

Abstract
To determine how pretreatment with sulphonylureas alters the β cell function, mouse islets were cultured (18–20 h) without (controls) or with (test) 0.01 μM glibenclamide. Acute responses to glucose were then determined in the absence of glibenclamide.
Test islets were insensitive to drugs (sulphonylureas and diazoxide) acting on K+-ATP channels, and their [Ca2+]i was already elevated in the absence of stimulation.
Insulin secretion was increased in the absence of glucose, and mainly stimulated between 0–10 instead of 7–20 mM glucose in controls. The maximum response was halved, but this difference disappeared after correction for the 45% decrease in the islet insulin content.
The first phase of glucose-induced insulin secretion was abrogated because of a paradoxical decrease of the high basal [Ca2+]i in β cells. The second phase was preserved but occurred with little rise of [Ca2+]i. These abnormalities did not result from alterations of glucose metabolism (NADPH fluorescence).
In islets cultured with 50 μM tolbutamide, glucose induced biphasic increases in [Ca2+]i and insulin secretion. The decrease in the secretory response was matched by the decrease in insulin content (45%) except at maximal glucose concentrations. Islets pretreated with tolbutamide, however, behaved like those cultured with glibenclamide if tolbutamide was also present during the acute functional tests.
In conclusion, treatment with a low glibenclamide concentration causes long-lasting blockade of K+-ATP channels and rise of [Ca2+]i in β cells. Glucose-induced insulin secretion occurs at lower concentrations, is delayed and is largely mediated by a modulation of Ca2+ action on exocytosis. It is suggested that glucose regulation of insulin secretion mainly depends on a K+-ATP channel-independent pathway during in vivo sulphonylurea treatment.
Keywords: Pancreatic islets, sulphonylureas, tolbutamide, glibenclamide, insulin secretion, β cell exhaustion, therapeutic failure, cytoplasmic Ca2+, K+-ATP channels
Introduction
Sulphonylureas remain a cornerstone of the treatment of type 2 diabetes (Groop, 1992; UK Prospective Diabetes Study Group, 1998). The mechanism by which they acutely increase insulin secretion is now well understood. They close ATP-sensitive K+ (K+-ATP) channels in β cells by binding to the sulphonylurea receptor SUR-1, which is a subunit of the channel itself (Aguilar-Bryan et al., 1998; Ashcroft & Gribble, 1998). This results in membrane depolarization, opening of voltage-dependent Ca2+ channels, Ca2+ influx, and rise in the cytoplasmic Ca2+ concentration ([Ca2+]i), which then triggers insulin secretion (Henquin, 1990; 1998; Satin, 1996; Panten et al., 1996). A similar sequence of events underlies glucose stimulation, with the major difference that it is initiated by the interaction of metabolic signals (presumably adenine nucleotides) with both SUR-1 and Kir 6.2, the pore-forming subunit of the channel (Dunne & Petersen, 1991; Misler et al., 1992; Henquin, 1994; Prentki, 1996; Ashcroft & Gribble, 1998). Glucose also increases insulin secretion by a second mechanism, known as the K+-ATP channel-independent pathway, which serves to increase the efficacy of Ca2+ on exocytosis (Gembal et al., 1992; 1993; Sato et al., 1992; Sato & Henquin, 1998).
Treatment of diabetic patients with sulphonylureas often causes secondary failure for reasons that could be linked to either the natural evolution of the disease or untoward effects of the drugs (Pontiroli et al., 1994; Matthews et al., 1998). Stimulation of insulin secretion by sulphonylureas is not compensated for by a stimulation of insulin biosynthesis and may lead to a progressive decrease of insulin stores (Borg & Andersson, 1980; Wilke et al., 1980; Gold et al., 1986; Hosokawa & Leahy, 1997). Depletion of insulin reserves by β cell-overworking is thought to impair the secretory process (Sako & Grill, 1990; Leahy, 1996). It has also been suggested that perturbations of glucose-induced insulin secretion provoked by sulphonylureas (Borg & Andersson, 1980; Wilke et al., 1980; Filipponi et al., 1983; Rabuazzo et al., 1992) might result from a dysfunction of K+-ATP channels (Rabuazzo et al., 1992), a target that the drugs share with glucose.
In the present study, normal mouse islets were cultured overnight with therapeutically relevant concentrations of glibenclamide or tolbutamide before their acute responsiveness to glucose was evaluated. The secretory response was studied under static and dynamic conditions and its changes were assessed as a function of the islet insulin content. It was also compared to the changes in β cell cytoplasmic Ca2+ concentration ([Ca2+]i) to evaluate how pretreatment with blockers of K+-ATP channels interferes with the action of glucose through the K+-ATP channel-dependent pathway.
Methods
Solutions and drugs
The medium used for islet isolation was a bicarbonate-buffered solution that contained (mM): NaCl 120, KCl 4.8, CaCl2 2.5, MgCl2 1.2 and NaHCO3 24. It was gassed with O2:CO2 (94:6) to maintain pH 7.4 and was supplemented with bovine serum albumin (1 mg ml−1). A similar medium was used for all experiments after culture. Tolbutamide and glibenclamide (Sigma, St Louis, MO, U.S.A.), meglitinide also known as compound HB 699 (a gift from Hoechst, Frankfurt/Main, Germany), and diazoxide (a gift from Schering-Plough, Avondale, Rathdrum, Ireland) were added to the medium from freshly prepared stock solutions in NaOH. Nimodipine (a gift from Bayer, Wuppertal, Germany) and forskolin (Calbiochem-Behring, San Diego, CA, U.S.A.) were added from stock solutions in DMSO. Adrenaline was from Parke-Davis (Detroit, MI, U.S.A.).
Preparation
Islets were isolated by collagenase digestion of the pancreas of fed male NMRI mice, followed by hand picking (Jonas et al., 1998). They were then cultured for 18–20 h at 37°C in a 95% air:5% CO2 atmosphere. Groups of about 15 islets were placed in 35 mm petri dishes containing 3 ml RPMI 1640 medium (Gibco BRL, Paisley, Scotland, U.K.) containing 10 mM glucose, 10% heat-inactivated foetal calf serum, 100 IU ml−1 penicillin, and 100 μg ml−1 streptomycin. The culture medium was supplemented or not with 0.01 μM glibenclamide or 50 μM tolbutamide.
