

Abstract
Renal vasoconstriction in response to angiotensin II (ANGII) is known to be modulated by nitric oxide (NO). Since shear stress stimulates the release of a variety of vasoactive compounds from endothelial cells, we studied the impact of shear stress on the haemodynamic effect of ANGII in isolated perfused kidneys of rats under control conditions and during NO synthase inhibition with L-NAME (100 μM).
Kidneys were perfused in the presence of cyclo-oxygenase inhibitor (10 μM indomethacin) with Tyrode's solution of relative viscosity ζ=1 (low viscosity perfusate, LVP) or, in order to augment shear stress, with Tyrode's solution containing 7% Ficoll 70 of relative viscosity ζ=2 (high viscosity perfusate, HVP).
Vascular conductance was 3.5±0.4 fold larger in HVP as compared with LVP kidneys, associated with an augmentation of overall wall shear stress by 37±5%. During NO inhibition, vascular conductance was only 2.5±0.2 fold elevated in HVP vs LVP kidneys, demonstrating shear stress-induced vasodilatation by NO and non-NO/non-prostanoid compound(s).
ANGII (10–100 pM) constricted the vasculature in LVP kidneys, but was without effect in HVP kidneys. During NO inhibition, in contrast, ANGII vasoconstriction was potentiated in HVP as compared with LVP kidneys.
The potentiation of ANGII vasoconstriction during NO inhibition has been shown to be mediated by endothelium-derived P450 metabolites and to be sensitive to AT2 receptor blockade in our earlier studies. Accordingly, in HVP kidneys, increasing concentrations of the AT2 receptor antagonist PD123319 (5 and 500 nM) gradually abolished the potentiation of ANGII vasoconstriction during NO inhibition, but did not affect vasoconstriction in response to ANGII in LVP kidneys.
Our results demonstrate, that augmentation of shear stress by increasing perfusate viscosity induces vasodilatation in the rat kidney, which is partially mediated by NO. Elevated levels of shear stress attenuate renal ANGII vasoconstriction through enhanced NO production and are required for AT2 sensitive potentiation during NO inhibition.
Keywords: Isolated perfused kidney, vascular resistance, nitric oxide, AT1 receptor, AT2 receptor, viscosity
Introduction
Many studies have shown that vasoactive factors derived from the endothelium enhance or blunt the pressor effect of any vasoconstrictor currently active in the renovascular system (Navar et al., 1996). This is particularly true for the action of angiotensin II (ANGII) on the renal microcirculation, which is modulated by endothelium-derived nitric oxide (NO), prostaglandins, and other eicosanoids (Arima et al., 1994; 1997; Ito et al., 1991; 1993; Navar et al., 1996; Oyekan et al., 1997; Sigmon et al., 1992). Among these compounds, NO importantly counter-balances the renal AT1 receptor-mediated vasoconstriction (Adachi et al., 1996; Madrid et al., 1997; Parekh et al., 1996). Interestingly, suppression of ANGII-induced renal vasoconstriction by NO involves further endothelial autacoids, as we have recently demonstrated that, during NO synthase inhibition, renal vasoconstriction in response to subnanomolar concentrations of ANGII is potentiated by an AT2 receptor antagonist-sensitive mechanism, which depends on intact endothelium and constrictory P450 metabolites (Muller et al., 1997; 1998).
Shear stress, which is exerted by the blood stream on the endothelial cell surface, regulates endothelial autacoid production and gene transcription (Davies, 1995; Takahashi et al., 1997). It has been demonstrated in many vascular preparations, that augmentation of shear stress leads to vasodilatation through the release of endothelial NO and/or prostaglandins (de Wit et al., 1997; Koller & Kaley, 1990; Koller et al., 1993; 1994; Pohl et al., 1991). In the kidney, however, the effect of shear stress on vasomotor tone has been addressed in only a few studies. In these studies, it has been demonstrated that renal blood flow remains constant despite changes of blood viscosity in vivo (Chen et al., 1989), and that pressure- and ANGII-induced constrictions in isolated afferent arterioles are enhanced under no-flow as compared to free-flow conditions (Juncos et al., 1995; 1996). However, whether basal tone or ANGII-induced constriction in renal vessels are intrinsically sensitive to shear stress mediated by changes in endogenous NO levels, has not yet been investigated in an experimental setup, in which shear stress was manipulated by viscosity. Since renal vessels exhibit a profound myogenic response, disturbances of pressure, caused by primary manipulation of flow in order to vary the level of shear stress, may confound analysis of shear stress-induced vasomotor responses. In the present study, we determined vascular conductance at a common perfusion pressure and manipulated shear stress through perfusate viscosity in the isolated rat kidney, thereby minimizing alterations of intravascular pressures. By this approach we were able to study the effect of shear stress on basal renovascular tone and on ANGII-induced renal vasoconstriction at subnanomolar ANGII concentrations, at which potentiation of the ANGII response depends on endogenous NO levels. Experiments were done in the absence and presence of the NO synthase inhibitor L-NAME to assess the role of endogenous NO production in mediating the effects of shear stress.
