl−) that is physiologically important. However, in this study cells were dialyzed with TEA and treated with nicardipine to isolate the Cl− current, emphasizing the effect of antagonists on VSOR Cl− current. It is important to remember that a mixture of ionic conductances determines the response of intact smooth muscle, and tamoxifen has much more potent inhibitory effects upon L-type Ca2+ and delayed rectifier K+ currents than does DIDS.
The multivalent cations, Gd3+ and La3+, had different effects on the VSOR Cl− current in colonic myocytes: Gd3+ strongly inhibited VSOR Cl− current, whereas La3+ did not. The reason for the different effects of these trivalent cations is unclear, but the difference could prove useful in future experiments. A major obstacle in studying the regulation of swelling-activated Cl− currents by agonists, neurotransmitters, and 2nd messenger systems is the fact that cells are normally dialyzed to control the intracellular anion composition. Cells could be studied with the amphotericin-perforated patch technique and K+ currents reduced with Cs+-containing pipette solutions; however, hypotonic solution (Waniishi et al., 1997) and membrane stretch (Davis et al., 1992; Kirber et al., 1988) are known to activate non-selective cation channels in some smooth muscle cells. Osmotic challenge also appears to activate K+ channels in colonic smooth muscle cells (Dick and Sanders, unpublished observation). Such a mixture of currents would be difficult to separate and characterize. However, La3+ is a blocker of non-selective cation channels and might be used to isolate the anion current when added to the extracellular solution. None of the pharmacological agents tested were specific in inhibiting VSOR Cl− channels. The non-specificity of tamoxifen is important because this anti-oestrogen compound is used in the treatment of breast cancer. Side effects of tamoxifen (which are numerous), therefore, could be related to the non-specific actions of the compounds on various ion channels. There may be other pharmacological agents that could provide more specific block of VSOR Cl− currents in smooth muscles. For example, mibefradil, an antagonist of low-threshold (T-type) Ca2+ channels, may be such a compound. Mibefradil appears to inhibit the swelling-activated anion currents of vascular endothelial cells (Nilius et al., 1997) as potently as T-type Ca2+ channels of smooth muscle (Mishra et al., 1994). Hopefully, new pharmacological agents will be found that specifically antagonize volume-sensitive Cl− channels. Such compounds would greatly facilitate studies of the physiological role of VSOR Cl− current in smooth muscle function.
It is likely that the VSOR Cl− current in colonic myocytes is encoded by ClC-3, as the mRNA is present in isolated cells. The pharmacology and properties of the VSOR Cl− conductance in canine smooth muscle closely match the properties of recombinant ClC-3 (Dick et al., 1998; Yamazaki et al., 1998). Canine colonic myocytes may be an ideal model to investigate the physiological role of VSOR Cl− channels in native cells because the pharmacological effects of anion transport inhibitors can be determined without contamination from other Cl− conductances, such as Ca2+-activated Cl− channels. Further, canine colonic smooth muscle is a well-established model and most of the conductances expressed in them have been characterized. Specifically, large conductance Ca2+-activated K+ (Carl & Sanders, 1989), delayed rectifier K+ (Carl, 1995), and L-type Ca2+ channels (Langton et al., 1989) are present and blockers of these conductances are available. In conclusion, our findings indicate that DIDS is the least non-specific antagonist of VSOR Cl− current in canine colonic myocytes. Thus, at present, DIDS appears to be the antagonist of choice for investigating the physiological role of VSOR Cl− channels in smooth muscle cells and tissues.
Figure 11.