Measurements of insulin secretion and islet insulin content
After culture, the islets were preincubated for 60 min at 37°C in a bicarbonate-buffered medium containing 10 mM glucose alone or with 0.01 μM glibenclamide or 50 μM tolbutamide, as during the culture period. Two techniques were then used to measure insulin secretion. In the first type of experiment (static incubations), the islets were distributed in batches of 3 in 1 ml of medium containing the appropriate concentrations of glucose and test substances. After 60 min at 37°C, a portion of the medium was withdrawn for insulin assay. In the second type of experiment (dynamic perifusions), batches of 25 islets were placed in parallel perifusion chambers and perifused at a flow rate of 1 ml min−1 (Henquin, 1978). Effluent samples were collected every 0.5 or 2 min. Between the end of the preincubation period and the application of high glucose, the islets were exposed to a sulphonylurea-free medium. This period lasted about 40–60 min for both static (washing and distribution into incubation tubes) and dynamic experiments (washing, distribution into chambers and initial 25 min of perifusion). In all experiments, islets were recovered from incubation tubes and perifusion chambers, and their insulin content was determined after extraction in acid ethanol (Detimary et al., 1995). Insulin was measured by a double antibody radioimmunoassay with rat insulin as the standard (Novo Research Institute, Bagsvaerd, Denmark).
Measurements of [Ca2+]i and NAD(P)H
After culture with or without glibenclamide or tolbutamide, the islets were preincubated for 90 min, again with or without sulphonylurea, in bicarbonate-buffered medium alone (NADPH measurements) or supplemented with the Ca2+-indicator fura PE3/AM (2 μM; Teflabs, Austin, TX, U.S.A.). After washing they were transferred into a perifusion chamber placed on the stage of a microscope, for recording of NAD(P)H or [Ca2+]i as described (Gilon & Henquin, 1992).
Statistical analysis
Results are presented as means±s.e.mean for the indicated number of islets or batches of islets, from the indicated number of different experiments. The statistical significance of differences between means was assessed by Student's t-test for unpaired data or by analysis of variance followed by Newman-Keuls test when more than two groups were compared. Differences were considered significant at P<0.05.
Results
Influence of glibenclamide pretreatment
In control islets, the concentration dependency of glucose-induced insulin secretion displayed a classical sigmoidal shape (Figure 1A). After 18–20 h of culture in the presence of 0.01 μM glibenclamide, insulin secretion in the absence of glucose was increased 2.5 fold (1.27±0.12 vs 0.50±0.09 ng islet−1 h−1), the dose-response curve was shifted to the left (EC50 values of ∼8 vs 14 mM glucose), and the maximum response was decreased by almost 45% (6.11±0.35 vs 11.1±0.81 ng islet−1 h−1). Because culture in the presence of glibenclamide lowered the islet insulin content by 45% as compared to controls (Figure 1B inset), insulin secretion was also expressed as a percentage of content. This exacerbated the differences in the response to 0–10 mM glucose and abrogated the differences in the maximal response (Figure 1B). Islets pretreated overnight with glibenclamide thus show an increased sensitivity to glucose, even when they are studied in the absence of the sulphonylurea.
Figure 1.
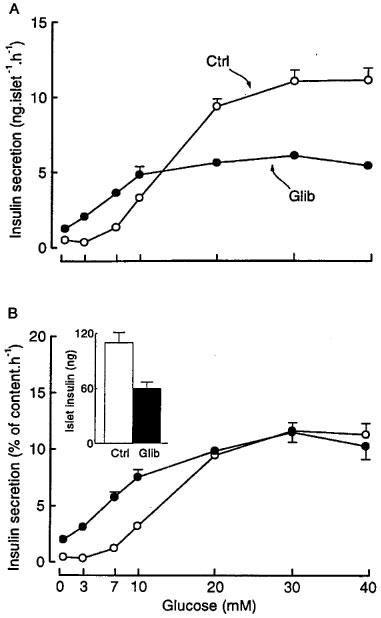
Influence of glibenclamide pretreatment of mouse islets on the concentration-dependence of glucose-induced insulin secretion. The islets were cultured for 18–20 h in a control medium (Ctrl) or in a medium containing 0.01 μM glibenclamide (Glib). After preincubation without glibenclamide (Ctrl) or with glibenclamide (Glib), batches of three islets were incubated in the presence of different glucose concentrations without glibenclamide. Results are presented as absolute values (A) or as a percentage of the islet insulin content (B), which is shown in the inset. Values are means±s.e.mean for 25 batches of islets from five separate experiments.
To elucidate the underlying mechanisms, control and glibenclamide-pretreated islets were incubated in the presence of 10 mM glucose and different test agents (Table 1). In control islets, tolbutamide, glibenclamide at two concentrations, and meglitinide, the non-sulphonylurea moiety of glibenclamide, three blockers of K+-ATP channels (Sturgess et al., 1998; Zünkler et al., 1988), increased insulin secretion, whereas diazoxide, an opener of K+-ATP channels (Sturgess et al., 1988; Zünkler et al., 1988), inhibited it by 90%. In contrast, the high rate of insulin secretion from glibenclamide-pretreated islets was unaffected by either of these four agents. The inability of sulphonylureas and meglitinide to further increase insulin secretion was not due to an already maximum rate of release because forskolin was very effective (Table 1). The lack of effect of diazoxide did not reflect any damage of the islets because blockade of Ca2+ channels by nimodipine and activation of α2-adrenoceptors by adrenaline inhibited insulin secretion by 80–90%. Glibenclamide-pretreated islets are thus unresponsive to drugs that normally close or open K+-ATP channels in β cells.
Table 1.