Methods
Drugs
Indomethacin, NG-nitro-L-arginine methyl ester (L-NAME) and balanced Tyrode's salt solution were obtained from Sigma (St Louis, MO, U.S.A.). ANGII was obtained from Neosystem Laboratory (Strasbourg, France), PD123319 from Research Biochemicals International (Natrick, U.S.A.) and L158809 from Merck (Rahway, U.S.A.). Ficoll®70 was from Pharmacia (Uppsala, Sweden).
Preparation of the isolated rat kidney
Male Wistar rats, weighing 170–220 g, with free access to standard food and water, were anaesthetized by intraperitoneal injection of sodium pentobarbital (65 mg kg−1) and used for the preparation of the isolated perfused kidney exactly as described previously (Muller et al., 1997; 1998). Briefly, the right kidney was perfused in an open single-pass circuit through the superior mesenteric artery. Perfusion was started in situ immediately after the suprarenal aorta had been tied, thereby protecting the kidney from any ischaemia. Heparin (1000 U) was injected into the left femoral artery. Perfusion pressure was monitored through the infrarenal aorta. While perfused, the kidney was excised from the animal body and transferred onto a heated holder. The kidney preparations were rendered non-filtering by tying the ureter in order to exclude tubular interferences with renal haemodynamics. The composition of the basic perfusion solution consisted of a commercially available Tyrode's solution supplemented extemporaneously with 17 mM sodium bicarbonate. This medium had an osmolality of 286 mosm kg−1, a relative viscosity of ζ=1.0 and was designated as ‘low viscosity perfusate' or LVP. The same medium containing 7% Ficoll had a relative viscosity of ζ=2.0, as determined by means of a Stoke's viscosimeter, and was designated as ‘high viscosity perfusate' or HVP. Ficoll 70 is a synthetic polymer of sucrose with a molecular weight of 60–80 kDa. Addition of 7% Ficoll 70 increased osmolality by about 7 mosm kg−1, according to the technical specifications supplied by the manufacturer. Kidneys were systematically perfused in the presence of 10 μM indomethacin to obviate the involvement of vasoactive prostaglandins (Muller et al., 1997; 1998). The perfusate was routinely thermostated at 37°C, was continuously filtered through a 1.2 μm filter and gassed with 95% O2–5% CO2. The pH was adjusted to 7.4 in the prewarmed, preoxygenated medium.
Experimental protocols
The vasoconstrictor responses to ANGII were measured as perfusion pressure changes under conditions of constant perfusate flow exactly as described before (Muller et al., 1997; 1998). The perfusate flows (expressed in ml min−1g−1) were adjusted during a 60 min equilibration period to achieve a common pressure baseline of 90 mmHg; thereafter, the flow thus adjusted was maintained constant. The resulting vascular conductance was determined at the end of the equilibration period. The kidney preparations were perfused with control LVP or HVP, or with these perfusates containing throughout 100 μM L-NAME alone, or in combination with the AT1 receptor antagonist L158809 (500 nM) or the AT2 receptor antagonist PD123319 (5 or 500 nM). The concentrations of L158809 and PD123319 employed in the present study have been demonstrated to be subtype-selective in a previous study (Muller et al., 1997). In this previous study, the concentration-dependent action of both antagonists on ANGII-induced renal vasoconstriction has been determined in our isolated kidney preparation, perfused with a high viscosity perfusate containing L-NAME. After the equilibration period, ANGII was infused at final perfusate concentrations of 10, 20, and 100 pM for periods of 6 min each, separated by recovery periods of 24 min. ANGII solutions were infused via a sideline at a rate of 0.5 ml min−1 by means of an automatically pushed syringe. ANGII was dissolved in an aliquot of current perfusion medium. Pressure values measured during ANGII infusions were corrected for a marginal rise in perfusion pressure (about 3–6 mmHg) due to the additionally infused volume of 0.5 ml min−1, representing about 5% of total perfusate flow.
Calculations and analysis of data
A computerized data acquisition system continuously collected pressure and flow values at 1 Hz throughout the experiment. Consecutive blocks of 15 data points were averaged to obtain four measurements per min for flow and pressure. The vasoconstrictor effect of ANGII was expressed as the maximum pressure increase over the 6 min of peptide infusion. Vascular conductance gv and overall wall shear stress τ were calculated from measured parameters, i.e. perfusion pressure p and perfusate flow Q, in order to quantify the effects of perfusate viscosity ζ on the renal vasculature. Conductance g is defined by Ohm's law as g=Q/p. According to Poiseuille's law, g is a function of viscosity ζ, vessel length l and radius r: g=(π·r4)/(8·ζ l). Thus, conductance g does not reflect solely changes in vasomotor tone in our experimental setting, since we modified viscosity. We therefore calculated ‘true' vascular conductance gv, which is independent from viscosity:
Note that, under the reasonable assumption of unchanged vessel length l, vascular conductance gv is a direct measure of changes in vessel radius r. Since viscosity was expressed for calculations in centiPoise (cP), vascular conductance gv is given as μl min−1 g−1 mmHg−1cP.
