

Abstract
RP58866 possesses a unique electrophysiological property: highly effective against various types of arrhythmias including ventricular fibrillation in animal models, noticeably those occurring during ischaemia with depolarized membrane due to elevated extracellular K+ concentrations. To understand the potential ionic mechanisms, we performed detailed studies on the effects of RP58866 on the HERG channels expressed in Xenopus oocytes, which are believed to be important compositions of the rapid component of delayed rectifier K+ current in the hearts.
RP58866 significantly inhibited the HERG channels in a concentration-dependent manner, with ∼50% decrease in the current amplitude at a concentration of 1 μM.
RP58866 produced more pronounced inhibition with voltage protocols which favoured inactivation of the HERG channels. It caused substantial negative shift of the inactivation curves but did not alter the activation properties. The inhibition was considerably relieved by elevating [K+]o from 5–20 mM, which weakened the channel inactivation. More importantly, the potency was reduced by ∼100 fold on the mutated HERG channels (S631A) in which the C-type inactivation was substantially weakened.
We conclude that blockade of the HERG channels by RP58866 is mainly associated with the binding of the drugs to the inactivated channels. This unique property of HERG blockade might explain some previously reported but unexplained observations: RP58866 maintains its efficacy in APD prolongation with depolarized membrane potential and in arrhythmias during ischaemia with manifested membrane depolarization.
Keywords: RP58866, HERG, Xenopus oocyte, two-electrode voltage-clamp, channel inactivation
Introduction
While the class III antiarrhythmic agents, K+ channel blockers, are effective against many types of arrhythmias, their usefulness seems rather limited in the setting of ischaemia-induced arrhythmias due largely to the fact that they lose their activity with increased heart excitation rate and with depolarized membrane potential which manifests during myocardial ischaemia. Recently, a new compound (RP58866, 1-[2-(3,4-dihydro-2H-1-benzopyran-4-yl)ethyl]-4-(3,4-dimethoxyphenyl)piperidine)), a member of a large series of benznopyran derivatives, was developed which demonstrated potent antiarrhythmic and antifibrillatory actions in various experimental models of arrhythmia without producing proarrhythmia (Escande et al., 1992; Rees & Curtis, 1996). For example, Escande et al. (1992) reported that RP58866 was effective in in vitro reentrant ventricular tachycardia in anisotropic myocardium, acute ventricular fibrillation in anaesthetized dogs and micropigs, as well as in a chronic model of atrial fibrillation. Subsequent studies further demonstrated that RP58866 significantly reduced the incidence of ischaemia-induced arrhythmias in isolated rat, rabbit, and marmoset hearts (Rees & Curtis, 1993). It was also found to be able to suppress reperfusion-induced arrhythmias in these species as well (Rees & Curtis, 1993). The effectiveness of RP58866 is attributed to its ability to prolong action potential duration (APD) and effective refractory period (ERP), properties of class III antiarrhythmic drugs. However, RP58866 varies from most of the other class III agents in that it maintains its effects in APD lengthening with depolarized membrane potentials and in arrhythmias during ischaemia (Escande et al., 1992; Rees & Curtis, 1996).
Potassium channels are a critical determinant of cardiac repolarization and APD/ERP thereby the likelihood of arrhythmias. RP58866 was initially identified as a specific blocker of the inward rectifier K+ current (IK1) in guinea-pig ventricular myocytes with patch-clamp studies (Escande et al., 1992). It was believed that RP58866 blockade of IK1 underlies the ability of this compound to prolong APD and refractoriness. However, a recent study reported by Jurkiewicz et al. (1996) clearly showed that RP58866 blocked IKr (the rapid component of the delayed rectifier K+ current) with IC50 of around 20 nM, about 250 fold more potent than it blocked IK1 (IC50=8 μM) in guinea-pig ventricular myocytes. These data argue strongly against RP58866 being a specific blocker of IK1 and blockade of IK1 being a mechanism for its efficacy. IKr is known to be a primary target for many antiarrhythmics, particular the class III drugs. A question yet to be answered is why unlike the other drugs which primarily act on IKr, RP58866 remains highly effective in prolonging APD/ERP and against arrhythmias even when the extracellular potassium concentration is elevated (Rees & Curtis, 1996), which may be of key importance for its unique efficacy against ischaemia-induced arrhythmias.
To understand the potential mechanisms for the efficacy of RP58866 against a variety of arrhythmias, it is of paramount importance to elucidate the drug-channel interactions between RP58866 and the channels. The gene encoding IKr has been cloned from the human heart. The HERG channels, when heterologously expressed in Xenopus oocytes generate a current that is essentially identical to IKr in cardiac myocytes (Sanguinetti et al., 1995). HERG clone provides an opportunity for studying drug effects on the IKr-equivalent channels in model systems. We therefore performed detailed studies to explore the interactions between RP58866 and the HERG channels expressed in Xenopus oocytes.