Influence of glibenclamide pretreatment of mouse islets on insulin secretion induced by various agents (ng insulin islet−1 h−1)

In control islets perifused with 10 or 15 mM glucose, [Ca2+]i displayed oscillations that are masked by averaging of the traces in Figure 2. As expected (Gilon et al., 1992), addition of tolbutamide caused a prompt increase in [Ca2+]i (Figure 2A), whereas addition of diazoxide caused a rapid return to basal values (Figure 2B). In glibenclamide-pretreated islets, [Ca2+]i was higher and more stable than in control islets. Tolbutamide was ineffective and diazoxide only had a marginal effect. The inability of the two drugs to influence insulin secretion can thus be explained by their inability to alter the already elevated [Ca2+]i islets. This interpretation is borne out by the observation that high K+ increased both [Ca2+]i and insulin secretion in glibenclamide-pretreated islets perifused with a medium containing 3 mM glucose. Thus, raising KCl from 4.8 to 15 and then 25 mM, increased the already elevated [Ca2+]i from 190±7 to 257±10 and 310±14 nM (n=12), and the already elevated insulin secretion rate from 25±3 to 53±7 and 76±5 pg islet−1 min−1 (n=4).
Figure 2.
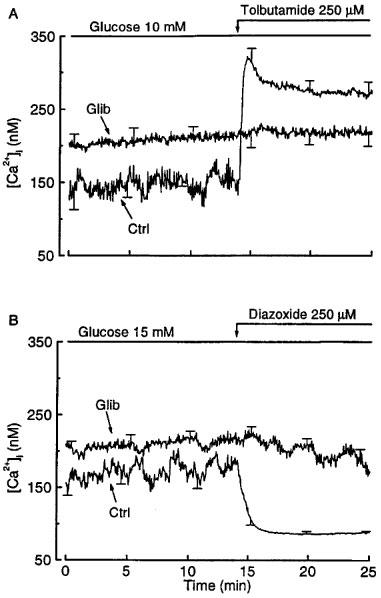
Influence of glibenclamide pretreatment of mouse islets on the changes in cytoplasmic [Ca2+]i produced by tolbutamide and diazoxide. The islets were cultured for 18–20 h in a control medium (Ctrl) or in a medium containing 0.01 μM glibenclamide (Glib). After preincubation without or with glibenclamide, the islets were then perifused with a medium that did not contain glibenclamide. Tolbutamide (A) and diazoxide (B) were added to a medium containing 10 and 15 mM glucose respectively. The traces correspond to means±s.e.mean for 11–12 islets from three separate experiments.
We next investigated the kinetics of glucose-induced insulin secretion from perifused islets. In controls, basal secretion in the presence of 3 mM glucose was low and stable, and 15 mM glucose triggered a biphasic response characterized by a large initial peak followed by a sustained second phase (Figure 3A). In glibenclamide-pretreated islets, insulin secretion was already elevated in the presence of 3 mM glucose, and 15 mM glucose produced a delayed, monophasic response that was of similar amplitude to that of the second phase observed in control islets (Figure 3A). When the rate of insulin secretion was expressed relative to the insulin content of the islets, the pattern of the response was not modified, but the sustained phase of release became larger in test than control islets (Figure 3A), in agreement with the results obtained with the incubation experiments (Figure 1B).
Figure 3.
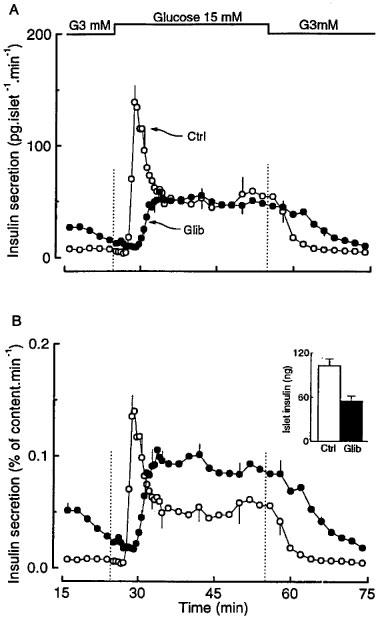
Influence of glibenclamide pretreatment of mouse islets on the kinetics of glucose-induced insulin secretion. The islets were cultured for 18–20 h in a control medium (Ctrl) or in a medium containing 0.01 μM glibenclamide (Glib). After preincubation without or with glibenclamide, batches of 25 islets were then perifused with a medium that did not contain glibenclamide. The glucose concentration was increased from 3 to 15 mM as indicated. Results are presented as absolute rates of secretion (A) or as a percentage of the islet insulin content (B), which is shown in the inset. Values are means±s.e.mean for five separate experiments.
To understand why the secretory response to glucose is delayed in glibenclamide-pretreated islets, the dynamics of the [Ca2+]i and metabolic changes was then studied (Figure 4). Raising the concentration of glucose from 3 to 15 mM caused a triphasic change in [Ca2+]i in control islets: the low basal [Ca2+]i first decreased slightly and transiently before increasing to a peak and eventually stabilizing at a steadily elevated level (Figure 4A). Note, however, that [Ca2+]i oscillations induced by glucose in individual islets (Gilon et al., 1992) are masked by averaging. In glibenclamide-pretreated islets, [Ca2+]i was already elevated in the presence of 3 mM glucose, and showed a biphasic change in response to 15 mM glucose. A marked and prolonged (∼2.5 min) drop initially occurred, and was followed by an increase to stable values (224±8 nM) that were slightly higher (P<0.01) than those measured in glibenclamide-pretreated islets perifused at 3 mM glucose (188±10 nM) and in control islets perifused at 15 mM glucose (184±8 nM). No oscillations of [Ca2+]i were present in individual islets pretreated with glibenclamide (not shown). This marked delay in the [Ca2+]i rise evoked by glucose may explain the lack of the first phase of insulin secretion.
Figure 4.