As we have not determined microvascular parameters, we can not calculate absolute values of wall shear stress τ=(4 ζ·Q)/(π·r3). However, if we assume that relative segmental changes of vessel radii are uniform throughout the renal vasculature, then relative changes in blood flow are identical in all vessels and relative changes of ‘overall' wall shear stress τ can be calculated. Though this assumption is often violated due to the heterogeneity of renal microvascular responses (Steinhausen & Endlich, 1996), calculation of overall wall shear stress nevertheless provides a useful estimate for the mean of segmental changes in shear stress. Solving equation (1) for vessel radius r, one obtains r=[(ζ·Q)/(c·p)]1/4. Substituting r in the equation for wall shear stress yields:
Equation (2) permits calculation of overall wall shear stress τ in arbitrary units, from perfusate viscosity ζ, perfusate flow Q, and perfusion pressure p. Means of relative segmental changes in wall shear stress during HVP and/or NO inhibition were estimated by calculating τ and setting the mean value of τ for LVP under control conditions to 100%.
All reported values are expressed as means±s.e.mean. ANOVA was performed on the absolute values of the various parameters. Differences were considered significant for P<0.05. When the ANOVA detected a significant effect, comparisons among individual means were based on the Student-Newman-Keul's test.
Results
Effect of perfusate viscosity on renal vascular tone and NO
Figure 1 shows the vascular conductance and the overall wall shear stress obtained from all the isolated kidney preparations performed in the present study. Consistent with our previous studies (Muller et al., 1997; 1998), none of the ANGII receptor antagonists used in L-NAME-perfused kidneys affected basal renal vascular conductance. Therefore, the data of L-NAME-perfused kidneys obtained in the absence or presence of ANGII receptor antagonists were pooled together. The 2 fold increase in relative perfusate viscosity ζ from LVP (ζ=1) to HVP (ζ=2) would halve flow for a given vascular conductance under conditions of constant perfusion pressure. However, perfusate flow in HVP kidneys was 58% higher than in LVP kidneys (14.9±1.2 vs 8.6± 0.7 ml min−1 g−1, P<0.005). Taking augmented viscosity into account (cf. Methods), this corresponds to a 3.5±0.4 fold increase in vascular conductance in HVP as compared with LVP kidneys (330±27 vs 95±7 μl min−1 g−1 mmHg−1cP). At the same time, overall wall shear stress was 37±5% higher in HVP than in LVP kidneys.
Figure 1.
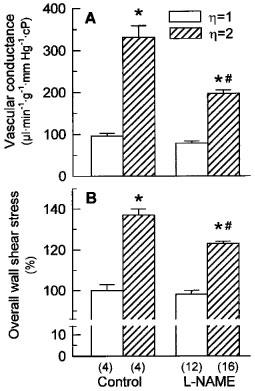
Vascular conductance (A) and relative changes of overall wall shear stress (B) in kidney preparations which have been perfused with low or high viscosity perfusate in the absence or presence of 100 μM L-NAME, as indicated. Perfusate flow was adjusted during an initial 60 min equilibration period to achieve a common perfusion pressure of 90.8±0.4 mmHg (n=36); thereafter, perfusate flow thus adjusted was maintained constant. Vascular conductance and relative changes of overall wall shear stress were calculated from perfusion pressure and perfusate flow (cf. Calculation and analysis of data in Methods) measured after equilibration and before administration of ANGII. The left kidney was used as a weight basis for calculations with a mean weight of 0.81±0.02 g (n=36). The results are presented as means±s.e.mean for the number of kidney preparations shown in parentheses. *P<0.05 high vs low viscosity perfusate. #P<0.05 L-NAME vs control perfusate.
Vascular conductance was not significantly diminished in L-NAME-perfused LVP kidneys as compared with control LVP kidneys (79±5 vs 95±7 μl min−1 g−1 mmHg−1cP), demonstrating little endogenous NO production under conditions of low shear stress. In L-NAME-perfused kidneys, HVP was still associated with a higher vascular conductance as compared with LVP (197±8 vs 79±5 μl min−1 g−1 mmHg−1cP). However, vascular conductance was by 40% significantly lower in L-NAME-perfused HVP kidneys than in control HVP kidneys, implicating NO in viscosity-induced renal vasodilatation. Overall wall shear stress was by 28±3% higher in HVP as compared with LVP kidneys during NO inhibition. Perfusion pressure did not significantly differ between HVP and LVP kidneys neither in absence (90.0±0.4 vs 89.6±1.1 mmHg) nor in presence of L-NAME (91.8±0.6 vs 90.6±0.8 mmHg).
Effect of perfusate viscosity on ANGII-induced vasoconstriction
Figure 2 shows averaged records of the changes in perfusion pressure in response to successive 6 min infusions of 10, 20, and 100 pM of ANGII in LVP and HVP kidneys. ANGII concentration-dependently constricted the renal vasculature in LVP kidneys, but was without effect in HVP kidneys. In marked contrast, when kidneys were perfused throughout with L-NAME, ANGII-induced vasoconstrictions in HVP kidneys exceeded those in LVP kidneys.
Figure 2.
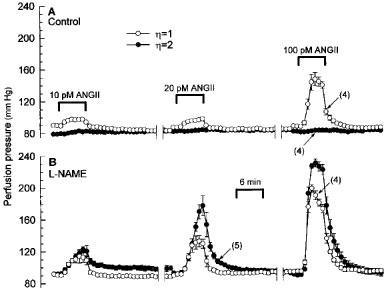
Changes in perfusion pressure depicting the vasoconstrictor effects of 10, 20, and 100 pM ANGII in isolated kidneys perfused at constant flow with low or high viscosity perfusate in the absence (A) or presence of 100 μM L-NAME (B). Pressure values are means±s.e.mean for the number of kidney preparations indicated in parentheses.