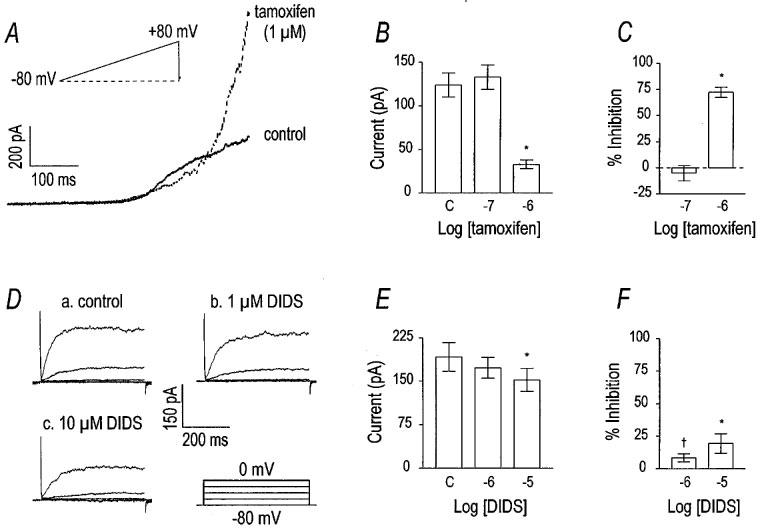
Tamoxifen and DIDS inhibit delayed rectifier K+ current. A set of representative current traces from a voltage ramp protocol is shown in panel (A). The cell was held at −80 mV and ramped to +80 mV before and after the addition of 1 μM tamoxifen. Tamoxifen decreased what appeared to be delayed rectifier K+ current, but increased outward current at more positive potentials. Panels (B) and (C) contain group data (n=7) for the effect of tamoxifen on current at 0 mV (a voltage step protocol identical to that in panel (D) was used). Asterisks indicate the significant inhibitory effect of tamoxifen detected by one-way repeated measures ANOVA. Panel (D) shows the inhibitory effect of DIDS on outward current at 0 mV in a representative cell. The cell was held at −80 mV and stepped to 0 mV in 20 mV increments at 5 s intervals. DIDS attenuated current in a concentration-dependent manner. Group data for the effect of DIDS on outward current at 0 mV are shown in panel (E) (asterisks indicate difference from control; detected by one-way repeated measures ANOVA with Tukey post-hoc analysis). This inhibition, however, was weak compared to tamoxifen. The cross symbol in panel (F) indicates that the inhibition of outward current at 0 mV by equal concentrations of DIDS and tamoxifen are significantly different (unpaired t-test).
Acknowledgments
The expert preparation of canine colonic myocytes by Nancy Horowitz, M.S. was appreciated. We are indebted to Dr James L Kenyon for his advice on the theoretical and practical considerations of dealing with liquid junction potentials. Financial support for this work was provided by a grant from the National Institutes of Health (DK41315).
Abbreviations
- BAPTA
1,2-bis[2-aminophenoxy]ethane-N,N,N′,N′-tetraacetic acid
- DIDS
4,4-diisothiocyanato-stilbene-2,2′-disulphonate
- EGTA
ethylene glycol-bis [β-aminoethyl ether] N,N,N′,N′-tetraacetic acid
- HEPES
N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid]
- SITS
4-acetamido-4′-isothiocyanato-stilbene-2,2′-disulphonate
- tamoxifen
[Z]-1-[p-dimethylaminoethoxy-phenyl]-1,2-diphenyl-1-butene
- TEA
tetraethylammonium
- TRIS
tris[hydroxymethyl]aminomethane
- VSOR
volume-sensitive, outwardly rectifying
References
- AICKIN C.C., BRADING A.F. Measurement of intracellular chloride in guinea-pig vas deferens by ion analysis, 36chloride efflux and micro-electrodes. J. Physiol. 1982;326:139–154. doi: 10.1113/jphysiol.1982.sp014182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKBARALI H.I., GILES W.R. Ca2+ and Ca2+-activated Cl− currents in rabbit oesophageal smooth muscle. J. Physiol. 1993;460:117–133. doi: 10.1113/jphysiol.1993.sp019462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAYLISS W.M. On the local reaction of the arterial wall to changes of internal pressure. J. Physiol. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BÜLBRING E. Correlation between membrane potential, spike discharge and tension in smooth muscle. J. Physiol. 1955;128:200–221. doi: 10.1113/jphysiol.1955.sp005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARL A. Multiple components of delayed rectifier K+ current in canine colonic myocytes. J. Physiol. 1995;484.2:339–353. doi: 10.1113/jphysiol.1995.sp020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARL A., SANDERS K.M. Ca2+-activated K channels of canine colonic myocytes. Am. J. Physiol. 1989;257:C470–C480. doi: 10.1152/ajpcell.1989.257.3.C470. [DOI] [PubMed] [Google Scholar]
- CLAPHAM D.E. The list of potential volume-sensitive chloride currents continues to swell (and shrink) J. Gen. Physiol. 1998;111:623–624. doi: 10.1085/jgp.111.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLE W.C., SANDERS K.M. Characterization of macroscopic outward currents of canine colonic myocytes. Am. J. Physiol. 1989;257:C461–C469. doi: 10.1152/ajpcell.1989.257.3.C461. [DOI] [PubMed] [Google Scholar]