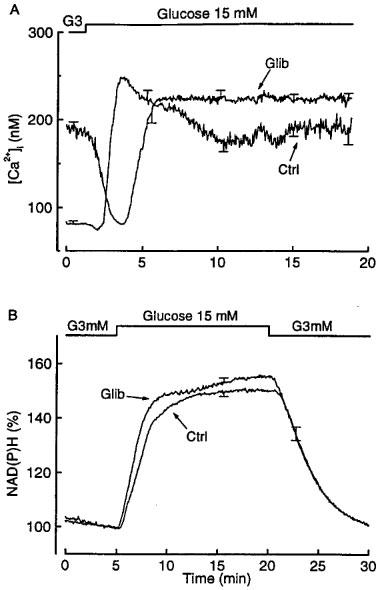
Influence of glibenclamide pretreatment of mouse islets on the changes in cytoplasmic [Ca2+]i (A) and NAD(P)H fluorescence (B) produced by glucose. The islets were cultured for 18–20 h in a control medium or in a medium containing 0.01 μM glibenclamide. After preincubation without or with glibenclamide, the islets were then perifused with a medium that did not contain glibenclamide. The glucose concentration was increased from 3 to 15 mM as indicated. The traces correspond to means±s.e.mean for 18–19 islets from four separate experiments. In (B) the changes in NAD(P)H fluorescence are expressed as a percentage of the signal recorded in the presence of 3 mM glucose. This basal fluorescence was similar in the two groups of islets.
The increase in NAD(P)H fluorescence brought about by a stimulation with 15 mM glucose had similar kinetics and magnitude in control and glibenclamide-pretreated islets (Figure 4B). The alterations of [Ca2+]i and insulin secretion are thus unlikely to result from a major perturbation of glucose metabolism.
Influence of tolbutamide pretreatment
After 18–20 h of culture in the presence of 50 μM tolbutamide, the insulin content of the islets was decreased by 45% as compared with controls (Figure 5B – inset). Although this decrease was similar to that measured in glibenclamide-pretreated islets, the concentration dependency of glucose-induced insulin secretion was different (Figure 5). Insulin secretion was similar in control and tolbutamide-pretreated islets after incubation at 0, 3 and 7 mM glucose. At all higher concentrations it was smaller in tolbutamide-pretreated islets (Figure 5A). The EC50 was similar (∼14–15 mM) in both groups, but the maximum response was decreased by 65% in test islets. This difference was larger than the fall in insulin content, so that the maximum secretory response remained smaller (P<0.001 at 30 and 40 mM glucose) even when the results were expressed as a percentage of insulin content (Figure 5B).
Figure 5.
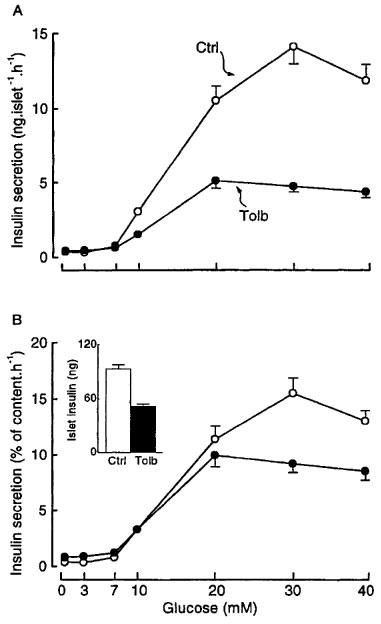
Influence of tolbutamide pretreatment of mouse islets on the concentration-dependence of glucose-induced insulin secretion. The islets were cultured for 18–20 h in a control medium (Ctrl) or in a medium containing 50 μM tolbutamide (Tolb). After preincubation without or with tolbutamide, batches of three islets were then incubated in the presence of different glucose concentrations without tolbutamide. Results are presented as absolute values (A) or as a percentage of the islet insulin content (B), which is shown in the inset. Values are means±s.e.mean for 20 batches of islets from four different cultures.
When tolbutamide-pretreated islets were perifused, basal insulin secretion, at 3 mM glucose, was low as in controls (Figure 6A). Subsequent stimulation with 15 mM glucose evoked a biphasic response with markedly attenuated first and second phases. The rate of rise in insulin secretion during the first phase was clearly less than in control islets, but the start of the rise was not delayed as it was after glibenclamide pretreatment (compare Figures 6A and 3A). When insulin secretion was expressed relative to the islet insulin content, the responses of control and tolbutamide-pretreated islets were almost superimposable (Figure 6B). The characteristics of this secretory response of tolbutamide-pretreated islets can be explained by the changes in [Ca2+]i (Figure 7A). Neither basal [Ca2+]i nor the rapidity and magnitude of the increase evoked by glucose was different between the test and control islets. The differences in insulin secretion between control and tolbutamide-pretreated islets can thus be ascribed to the differences in insulin stores.
Figure 6.
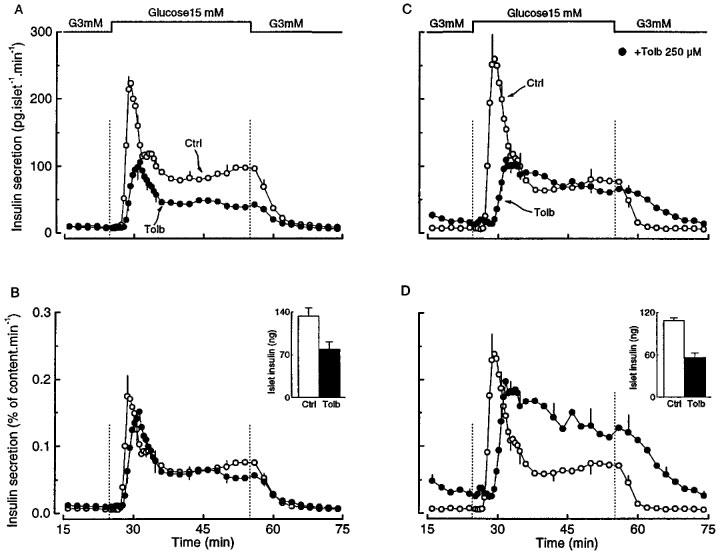
Influence of tolbutamide pretreatment of mouse islets on the kinetics of glucose-induced insulin secretion. The islets were cultured for 18–20 h in a control medium (Ctrl) or in a medium containing 50 μM tolbutamide (Tolb). After preincubation without or with tolbutamide, batches of 25 islets were then perifused with a medium that did not contain tolbutamide except for one series of tolbutamide-pretreated islets (C and D) which were perifused with 250 μM tolbutamide (•). The glucose concentration was increased from 3 to 15 mM as indicated. Results are presented as absolute rates of secretion (A and C) or as a percentage of the islet insulin content (B and D), which is shown in the insets. Values are means±s.e.mean for five separate experiments.