These observations have been quantified in Figure 3, in which the vasoconstrictor responses to increasing concentrations of ANGII (10–100 pM) are presented as the maximum pressure increase during the 6 min period of ANGII infusion. While ANGII induced significant vasoconstriction in LVP kidneys (63.1±5.7 mmHg at 100 pM), ANGII had no effect in HVP kidneys (5.4±1.3 mmHg at 100 pM) at these low concentrations employed in the present study. Vasoconstrictions in response to ANGII were modestly augmented in L-NAME-perfused as compared with control LVP kidneys. In contrast, ANGII-induced vasoconstrictions in L-NAME-perfused HVP kidneys were markedly elevated as compared with those in L-NAME-perfused LVP kidneys. Thus, during NO inhibition, ANGII-induced vasoconstrictions in HVP kidneys were 2.7, 2.9, and 1.4 fold higher than those in LVP kidneys at 10, 20, and 100 pM, respectively. Since enhancement of ANGII-induced vasoconstriction was maximal at 10 and 20 pM, HVP as compared with LVP mainly resulted in potentiation of ANGII-induced vasoconstriction during NO inhibition.
Figure 3.
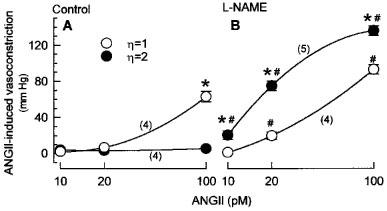
Effects of viscosity on vasoconstrictions induced by 10, 20, and 100 pM ANGII in isolated kidneys. Kidneys were perfused with low or high viscosity perfusate in the absence (A) or presence of 100 μM L-NAME (B). Values are means±s.e.mean for the number of kidney preparations indicated in parentheses. *P<0.05 high vs low viscosity perfusate. #P<0.05 L-NAME vs control perfusate.
We recently demonstrated (Muller et al., 1997) that NO synthesis inhibition potentiates (pD2 increase) and augments (Emax increase) the AT1-mediated vasoconstriction in isolated rat kidneys, which were perfused with a medium of high viscosity comparable to that of the HVP used in the present study. Since AT2 antagonists inhibited the L-NAME-induced pD2 increase without affecting the L-NAME-induced Emax increase in these earlier studies, we examined in the present study, whether the viscosity-dependent potentiation of the ANGII-induced vasoconstriction during NO inhibition were sensitive to AT2 antagonist. In the absence of endogenous NO, ANGII-induced renal vasoconstrictions were not affected by the AT2 receptor antagonist PD123319 (500 nM) in LVP kidneys (Figure 4A). In HVP kidneys, by contrast, PD123319 (500 nM) markedly blunted ANGII-induced vasoconstrictions by 73, 83, and 18% at 10, 20, and 100 pM, respectively (Figure 4B). As a result, ANGII-induced vasoconstriction was identical in L-NAME-perfused LVP and HVP kidneys in the presence of AT2 antagonist. As expected, L158809, an AT1 receptor antagonist, abolished ANGII-induced vasoconstrictions in both LVP and HVP kidneys. In order to confirm that the inhibitory action of PD123319 on ANGII-induced vasoconstriction in L-NAME-perfused HVP kidneys occurred via antagonized AT2 receptors, a lower concentration of PD123319 was used. A concentration of 5 nM PD123319 already attenuated vasoconstrictions in L-NAME-perfused HVP kidneys in response to 10, 20, and 100 pM ANGII by 37, 52, and 5%, respectively (Figure 5).
Figure 4.
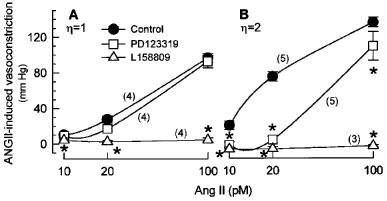
Effects of AT1 and AT2 receptor antagonists on vasoconstrictions induced by 10, 20, and 100 pM ANGII in isolated kidneys perfused with low (A) or high viscosity perfusate (B) containing 100 μM L-NAME alone, or in combination with the AT1 antagonist L158809 (500 nM) or the AT2 antagonist PD123319 (500 nM). Values are means±s.e.mean for the number of kidney preparations indicated in parentheses. *P<0.05 significant inhibitory effect by antagonist.
Figure 5.
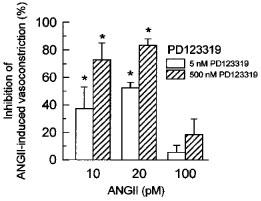
Concentration-dependent inhibition of ANGII-induced vasoconstriction by 5 and 500 nM of the AT2 receptor antagonist PD123319 in isolated kidneys perfused with high viscosity perfusate containing 100 μM L-NAME. Values are means±s.e.mean of 3–5 kidney preparations. *P<0.05 significant inhibitory effect of PD123319.