Methods
In vitro transcription and functional expression in Xenopus laevis oocytes
Procedures for in vitro transcription and oocyte injection have been previously described in detail (De Biasi et al., 1997; Wang, 1998; Wang et al., 1996a; 1998b). HERG (a kind gift from Dr Mark Keating) and S631A mutant of HERG (a kind gift from Dr Terrence E. Hebert) were subcloned into pSP64 vector. cRNAs were prepared with the mMESSAGE mMACHINE kit (Ambion, Austin, TX, U.S.A.) using SP6 RNA polymerase after linearization of the plasmid with EcoRI, according to manufacturer's protocols. cRNAs were dissolved in DEPC (diethyl pyrocarbonate)-treated sterile water, stored at −80°C, and diluted immediately prior to injection. Stage V–VI Xenopus oocytes were injected with 46 nl of cRNA.
Two-electrode voltage-clamp recording
Whole-cell macroscopic currents were recorded with conventional two-electrode techniques (Wang et al., 1996b). Electrodes filled with 3 M KCl and 10 mM HEPES had resistance of approximately 1.0–1.5 MΩ when measured in the bath solution containing (mM): 100 NaCl, 5 KCl, 0.3 CaCl2, 2 MgCl2, and 10 HEPES (pH 7.4). The electrodes were connected to a GeneClamp-500 amplifier (Axon Instrument, Burlingame, CA, U.S.A.). The pClamp suite of programs was employed for data acquisition and analysis. Records were digitized at 5 kHz and filtered at 2 kHz. Experiments were conducted at room temperature (22–23°C). Currents were measured before and 10 min after drug application to the bath.
Data analysis
Group data are expressed as mean±s.e.mean. Statistical comparisons among groups were performed by analysis of variance (ANOVA). If significant effects were indicated by ANOVA, a t-test with Bonferroni correction or a Dunnett's test was used to evaluate the significance of differences between individual means. Otherwise, baseline and drug data were compared by Student's t-test. A two-tailed P<0.05 was taken to indicate a statistically significant difference. A nonlinear least-square curve-fitting program (CLAMPFIT in pCLAMP 6.0 or Graphpad Prism) was used to perform curve fitting procedures.
Results
Concentration-dependence of HERG blockade by RP58866
Expression of the HERG channels resulted in the induction of a K+ conductance with characteristic activation and rectification properties. With standard depolarizing voltage steps (from −60 to +50 mV), time-dependent activation of outward currents was elicited, followed by large tail currents upon repolarization to −50 mV. Because of the rapid C-type inactivation of the HERG channels compared to their activation, outward currents at potentials positive to 0 mV became smaller (Smith et al., 1996; Spector et al., 1996; Wang et al., 1998a). Application of RP58866 to the bath produced concentration-dependent suppression of the HERG-expressed currents. The inhibitory potency of RP58866 was evaluated with standard depolarizing pulse protocols, as illustrated in the inset of Figure 1A. The dose-response curves were constructed by plotting the per cent block of the step currents at a TP (test potential) of 0 mV. The IC50 value calculated from the Hill equation was 0.8±0.2 μM with a Hill coefficient of 1.3. It should be noted that different voltage protocols yielded different IC50 values, as described below.
Figure 1.
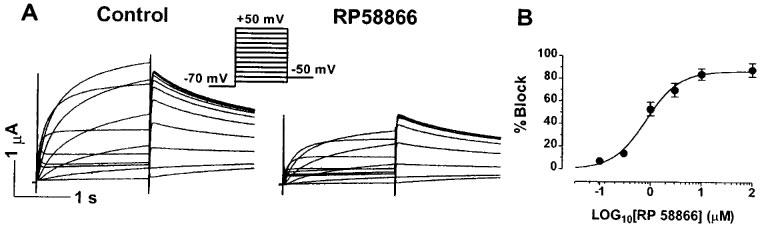
Concentration-dependence of the HERG channel blockade by RP58866 in Xenopus oocytes. Currents were elicited by 2 s pulses to test potentials ranging from −60 to +50 mV with 10-mV increments from an HP of −70 mV at an interpulse interval of 10 s. (A) Typical examples of the HERG-expressed currents recorded before and after RP58866 (1 μM). (B) Dose-response curves of the HERG channel blockade by RP58866 (n=7), determined from the step currents at 0 mV. Shown are averaged data and fits to the Hill equation: B(%)=100/[1+(IC50/D)n], where B(%) is the per cent changes of HERG-expressed current at a drug concentration D, IC50 is the concentration of RP58866 that produces 50% effects, and n is the Hill coefficient.