Figure 7.
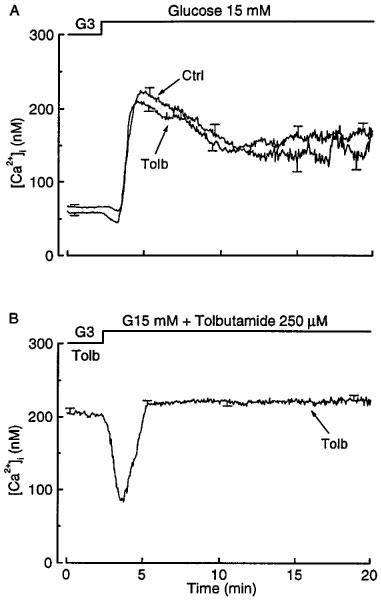
Influence of tolbutamide pretreatment of mouse islets on the changes in cytoplasmic [Ca2+]i produced by glucose. The islets were cultured for 18–20 h in a control medium (Ctrl) or in a medium containing 50 μM tolbutamide (Tolb). After preincubation without or with tolbutamide, the islets were then perifused with a medium which contained either no tolbutamide (A) or 250 μM tolbutamide (B), and in which the concentration of glucose was increased from 3 to 15 mM as indicated. The traces correspond to means±s.e.mean for 11–12 islets from three separate experiments.
The different behaviour of tolbutamide- and glibenclamide-pretreated islets could be explained by a longer persistence of the latter in β cells. If the hypothesis is correct, islets pretreated with tolbutamide should behave like glibenclamide-pretreated islets when tolbutamide is also present during the acute functional tests. In a first series of experiments, 50 μM tolbutamide was thus added to the perifusion medium. In 3 mM glucose this resulted in a [Ca2+]i rise from 58±4 to 102±6 nM. Tolbutamide-pretreated islets thus remain stimulatable by an agent that blocks K+-ATP channels. Subsequent stimulation with 15 mM glucose was followed by a prompt rise in [Ca2+]i and biphasic secretion of insulin (data not shown). Because the [Ca2+]i rise produced by 50 μM tolbutamide remained smaller than that measured in islets cultured with glibenclamide (102±6 vs 188±10 nM), a second series of experiments was performed with 250 μM tolbutamide. With this concentration of the drug in the medium used to perifuse tolbutamide-pretreated islets, [Ca2+]i and insulin secretion were elevated even in the presence of 3 mM glucose (Figures 7B and 6C). Upon stimulation with 15 mM glucose, [Ca2+]i initially decreased before increasing again to a stable level that was slightly higher than in the presence of 3 mM glucose (228±8 vs 211±8 nM, P<0.01 by paired t-test) (Figure 7B). The insulin secretion rate increased with a delay but was not different from that in control islets during sustained stimulation. Because of the lower insulin content of these tolbutamide-pretreated islets, this corresponded to a higher fractional release of insulin than in controls (Figure 6D). These characteristics are superimposable on those of the [Ca2+]i and insulin responses recorded in glibenclamide-pretreated islets (Figures 4A and 3B).
Discussion
Under hyperglycaemic conditions, the increase in plasma insulin levels induced by glibenclamide occurs over a narrow range of drug concentrations (0.05–0.1 μM) (Groop, 1992). In most patients treated with glibenclamide, plasma concentrations of the drug are below 0.4 μM (Heptner et al., 1984; Jaber et al., 1994). However, many in vitro studies of glibenclamide effects on β cell function have used the drug at substantially higher concentrations, and neglected its strong binding to albumin, that is present in lower concentrations in culture or incubation media than in plasma. In the present study glibenclamide was used at the concentration of 0.01 μM which, in the presence of 10% serum in the culture medium, should give a free drug concentration in the range of those found in vivo (Panten et al., 1989).
Culture of normal mouse islets for 20 h in the presence of 0.01 μM glibenclamide profoundly altered several aspects of glucose-induced insulin secretion in spite of the removal of the drug during the tests. Perturbations of [Ca2+]i regulation in β cells can account for several of these alterations. First, insulin secretion was increased even at low glucose concentrations (<7 mM) that are ineffective in control islets. This can be attributed to an elevation of basal [Ca2+]i resulting from a persistent closure of K+-ATP channels. Thus, glibenclamide-pretreated islets were unresponsive to pharmacological openers or inhibitors of K+-ATP channels, but still inhibitable by blockers of voltage-dependent Ca2+ channels. It has also been reported that neither glucose nor glibenclamide is able to decrease K+ (86Rb) efflux from islets cultured for 24 h with glibenclamide (Rabuazzo et al., 1992). Second, no first phase insulin secretion occurred in response to 15 mM glucose. This cannot be attributed to a delay or major impairment in the acceleration of glucose metabolism but can be explained by the lack of a rapid rise in [Ca2+]i. Instead, glucose caused a marked drop of the already elevated [Ca2+]i, followed by a monophasic climb slightly above initial values. This paradoxical drop in [Ca2+]i probably reflects sequestration of the ion within cellular organelles (Hellman et al., 1990; Roe et al., 1994).