Discussion
The present study provides clear evidence that shear stress can modulate renal vascular tone. Doubling perfusate viscosity increased overall shear stress by 37% and vascular conductance 3.5 fold. In non-renal preparations, it is a well-established fact that shear stress induces vasodilatation, which has been demonstrated by variation of flow and/or viscosity in vivo and in vitro (de Wit et al., 1997; Koller et al., 1993; Melkumyants et al., 1989; Pohl et al., 1991). However, only a few studies have so far examined the impact of acute changes in shear stress on the renal vasculature (Chen et al., 1989; Juncos et al., 1995; 1996). Chen and coworkers measured regional blood flows, including renal blood flow, with the microsphere technique before and after increasing blood viscosity by addition of high molecular weight dextrans in anaesthetized dogs (Chen et al., 1989). They reported a 2 fold increase in renal vascular conductance, after having raised blood viscosity by a factor of two from a control value of 4 cP (Chen et al., 1989). Juncos and coworkers measured vascular diameters in a pair of isolated afferent arterioles of the rabbit, which were microperfused through the interlobular artery at constant pressure. Flow in one of the two afferent arterioles was halted by occlusion of the efferent arteriole (Juncos et al., 1995; 1996). Surprisingly, Juncos and coworkers did not observe any difference in diameter between free-flow and non-flow afferent arterioles under control conditions (Juncos et al., 1995; 1996). However, flow modulated the myogenic response to perfusion pressures ⩾90 mmHg and the ANGII-induced constriction in afferent arterioles (Juncos et al., 1995; 1996). Finally, the study of Wilcox and coworkers provides further support that shear stress might alter renovascular tone (Wilcox et al., 1993). After chronic application of erythropoietin over 2–5 weeks, which raised hematocrit to 70%, they measured an increase in renal blood flow in rats. Thus, renal vasodilatation even exceeded the level of dilatation necessary to compensate for elevated blood viscosity in this study.
Shear stress-induced vasodilatation has been shown to be endothelium-dependent in non-renal vascular preparations (Koller et al., 1993; Kuo et al., 1990; Melkumyants et al., 1989). NO and/or prostaglandins have been identified as mediators of shear stress-induced dilatation in non-renal vasculature (de Wit et al., 1997; Koller & Kaley, 1990; Koller et al., 1993; 1994; Pohl et al., 1991). In the kidney, virtually nothing is known about mediators of shear stress-induced vasodilatation. Since L-NAME abolished the difference in renal blood flow between control and polycythemic rats, the study of Wilcox et al. points to NO as a possible mediator of shear stress-induced dilatation in the kidney (Wilcox et al., 1993). Though L-NAME decreased afferent arteriolar diameter under control conditions in the studies of Juncos and coworkers, there was no difference between free-flow and non-flow vessels (Juncos et al., 1995; 1996). However, L-NAME as well as endothelium damage abolished the flow modulation on myogenic and ANGII-induced afferent arteriolar constriction (Juncos et al., 1995; 1996). Our results clearly demonstrate that shear stress induces renal vasodilatation via NO. Furthermore, our findings indicate that non-NO/non-prostanoid compound(s) are involved in shear stress-induced renal vasodilatation, since the experiments were carried out in the presence of indomethacin, and L-NAME blocked only a part of the vasodilatation in kidneys that were exposed to augmented shear stress. At present, we can only speculate about the nature of the non-NO/non-prostanoid compound(s). It has been shown that, after NO synthase and cyclooxygenase inhibition, acetylcholine still induces dilatation in the renal vasculature, which is sensitive to K+ channel blockers and inhibitors of cytochrome P450 (Mieyal et al., 1998; Vargas et al., 1994). Thus, an endothelium-derived hyperpolarizing factor (EDHF) is an attractive candidate to be involved in renal shear stress-induced vasodilatation.
ANGII-induced vasoconstriction is importantly modulated by NO, as it has been demonstrated by means of NO donors and blockade of endogenous NO (Adachi et al., 1996; Madrid et al., 1997; Parekh et al., 1996). Our present results extend these findings, showing that vasoconstriction in response to ANGII can be strongly attenuated by shear stress-induced NO production. In agreement with our observation, flow attenuated ANGII-induced constriction of afferent arterioles via the endothelial NO synthase and cyclo-oxygenase pathway in isolated afferent arterioles (Juncos et al., 1996). Though shear stress-induced vasodilatation also involved non-NO/non-PG compound(s) in our experiments, it should be noted that the strong attenuation of vasoconstriction to subnanomolar ANGII concentrations under conditions of elevated shear stress were primarily mediated by NO.
In earlier studies we found that NO inhibition enhances renal ANGII vasoconstriction by two distinct mechanisms (Muller et al., 1997). On the one hand, NO inhibition increased Emax, on the other hand it potentiated renal ANGII vasoconstriction by one order of magnitude (pD2 value increased from 9.4 to 10.4). In contrast to the Emax increase, the potentiation was sensitive to AT2 receptor antagonists (Muller et al., 1997), and it further depended on intact endothelium and eicosanoids, most likely P450 metabolites (Muller et al., 1998). In agreement with these earlier studies, that were performed with a gelatine-containing perfusate of similar viscosity as the HVP employed in the present study, vasoconstriction in response to ANGII was potentiated by NO inhibition in HVP kidneys in the present study. In further agreement with earlier results, the potentiation was abolished in a concentration-dependent manner by the AT2 receptor antagonist PD123319. Besides the concentration-dependent effect of PD123319, the differential antagonistic activity of PD123319 in LVP and HVP kidneys and on renal vasoconstriction induced by 20 and 100 pM ANGII provides further evidence that PD123319 acted specifically on AT2 receptors. Intriguingly, in LVP kidneys, ANGII-induced vasoconstriction was not potentiated during NO inhibition and was insensitive to AT2 receptor antagonist. Therefore, one is led to the conclusion that a certain level of shear stress is needed for the potentiating mechanism to be operative.