- DAVIS M.J., DONOVITZ J.A., HOOD J.D. Stretch-activated single-channel and whole cell currents in vascular smooth muscle cells. Am. J. Physiol. 1992;262:C1083–C1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- DICK G.M., BRADLEY K.K., HOROWITZ B., HUME J.R., SANDERS K.M. Functional and molecular identification of a novel chloride conductance in canine colonic myocytes. Am. J. Physiol. 1998;275:C940–C950. doi: 10.1152/ajpcell.1998.275.4.C940. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., MILLER A.L., LANGTON P.D. Non-specificity of chloride channel blockers in rat cerebral arteries: block of L-type calcium channels. J. Physiol. 1998;507.2:433–439. doi: 10.1111/j.1469-7793.1998.433bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUAN D., COWLEY S., HOROWITZ B., HUME J.R. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J. Gen. Physiol. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUAN D., HUME J.R., NATTEL S. Evidence that outwardly rectifying Cl− channels underlie volume-regulated Cl− currents in heart. Circulation Res. 1997a;80:103–113. doi: 10.1161/01.res.80.1.103. [DOI] [PubMed] [Google Scholar]
- DUAN D., WINTER C., COWLEY S., HUME J.R., HOROWITZ B. Molecular identification of a volume-regulated chloride channel. Nature. 1997b;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- GREENWOOD I.A., LARGE W.A. Properties of a Cl− current activated by cell swelling in rabbit portal vein vascular smooth muscular cells. Am. J. Physiol. 1998;275:H1524–H1532. doi: 10.1152/ajpheart.1998.275.5.H1524. [DOI] [PubMed] [Google Scholar]
- HARDER D.R. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circulation Res. 1984;55:197–202. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- JACKSON P.S., STRANGE K. Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J. Gen. Physiol. 1995;105:661–676. doi: 10.1085/jgp.105.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENTSCH T.J., GÜNTHER W., PUSCH M., SCHWAPPACH B. Properties of voltage-gated chloride channels of the ClC gene family. J. Physiol. 1995;482.P:19S–25S. doi: 10.1113/jphysiol.1995.sp020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIRBER M.T., WALSH J.V., JR, SINGER J.J. Stretch-activated ion channels in smooth muscle: mechanism for the initiation of stretch-induced contraction. Pflügers Arch. 1988;412:339–345. doi: 10.1007/BF01907549. [DOI] [PubMed] [Google Scholar]
- LANGTON P.D., BURKE E.P., SANDERS K.M. Participation of Ca currents in colonic electrical activity. Am. J. Physiol. 1989;257:C451–C460. doi: 10.1152/ajpcell.1989.257.3.C451. [DOI] [PubMed] [Google Scholar]
- LARGE W.A., WANG Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am. J. Physiol. 1996;271:C435–C454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- LEVITAN I., GARBER S.S. Voltage-dependent inactivation of volume-regulated Cl− current in human T84 colonic and B-cell myeloma cell lines. Pflügers Arch. 1997;431:297–299. doi: 10.1007/BF00410203. [DOI] [PubMed] [Google Scholar]
- MEININGER G.A., DAVIS M.J. Cellular mechanisms involved in the vascular myogenic response. Am. J. Physiol. 1992;263:H647–H659. doi: 10.1152/ajpheart.1992.263.3.H647. [DOI] [PubMed] [Google Scholar]
- MISHRA S.K., HERMSMEYER K. Selective inhibition of T-type Ca2+ channels by Ro 40-5967. Circulation Res. 1994;75:144–148. doi: 10.1161/01.res.75.1.144. [DOI] [PubMed] [Google Scholar]
- NELSON M.T. Bayliss, myogenic tone and volume-regulated chloride channels in arterial smooth muscle. J. Physiol. 1998;507:629. doi: 10.1111/j.1469-7793.1998.629bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON M.T., CONWAY M.A., KNOT H.J., BRAYDEN J.E. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J. Physiol. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILIUS B., PRENEN J., KAMOUCHI M., VIANA F., VOETS T., DROOGMANS G. Inhibition by mibefradil, a novel calcium channel antagonist, of Ca2+- and volume-activated Cl− channels in macrovascular endothelial cells. Br. J. Pharmacol. 1997;121:546–555. doi: 10.1038/sj.bjp.0701140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILIUS B., SEHRER J., VIANA F., DE GREEF C., RAEYMAEKERS L., EGGERMONT J., DROOGMANS G. Volume-activated Cl− currents in different mammalian non-excitable cell types. Pflügers Arch. 1994;428:364–371. doi: 10.1007/BF00724520. [DOI] [PubMed] [Google Scholar]