To determine whether these perturbations of β cell function are specific effects of glibenclamide, islets were also cultured in the presence of 50 μM tolbutamide. This concentration was selected to achieve a similar stimulation of secretion and decrease (45%) in islet insulin stores as those produced by glibenclamide. It is, even when protein binding is taken into account (Panten et al., 1989), within the range of tolbutamide plasma levels in patients under chronic treatment (Cattaneo et al., 1990; Sartor et al., 1980). The behaviour of tolbutamide-pretreated islets strikingly differed from that of glibenclamide-pretreated islets in several respects. [Ca2+]i was low in the basal state, and it normally increased in response to glucose, which explains that basal insulin secretion was low and that stimulated secretion displayed a biphasic pattern. It is thus clear that treatment of the islets with a sulphonylurea for 20 h does not automatically put K+-ATP channels out of action. This conclusion is borne out by our observations that tolbutamide-pretreated islets still responded by [Ca2+]i and insulin secretion changes to acute stimulation with tolbutamide. Others have reported that a high concentration of tolbutamide (1 mM) caused a normal inhibition of K+ (86Rb) efflux from islets cultured for 24 h with tolbutamide (Rabuazzo et al., 1992). However, when the effects of glucose on [Ca2+]i and insulin secretion were evaluated in the presence of tolbutamide, tolbutamide-pretreated islets displayed the abnormalities characteristic of glibenclamide-pretreated islets. The behaviour of the latter can thus be explained by the persistence of the drug in β cells even after its removal from the medium. It is known that, unlike other sulphonylureas, glibenclamide accumulates within β cells (Hellman et al., 1984).
The overworked-β cell hypothesis suggests that chronic hyperstimulation leads to β cell secretory dysfunction because of a decrease in insulin stores (Leahy, 1996). Islets cultured with tolbutamide, but not those cultured with glibenclamide, are a good model to evaluate the consequences of β cell-overworking. The secretory response to glucose was quantitatively smaller than that of control islets, but the decrease matched the ∼50% lowering of insulin stores except for glucose concentrations exceeding 20 mM. The kinetics of glucose-induced insulin secretion and [Ca2+]i rise were unaltered, which suggests that the characteristics of the [Ca2+]i changes are more important than the size of insulin stores in determining the biphasic pattern of secretion. The sensitivity to glucose of the insulin release process was unchanged after tolbutamide pretreatment. This indicates that, when an increased sensitivity to glucose is observed after in vitro or in vivo islet-overworking (Leahy, 1996), it results from changes other than the decrease in insulin stores, probably from nutrient-mediated metabolic changes.
Glucose-induced insulin secretion normally depends on a rise in β cell [Ca2+]i, mediated by closure of K+-ATP channels and membrane depolarization (the K+-ATP channel-dependent pathway) (Dunne & Petersen, 1991; Misler et al., 1992; Henquin, 1994; Prentki, 1996). When islets, that have not been pretreated with sulphonylureas during culture, are acutely depolarized by a high concentration of K+ or a maximally effective concentration of sulphonylurea, they remain able to secrete more insulin in response to glucose (Panten et al., 1988; Best et al., 1992; Gembal et al., 1992). This K+-ATP channel-independent pathway involves potentiation of the action of Ca2+ on exocytosis (Gembal et al., 1993; Eliasson et al., 1997; Sato et al., 1999). It is illustrated here by the following comparison: although [Ca2+]i was almost identical in glibenclamide-pretreated islets maintained at 3 mM glucose and in control islets stimulated by 15 mM glucose alone, insulin secretion was greater in the latter. This pathway also largely contributes to the stimulation of secretion by glucose in glibenclamide-pretreated islets and in tolbutamide pretreated islets tested in the presence of tolbutamide. Thus, the large increase in secretion occurred with a minimal change in [Ca2+]i. As these observations were made after pretreatment of the islets with therapeutic concentrations of the drugs, a similar situation is likely to prevail in vivo. During continuous sulphonylurea treatment, glucose regulation of insulin secretion may thus depend more on the K+-ATP channel-independent pathway than on changes in the β cell membrane potential via the K+-ATP channel-dependent pathway. As the former pathway has a higher sensitivity to glucose than the latter (Gembal et al., 1993), secretion may be stimulated at low glucose concentrations as shown here in vitro. The same mechanism may explain the inappropriate secretion of insulin at low blood glucose levels in vivo and the hypoglycaemic episodes sometimes complicating sulphonylurea treatment. One should, however, remain cautious in these extrapolations and bear in mind that the β cells of those patients receiving sulphonylureas have intrinsically abnormal glucose responsiveness. For the same reasons and because no available experimental studies in normal subjects can be strictly compared to our in vitro experiments, it is very difficult to predict that chronic sulphonylurea treatment will exert consistent effects on the first phase insulin secretion (Groop, 1992). The K+-ATP channel-independent pathway may also be the major mechanism by which glucose influences insulin secretion in infants suffering from persistent hyperinsulinaemic hypoglycaemia because of a loss of functional K+-ATP channels in their β cells (Kane et al., 1996).
Acknowledgments
This work was supported by grants 3.4552.98 from the Fonds de la Recherche Scientifique Médicale, Brussels; by grant ARC 95/00-188 from the General Direction of Scientific Research of the French Community of Belgium; and by the Interuniversity Poles of Attraction Programme (P4/21), Belgian State, Prime Minster's Office. We are grateful to F. Knockaert for technical help and to M. Nenquin and S. Roiseux for editorial assistance.