Shear stress importantly affected basal renovascular tone, NO release, and ANGII vasoconstriction in the present study. Therefore, it is worthwhile to discuss whether LVP or HVP resembles more the physiological situation encountered in vivo, and whether shear stress is likely to be involved in the physiological regulation of renal hemodynamics. Studies on glass capillaries have shown that the relative apparent viscosity of blood varies between 1 and 3 in 10–400 μm tubes due to the Fahraeus-Lindqvist effect (Pries et al., 1992). However, by comparison of in vivo data and network simulations, Pries and coworkers concluded that relative apparent viscosity probably lies in the range of 2–5 in 10–400 μm vessels in vivo (Pries et al., 1994). Therefore, HVP appears to match the real physiological situation better than LVP.
Regarding the function of shear stress as a regulator of renal vascular tone, the interpretation of the data in the literature is intricate. On the one hand, the present study and other studies demonstrate that augmentation of shear stress leads to a decrease of renal vascular resistance (Chen et al., 1989; Wilcox et al., 1993). On the other hand, if NO release were controlled by changes in shear stress, one would expect that myogenic autoregulation of renal blood flow during increasing renal perfusion pressure, which is associated with an increase in shear stress (constant flow and vasoconstriction), should be counteracted by a concomitantly increasing level of vasodilating NO (Navar et al., 1996). Furthermore, NO donors have been found to directly interfere with the pressure-dependent constriction of renal microvessels (Bouriquet & Casellas, 1995). However, improvement of renal blood flow autoregulation during NO inhibition has not been observed (Beierwaltes et al., 1992; Just, 1997; Majid et al., 1993), questioning the role of shear stress-induced renal vasodilatation at least during renal blood flow autoregulation. Recently, it has been shown that vimentin knock-out mice, which possess an impaired shear stress-induced vasodilatation (Henrion et al., 1997), die of renal failure after reduction of renal mass, pointing to an important role of shear stress for renal vascular adaptation (Terzi et al., 1997). Mechanical stability of cells, which is importantly diminished in mice lacking the intermediate filament vimentin (Eckes et al., 1998), appears to be a crucial element in endothelial mechanotransduction. This has also been demonstrated in acute experiments on isolated arterial rings, where interference with actin as well as tubulin polymerization attenuated shear stress-induced vasodilatation (Hutcheson & Griffith, 1996). Obviously, further studies are required to fully understand the role of shear stress in the physiological regulation of renal haemodynamics.
Our results demonstrate that shear stress increases endogenous levels of NO, which strongly attenuate ANGII-induced renal vasoconstriction. In addition, a certain level of shear stress is required for AT2-sensitive potentiation of ANGII-induced renal vasoconstriction, which however is completely suppressed by the enhanced NO production. As a result, modulation of ANGII-induced renal vasoconstriction by endogenous NO is low under conditions of low shear stress and high under conditions of high shear stress. Shear stress has therefore to be considered a critical determinant of the NO/ANGII interaction.
Acknowledgments
We thank Mrs Jeannine Krill and Suzanne Wendling for skilled technical assistance. K. Endlich and C. Muller contributed equally to this work, which is part of the Ph.D. thesis of C. Muller. This work was supported by the French National Institute of Health (INSERM) grant CJF 9409, and the French Ministry of Higher Education (EA MENRT 2307).
Abbreviations
- ANGII
angiotensin II
- AT1
angiotensin II type 1 receptor
- AT2
angiotensin II type 2 receptor
- cP
centiPoise
- EDHF
endothelium-derived hyperpolarizing factor
- HVP
high viscosity perfusate
- L-NAME
NG-nitro-L-arginine methyl ester
- LVP
low viscosity perfusate
- NO
nitric oxide
References
- ADACHI Y., HASHIMOTO K., HISA H., YOSHIDA M., SUZUKIKU-SABA M., SATOH S. Angiotensin II-induced renal responses in anesthetized rabbits. Effects of N(omega)-nitro-L-arginine methyl ester and losartan. Eur. J. Pharmacol. 1996;308:165–171. doi: 10.1016/0014-2999(96)00298-1. [DOI] [PubMed] [Google Scholar]