- NOMA A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- OKADA Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am. J. Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- OKADA Y., OIKI S., HAZAMA A., MORISHIMA S. Criteria for the molecular identification of the volume-sensitive outwardly rectifying Cl− channel. J. Gen. Physiol. 1998;112:365–367. doi: 10.1085/jgp.112.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OZAKI H., STEVENS R.J., BLONDFIELD D.P., PUBLICOVER N.G., SANDERS K.M. Simultaneous measurement of membrane potential, cytosolic Ca2+, and tension in intact smooth muscles. Am. J. Physiol. 1991;260:C917–C925. doi: 10.1152/ajpcell.1991.260.5.C917. [DOI] [PubMed] [Google Scholar]
- RICH A., KENYON J.L., HUME J.R., OVERTURF K., HOROWITZ B., SANDERS K.M. Dihydropyridine-sensitive calcium channels expressed in canine colonic smooth muscle cells. Am. J. Physiol. 1993;264:C745–C754. doi: 10.1152/ajpcell.1993.264.3.C745. [DOI] [PubMed] [Google Scholar]
- SAKAGUCHI M., MATSUURA H., EHARA T. Swelling-induced Cl− current in guinea-pig atrial myocytes: inhibition by glibenclamide. J. Physiol. 1997;505:41–52. doi: 10.1111/j.1469-7793.1997.041bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEPPARD D.N., WELSH M.J. Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J. Gen. Physiol. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH T.K., REED J.B., SANDERS K.M. Origin and propagation of electrical slow waves in circular muscle of canine proximal colon. Am. J. Physiol. 1987;252:C215–C224. doi: 10.1152/ajpcell.1987.252.2.C215. [DOI] [PubMed] [Google Scholar]
- STRANGE K. Molecular identity of the outwardly rectifying, swelling-activated anion channel: Time to reevaluate pICln. J. Gen. Physiol. 1998;111:617–622. doi: 10.1085/jgp.111.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRANGE K., EMMA F., JACKSON P.S. Cellular and molecular physiology of volume-sensitive anion channels. Am. J. Physiol. 1996;270:C711–C730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- SUN X.P., SUPPLISSON S., TORRES R., SACHS G., MAYER E. Characterization of large-conductance chloride channels in rabbit colonic smooth muscle. J. Physiol. 1992;448:355–382. doi: 10.1113/jphysiol.1992.sp019046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOKIMASA T., NORTH R.A. Effects of barium, lanthanum and gadolinium on endogenous chloride and potassium currents in Xenopus oocytes. J. Physiol. 1996;496:677–686. doi: 10.1113/jphysiol.1996.sp021718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANDENBERG J.I., BETT G.C., POWELL T. Contribution of a swelling-activated chloride current to changes in the cardiac action potential. Am. J. Physiol. 1997;273:C541–C547. doi: 10.1152/ajpcell.1997.273.2.C541. [DOI] [PubMed] [Google Scholar]
- VOETS T., DROOGMANS G., NILIUS B. Modulation of voltage-dependent properties of a swelling-activated Cl− current. J. Gen. Physiol. 1997;110:313–325. doi: 10.1085/jgp.110.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANIISHI Y., INOUE R., ITO Y. Preferential potentiation by hypotonic cell swelling of muscarinic cation current in guinea pig ileum. Am. J. Physiol. 1997;272:C240–C253. doi: 10.1152/ajpcell.1997.272.1.C240. [DOI] [PubMed] [Google Scholar]
- WARD S.M., SANDERS K.M. Upstroke component of electrical slow waves in canine colonic smooth muscle is due to nifedipine-resistant calcium current. J. Physiol. 1992;455:321–337. doi: 10.1113/jphysiol.1992.sp019304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU W.X., KIM S.J., SO I., KANG T.M., RHEE J.C., KIM K.W. Volume-sensitive chloride current activated by hyposmotic swelling in antral gastric myocytes of the guinea-pig. Pflügers Arch. 1997;435:9–19. doi: 10.1007/s004240050478. [DOI] [PubMed] [Google Scholar]
- YAMAZAKI J., DUAN D., JANIAK R., KUENZLI K., HOROWITZ B., HUME J.R. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. J. Physiol. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAZAKI J., HUME J.R. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, and Ca2+-activated Cl− channels in mammalian cardiac myocytes. Circulation Res. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]