Abbreviations
- [Ca2+]i
cytoplasmic Ca2+ concentration
- K+-ATP channels
ATP-sensitive K+ channels
References
- AGUILAR-BRYAN L., CLEMENT J.P., GONZALEZ G., KUNJILWAR K., BABENKO A., BRYAN J. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., GRIBBLE F.M. Correlating structure and function in ATP-sensitive K+ channels. TINS. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- BEST L., YATES A.P., TOMLINSON S. Stimulation of insulin secretion by glucose in the absence of diminished potassium (86Rb+) permeability. Biochem. Pharmacol. 1992;43:2483–2485. doi: 10.1016/0006-2952(92)90330-l. [DOI] [PubMed] [Google Scholar]
- BORG L.A.H., ANDERSSON A. Long-term effects of glibenclamide on the insulin production, oxidative metabolism and quantitative ultrastructure of mouse pancreatic islets maintained in tissue culture at different glucose concentrations. Acta Diabetol. Lat. 1980;18:65–83. doi: 10.1007/BF02056108. [DOI] [PubMed] [Google Scholar]
- CATTANEO A.G., CAVIEZEL F., POZZA G. Pharmacological interaction between tolbutamide and acetylsalicylic acid: study on insulin secretion in man. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990;28:229–234. [PubMed] [Google Scholar]
- DETIMARY P., JONAS J.C., HENQUIN J.C. Possible links between glucose-induced changes in the energy state of pancreatic B-cells and insulin release: unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J. Clin. Invest. 1995;96:1738–1745. doi: 10.1172/JCI118219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNNE M.J., PETERSEN O.H. Potassium selective ion channels in insulin-secreting cells: physiology, pharmacology and their role in stimulus-secretion coupling. Biochim. Biophys. Acta. 1991;1070:67–82. doi: 10.1016/0304-4157(91)90012-l. [DOI] [PubMed] [Google Scholar]
- ELIASSON L., RENSTROM E., DING W.G., PROKS P., RORSMAN P. Rapid ATP-dependent priming of secretory granules precedes Ca2+-induced exocytosis in mouse pancreatic β-cells. J. Physiol. 1997;503:399–412. doi: 10.1111/j.1469-7793.1997.399bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIPPONI P., MARCELLI M., NICOLETTI I., PACIFICI R., SANTEUSANIO F., BRUNETTI P. Suppressive effect of long term sulphonylurea treatment on A, B, and D cells of normal rat pancreas. Endocrinology. 1983;113:1972–1979. doi: 10.1210/endo-113-6-1972. [DOI] [PubMed] [Google Scholar]
- GEMBAL M., DETIMARY P., GILON P., GAO Z.Y., HENQUIN J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channel-independent in mouse cells. J. Clin. Invest. 1993;91:871–880. doi: 10.1172/JCI116308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEMBAL M., GILON P., HENQUIN J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse cells. J. Clin. Invest. 1992;89:1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILON P., HENQUIN J.C. Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin secreting pancreatic B-cell. J. Biol. Chem. 1992;267:20713–20720. [PubMed] [Google Scholar]
- GOLD G., POU J., GISHIZKY M.L., LANDAHL H.D., GRODSKY G.M. Effects of tolbutamide pretreatment on the rate of conversion of newly synthesized proinsulin to insulin and the compartmental characteristics of insulin storage in isolated rat islets. Diabetes. 1986;35:6–12. doi: 10.2337/diab.35.1.6. [DOI] [PubMed] [Google Scholar]
- GROOP L.C. Sulfonylureas in NIDDM. Diabetes Care. 1992;15:737–754. doi: 10.2337/diacare.15.6.737. [DOI] [PubMed] [Google Scholar]
- HELLMAN B., BERNE C., GRAPENGIESSER E., GRILL V., GYLFE E., LUND P.E. The cytoplasmic Ca2+ response to glucose as an indicator of impairment of the pancreatic β-cell function. Eur. J. Clin. Invest. 1990;20 Suppl 1:S10–S17. doi: 10.1111/j.1365-2362.1990.tb01771.x. [DOI] [PubMed] [Google Scholar]
- HELLMAN B., SEHLIN J., TALJEDAL I.B. Glibenclamide is exceptional among hypoglycaemic sulphonylureas in accumulating progressively in β-cell-rich pancreatic islets. Acta Endocrinol. 1984;105:389–390. doi: 10.1530/acta.0.1050385. [DOI] [PubMed] [Google Scholar]
- HENQUIN J.C. D-Glucose inhibits potassium efflux from pancreatic islet cells. Nature. 1978;271:271–273. doi: 10.1038/271271a0. [DOI] [PubMed] [Google Scholar]
- HENQUIN J.C.Established, unsuspected and novel pharmacological insulin secretagogues New Antdiabetic Drugs 1990London: Smith-Gordon; 93–106.Bailey, C.J., Flatt, P.R., eds [Google Scholar]
- HENQUIN J.C.The cell biology of insulin secretion The Joslin's Diabetes Mellitus 1994Philadelphia: Lea & Febiger; 56–80.13th edn. Kahn, C.R., Weir, G.C., eds [Google Scholar]
- HENQUIN J.C. A minimum of fuel is necessary for tolbutamide to mimic the effects of glucose on electrical activity in pancreatic β-cells. Endocrinology. 1998;139:993–998. doi: 10.1210/endo.139.3.5783. [DOI] [PubMed] [Google Scholar]
- HEPTNER W., BADIAN M., BAUDNER S., HELLSTERN C., IRMISCH R., RUPP W., WEIMER K., WISSMANN H. A radioimmunoassay for determination of glibenclamide and other sulphonylureas. Pharmacol. Res. 1984;1:215–220. doi: 10.1023/A:1016321313510. [DOI] [PubMed] [Google Scholar]
- HOSOKAWA Y.A., LEAHY J.L. Parallel reduction of pancreas insulin content and insulin secretion in 48-h tolbutamide-infused normoglycemic rats. Diabetes. 1997;46:808–813. doi: 10.2337/diab.46.5.808. [DOI] [PubMed] [Google Scholar]
- JABER L.A., SLAUGHTER R.L., ANTAL E.J., WELSHMAN I.R. Comparison of pharmacokinetics and pharmacodynamics of short- and long-term glyburide therapy in NIDDM. Diabetes Care. 1994;17:1300–1306. doi: 10.2337/diacare.17.11.1300. [DOI] [PubMed] [Google Scholar]