- ARIMA S., ENDO Y., YAOITA H., OMATA K., OGAWA S., TSUNODA K., ABE M., TAKEUCHI K., ABE K., ITO S. Possible role of P-450 metabolite of arachidonic acid in vasodilator mechanism of angiotensin II type 2 receptor in the isolated microperfused rabbit afferent arteriole. J. Clin. Invest. 1997;100:2816–2823. doi: 10.1172/JCI119829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARIMA S., REN Y.L., JUNCOS L.A., CARRETERO O.A., ITO S. Glomerular prostaglandins modulate vascular reactivity of the downstream efferent arterioles. Kidney Int. 1994;45:650–658. doi: 10.1038/ki.1994.87. [DOI] [PubMed] [Google Scholar]
- BEIERWALTES W.H., SIGMON D.H., CARRETERO O.A. Endothelium modulates renal blood flow but not autoregulation. Am. J. Physiol. 1992;262:F943–F949. doi: 10.1152/ajprenal.1992.262.6.F943. [DOI] [PubMed] [Google Scholar]
- BOURIQUET N., CASELLAS D. Interaction between cGMP-dependent dilators and autoregulation in rat preglomerular vasculature. Am. J. Physiol. 1995;268:F338–F346. doi: 10.1152/ajprenal.1995.268.2.F338. [DOI] [PubMed] [Google Scholar]
- CHEN R.Y.Z., CARLIN R.D., SIMCHON S., JAN K.-M., CHIEN S. Effects of dextran-induced hyperviscosity on regional blood flow and hemodynamics in dogs. Am. J. Physiol. 1989;256:H898–H905. doi: 10.1152/ajpheart.1989.256.3.H898. [DOI] [PubMed] [Google Scholar]
- DAVIES P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE WIT C., SCHÄFER C., VON BISMARCK P., BOLZ S.-S., POHL U. Elevation of plasma viscosity induces sustained NO-mediated dilation in the hamster cremaster microcirculation in vivo. Pflügers Arch. 1997;434:354–361. doi: 10.1007/s004240050408. [DOI] [PubMed] [Google Scholar]
- ECKES B., DOGIC D., COLUCCI-GUYON E., WANG N., MANIOTIS A., INGBER D., MERCKLING A., LANGA F., AUMAILLEY M., DELOUVEE A., KOTELIANSKY V., BABINET C., KRIEG T. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 1998;111:1897–1907. doi: 10.1242/jcs.111.13.1897. [DOI] [PubMed] [Google Scholar]
- HENRION D., TERZI F., MATROUGUI K., DURIEZ M., BOULANGER C.M., COLUCCI-GUYON E., BABINET C., BRIAND P., FRIEDLANDER G., POITEVIN P., LEVY B.I. Impaired flow-induced dilation in mesenteric resistance arteries from mice lacking vimentin. J. Clin. Invest. 1997;100:2909–2914. doi: 10.1172/JCI119840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUTCHESON I.R., GRIFFITH T.M. Mechanotransduction through the endothelial cytoskeleton: mediation of flow- but not agonist-induced EDRF release. Br. J. Pharmacol. 1996;118:720–726. doi: 10.1111/j.1476-5381.1996.tb15459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO S., ARIMA S., REN Y.L., JUNCOS L.A., CARRETERO O.A. Endothelium-derived relaxing factor/nitric oxide modulates angiotensin II action in the isolated microperfused rabbit afferent but not efferent arteriole. J. Clin. Invest. 1993;91:2012–2019. doi: 10.1172/JCI116423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO S., JOHNSON C.S., CARRETERO O.A. Modulation of angiotensin II-induced vasoconstriction by endothelium-derived relaxing factor in the isolated microperfused rabbit afferent arteriole. J. Clin. Invest. 1991;87:1656–1663. doi: 10.1172/JCI115181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUNCOS L.A., GARVIN J., CARRETERO O.A., ITO S. Flow modulates myogenic responses in isolated microperfused rabbit afferent arterioles via endothelium-derived nitric oxide. J. Clin. Invest. 1995;95:2741–2748. doi: 10.1172/JCI117977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUNCOS L.A., REN Y., ARIMA S., GARVIN J., CARRETERO O.A., ITO S. Angiotensin II action in isolated microperfused rabbit afferent arterioles is modulated by flow. Kidney Int. 1996;49:374–381. doi: 10.1038/ki.1996.55. [DOI] [PubMed] [Google Scholar]
- JUST A. Nitric oxide and renal autoregulation. Kidney Blood Press. Res. 1997;20:201–204. doi: 10.1159/000174145. [DOI] [PubMed] [Google Scholar]
- KOLLER A., KALEY G. Prostaglandins mediate arteriolar dilation to increased blood flow velocity in skeletal muscle microcirculation. Circ. Res. 1990;67:529–534. doi: 10.1161/01.res.67.2.529. [DOI] [PubMed] [Google Scholar]
- KOLLER A., SUN D., HUANG A., KALEY G. Corelease of nitric oxide and prostaglandins mediates flow-dependent dilation of rat gracilis muscle arterioles. Am. J. Physiol. 1994;267:H326–H332. doi: 10.1152/ajpheart.1994.267.1.H326. [DOI] [PubMed] [Google Scholar]
- KOLLER A., SUN D., KALEY G. Role of shear stress and endothelial prostaglandins in flow- and viscosity-induced dilation of arterioles in vitro. Circ. Res. 1993;72:1276–1284. doi: 10.1161/01.res.72.6.1276. [DOI] [PubMed] [Google Scholar]