- JONAS J.C., GILON P., HENQUIN J.C. Temporal and quantitative correlations between insulin secretion and stably elevated or oscillatory cytoplasmic Ca2+ in mouse pancreatic β cells. Diabetes. 1998;47:1266–1273. doi: 10.2337/diab.47.8.1266. [DOI] [PubMed] [Google Scholar]
- KANE C., SHEPHERD R.M., SQUIRES P.E., JOHNSON P.R.V., JAMES R.F.L., MILLA P.J., AYNSLEY-GREEN A., LINDLEY K.J., DUNNE M.J. Loss of functional KATP channels in pancreatic β-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nature Med. 1996;2:1344–1347. doi: 10.1038/nm1296-1344. [DOI] [PubMed] [Google Scholar]
- LEAHY J.L. Impaired β-cell function with chronic hyperglycemia: ‘overworked β-cell' hypothesis. Diabetes Rev. 1996;4:298–319. [Google Scholar]
- MATTHEWS D.R., CULL C.A., STRATTON I.M., HOLMAN R.R., TURNER R.C. Sulphonylurea failure in non-insulin-dependent diabetic patients over six years. Diabet. Med. 1998;15:297–303. doi: 10.1002/(SICI)1096-9136(199804)15:4<297::AID-DIA572>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- MISLER S., BARNETT D.W., GILLIS K.D., PRESSEL D.M. Electrophysiology of stimulus-secretion coupling in human β-cell. Diabetes. 1992;41:1221–1228. doi: 10.2337/diab.41.10.1221. [DOI] [PubMed] [Google Scholar]
- PANTEN U., BURGFELD J., GOERKE F., RENNICKE M., SCHWANSTECHER M., WALLASCH A., ZÜNKLER B.J., LENZEN S. Control of insulin secretion by sulphonylureas, meglitinide and diazoxide in relation to their binding to the sulphonylurea receptor in pancreatic islets. Biochem. Pharmacol. 1989;38:1217–1229. doi: 10.1016/0006-2952(89)90327-4. [DOI] [PubMed] [Google Scholar]
- PANTEN U., SCHWANSTECHER M., SCHWANSTECHER C.Mode of action of sulphonylureas Oral Antidiabetics 1996Springer Verlag: Berlin, Heidelberg; 129–159.Kuhlmann J., Puls, W., eds [Google Scholar]
- PANTEN U., SCHWANSTECHER M., WALLASCH A., LENZEN S. Glucose both inhibits and stimulates insulin secretion from isolated pancreatic islets exposed to maximally effective concentrations of sulphonylureas. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;338:459–462. doi: 10.1007/BF00172128. [DOI] [PubMed] [Google Scholar]
- PONTIROLI A.E., CALDERARA A., POZZA G. Secondary failure of oral hypoglycaemic agents: frequency, possible causes, and management. Diabetes Metab. Rev. 1994;10:31–43. doi: 10.1002/dmr.5610100104. [DOI] [PubMed] [Google Scholar]
- PRENTKI M. New insights into pancreatic β-cell metabolic signaling in insulin secretion. Eur. J. Endocrinol. 1996;134:272–286. doi: 10.1530/eje.0.1340272. [DOI] [PubMed] [Google Scholar]
- RABUAZZO A.M., BUSCEMA M., VINCI C., CALTABIANO V., VETRI M., FORTE F., VIGNERI R., PURRELLO F. Glyburide and tolbutamide induce desensitization of insulin release in rat pancreatic islets by different mechanisms. Endocrinology. 1992;131:1815–1820. doi: 10.1210/endo.131.4.1396327. [DOI] [PubMed] [Google Scholar]
- ROE M.W., MERTZ R.J., LANCASTER M.E., WORLEY J.F., III, DUKES I.D. Thapsigargin inhibits the glucose-induced decrease of intracellular Ca2+ in mouse islets of Langerhans. Am. J. Physiol. 1994;266:E852–E862. doi: 10.1152/ajpendo.1994.266.6.E852. [DOI] [PubMed] [Google Scholar]
- SAKO Y., GRILL V.E. Coupling of β-cell desensitization by hyperglycemia to excessive stimulation and circulating insulin in glucose-infused rats. Diabetes. 1990;39:1580–1583. doi: 10.2337/diab.39.12.1580. [DOI] [PubMed] [Google Scholar]
- SARTOR G., MELANDER A., SCHERSTEN B., WAHLIN-BOLL E. Influence of food and age on the single-dose kinetics and effects of tolbutamide and chlorpropamide. Eur. J. Clin. Pharmacol. 1980;17:285–293. doi: 10.1007/BF00625802. [DOI] [PubMed] [Google Scholar]
- SATIN L.S. New mechanisms for sulphonylurea control of insulin secretion. Endocrine. 1996;4:191–198. doi: 10.1007/BF02738684. [DOI] [PubMed] [Google Scholar]
- SATO Y., AIZAWA T., KOMATSU M., OKADA N., YAMADA T. Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic β-cell. Diabetes. 1992;41:438–443. doi: 10.2337/diab.41.4.438. [DOI] [PubMed] [Google Scholar]
- SATO Y., ANELLO M., HENQUIN J.C. The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose during opening and closure of the channel. Endocrinology. 1999;140:2252–2257. doi: 10.1210/endo.140.5.6729. [DOI] [PubMed] [Google Scholar]
- SATO Y., HENQUIN J.C. The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose. In search of the underlying mechanism. Diabetes. 1998;47:1713–1721. doi: 10.2337/diabetes.47.11.1713. [DOI] [PubMed] [Google Scholar]
- STURGESS N.C., KOZLOWSKI R.Z., CARRINGTON C.A., HALES C.N., ASHFORD M.L.J. Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. Br. J. Pharmacol. 1988;95:83–94. doi: 10.1111/j.1476-5381.1988.tb16551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK PROSPECTIVE DIABETES STUDY GROUP Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- WILKE B., ZIEGLER B., SCHMIDT S., WOLTANSKI P., ZÜHLKE H. The influence of cultivation on proinsulin biosynthesis and secretion of isolated pancreatic islets of C57BL-mice, long-term treated with glibenclamide in vivo. Endokrinologie. 1980;76:S357–S364. [PubMed] [Google Scholar]
- ZÜNKLER B.J., LENZEN S., MANNER K., PANTEN U., TRUBE G. Concentration-dependent effects of tolbutamide, meglitinide, glipizide, glibenclamide and diazoxide on ATP-regulated K+ currents in pancreatic B-cells. Naunyn Schmiedeberg's Arch. Pharmacol. 1988;337:225–230. doi: 10.1007/BF00169252. [DOI] [PubMed] [Google Scholar]