- KUO L., DAVIS M.J., CHILIAN W.M. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am. J. Physiol. 1990;259:H1063–H1070. doi: 10.1152/ajpheart.1990.259.4.H1063. [DOI] [PubMed] [Google Scholar]
- MADRID M.I., GARCIA-SALOM M., TORNEL J., DE GASPARO M., FENOY F.J. Interactions between nitric oxide and angiotensin II on renal cortical and papillary blood flow. Hypertension. 1997;30:1175–1182. doi: 10.1161/01.hyp.30.5.1175. [DOI] [PubMed] [Google Scholar]
- MAJID D.S.A., WILLIAMS A., KADOWITZ P.J., NAVAR L.G. Renal responses to intra-arterial administration of nitric oxide donor in dogs. Hypertension. 1993;22:535–541. doi: 10.1161/01.hyp.22.4.535. [DOI] [PubMed] [Google Scholar]
- MELKUMYANTS A.M., BALASHOV S.A., KHAYUTIN V.M. Endothelium dependent control of arterial diameter by blood viscosity. Cardiovasc. Res. 1989;23:741–747. doi: 10.1093/cvr/23.9.741. [DOI] [PubMed] [Google Scholar]
- MIEYAL P., FULTON D., MCGIFF J.C., QUILLEY J. NO-independent vasodilation to acetylcholine in the rat isolated kidney utilizes a charybdotoxin-sensitive, intermediate-conductance Ca++-activated K+ channel. J. Pharmacol. Exp. Ther. 1998;285:659–664. [PubMed] [Google Scholar]
- MULLER C., ENDLICH K., BARTHELMEBS M., HELWIG J.-J. AT2-antagonist sensitive potentiation of angiotensin II-induced vasoconstrictions by blockade of nitric oxide synthesis in rat renal vasculature. Br. J. Pharmacol. 1997;122:1495–1501. doi: 10.1038/sj.bjp.0701505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER C., ENDLICH K., HELWIG J.-J. AT2 antagonist-sensitive potentiation of angiotensin II-induced constriction by NO blockade and its dependence on endothelium and P450 eicosanoids in rat renal vasculature. Br. J. Pharmacol. 1998;124:946–952. doi: 10.1038/sj.bjp.0701906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAVAR L.G., INSCHO E.W., MAJID D.S.A., IMIG J.D., HARRISON-BERNARD L.M., MITCHELL K.D. Paracrine regulation of the renal microcirculation. Physiol. Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- OYEKAN A., BALAZY M., MCGIFF J.C. Renal oxygenases: differential contribution to vasoconstriction induced by ET-1 and ANGII. Am. J. Physiol. 1997;42:R293–R300. doi: 10.1152/ajpregu.1997.273.1.R293. [DOI] [PubMed] [Google Scholar]
- PAREKH N., DOBROWOLSKI L., ZOU A.P., STEINHAUSEN M. Nitric oxide modulates angiotensin II- and norepinephrine-dependent vasoconstriction in rat kidney. Am. J. Physiol. 1996;270:R630–R635. doi: 10.1152/ajpregu.1996.270.3.R630. [DOI] [PubMed] [Google Scholar]
- POHL U., HERLAN K., HUANG A., BASSENGE E. EDRF-mediated shear-induced dilation opposes myogenic vasoconstriction in small rabbit arteries. Am. J. Physiol. 1991;261:H2016–H2023. doi: 10.1152/ajpheart.1991.261.6.H2016. [DOI] [PubMed] [Google Scholar]
- PRIES A.R., NEUHAUS D., GAEHTGENS P. Blood viscosity in tube flow: dependence on diameter and hematocrit. Am. J. Physiol. 1992;263:212–222. doi: 10.1152/ajpheart.1992.263.6.H1770. [DOI] [PubMed] [Google Scholar]
- PRIES A.R., SECOMB T.W., GESSNER T., SPERANDIO M.B., GROSS J.F., GAEHTGENS P. Resistance to blood flow in microvessels in vivo. Circ. Res. 1994;75:904–915. doi: 10.1161/01.res.75.5.904. [DOI] [PubMed] [Google Scholar]
- SIGMON D.H., CARRETERO O.A., BEIERWALTES W.H. Angiotensin dependence of endothelium-mediated renal hemodynamics. Hypertension. 1992;20:643–650. doi: 10.1161/01.hyp.20.5.643. [DOI] [PubMed] [Google Scholar]
- STEINHAUSEN M., ENDLICH K. Controversies on glomerular filtration from Ludwig to the present. Pflügers Arch. 1996;432 Suppl.:R73–R81. [PubMed] [Google Scholar]
- TAKAHASHI M., ISHIDA T., TRAUB O., CORSON M.A., BERK B.C. Mechanotransduction in endothelial cells: temporal signaling events in response to shear stress. J. Vasc. Res. 1997;34:212–219. doi: 10.1159/000159225. [DOI] [PubMed] [Google Scholar]
- TERZI F., HENRION D., COLUCCI-GUYON E., FEDERICI P., BABINET C., LEVY B.I., BRIAND P., FRIEDLANDER G. Reduction of renal mass is lethal in mice lacking vimentin. Role of endothelin-nitric oxide balance. J. Clin. Invest. 1997;100:1520–1528. doi: 10.1172/JCI119675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VARGAS F., SABIO J.M., LUNA J.D. Contribution of endothelium derived relaxing factors to acetylcholine induced vasodilatation in the rat kidney. Cardiovasc. Res. 1994;28:1373–1377. doi: 10.1093/cvr/28.9.1373. [DOI] [PubMed] [Google Scholar]
- WILCOX C.S., DENG X., DOLL A.H., SNELLEN H., WELCH W.J. Nitric oxide mediates renal vasodilation during erythropoietin-induced polycythemia. Kidney Int. 1993;44:430–435. doi: 10.1038/ki.1993.261. [DOI] [PubMed] [Google Scholar]