Voltage-dependence of HERG blockade by RP58866
Voltage-dependence of HERG blockade by RP58866 was assessed with three different voltage protocols: the standard I–V protocols, the fully activated I–V protocols, and the instantaneous I–V protocols. With the standard I–V protocols, the activating currents were elicited by 2 s depolarizing pulses ranging from −60 to +50 mV and the tail currents by 1 s repolarizing pulses to −50 mV. The voltage steps were delivered from an HP (holding potential) of −70 mV at an interpulse interval of 10 s. The standard I–V relationship demonstrated a strong inward rectification at potentials positive to −10 mV. RP58866 at a concentration of 1 μM substantially diminished the currents at varying test potentials: the blockade was slightly enhanced with stronger depolarization between −40 and 0 mV but declined thereafter at more positive potentials up to +50 mV (see Figure 2A). To determine the fully activated I–V relationships, a 1 s prepulse to +20 mV was applied before each of the repolarizing pulses to test potentials between −140 mV and +20 mV. Note that the prepulse potential at +20 mV was positive enough to induce a full conductance of the HERG channels but also rendered a large amount of the channels into the inactivated state. Thus, the fully activated I–V also demonstrated striking inward rectification with a negative slope across a voltage range between −20 mV and +50 mV. RP58866 (1 μM) produced most pronounced inhibition of the outward HERG currents elicited at potentials between −60 to 0 mV. The effects were accentuated with increasing test potentials up to −20 mV from −140 mV and relieved drastically at more positive potentials within the range where the negative slope occurred. Combined with data from the standard I–V data, it appeared that in both cases the inhibitory effects of RP58866 were more pronounced within the physiological range of membrane potentials for cardiac cells but declined as the channel rectification (inactivation) became manifested at more positive potentials. This observation was unlikely due to a relative larger contribution of leak currents at more positive potentials because the same results held true whether the leak was subtracted or not. We then turned to investigate whether removal of the HERG rectification could eliminate the negative correlation between the blockade and the voltages at potentials positive to 0 mV. The channels were first inactivated by clamping the membrane at +20 mV for 1 s followed by a prepulse to −100 mV for 20 ms. This prepulse was sufficiently long for rapid recovery of channels from inactivation but short enough to prevent significant channel deactivation. Following the recovery prepulse, a family of test pulses were delivered to potentials ranging from −140 to +20 mV. The currents recorded were fitted by the single exponential function with extrapolation to the initial point of the test pulse and the amplitude was plotted against test potentials to construct the instantaneous I–V curves. The I–V relationships from such protocols were linear because no inactivation occurred during the prepulse. This protocol revealed a voltage-dependence of RP58866 blockade of the HERG channels different from the other two protocols: the inhibitory effects enhanced linearly with stronger depolarization over the whole voltage range (between −140 and +20 mV). However, when compared with the standard I–V and the fully activated I–V protocols, the per cent reduction at a given TP within the plateau range (−30 to +10 mV) was smaller with the instantaneous I–V protocols. For example, the reduction of the HERG currents (at a TP of 0 mV) by RP58866 (1 μM) was 53.1±6.0% with the standard I–V protocols, 58.4±2.7% with the fully activated I–V protocols, and 40.3±4.8% with the instantaneous I–V protocols.
Figure 2.
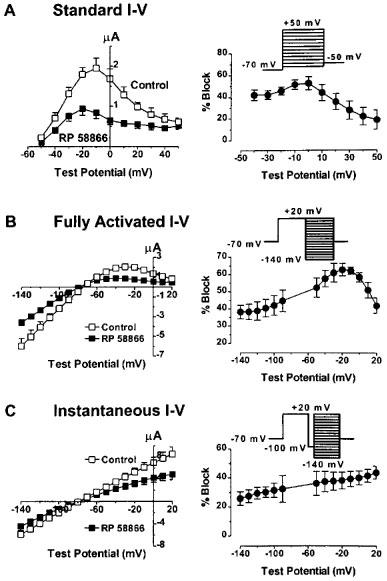
Voltage-dependence of HERG channel blockade by RP58866 (1 μM) in Xenopus oocytes. (A, B and C) I–V relationships of the HERG channels before and after RP58866, as determined by the standard I–V protocols (shown in the inset of (A)), the fully activated I–V protocols (the inset in (B)) and the instantaneous I–V protocols (C). The right panels show the per cent block of the HERG channels vs test potentials for the three voltage protocols. Data are averaged from seven oocytes. All three protocols were done in the same oocytes.
Diminished potency of block at potentials where apparent inward rectification is seen would suggest that the drugs bind to the open channels and unbind from the inactivated channels. However, this does not explain why the drug effects were smaller with the instantaneous I–V protocols relative to the other two protocols. The relief of block at the potentials with rapid channel inactivation could also mean that the drugs bind preferentially to the inactivated channels with an association rate faster than the intrinsic inactivation gating at less positive potentials but slower than the intrinsic inactivation of the HERG channels at positive potentials with apparent rectification. To clarify this issue, we performed detailed analysis on the voltage-dependent activation and inactivation gating parameters. The activation curves were constructed by normalizing the tail currents recorded with the standard I–V protocols. The normalized data (or conductance) were plotted against the prepulse potentials and fitted to the Boltzmann distribution. As shown in Figure 3A, the activation curves with and without RP58866 are superimposed, indicating that RP58866 (1 μM) did not alter the activation properties: the V1/2 values (the half-maximal activation voltage) and the slope factor (k) were −36.6±4.3 mV and 8.3±1.5 mV before, and −36.9±5.1 mV (P>0.05, n=5) and 7.4±1.4 mV (P>0.05, n=5) after RP58866 (1 μM). The per cent block did not depend on the voltages where the conductance of the channels increased steeply (Figure 3C, open triangles). Note that at potentials positive to −10 mV, the activation curve in the presence of RP58866 began to decline with increasing depolarization (Figure 3A). This resulted in strong voltage-dependent inhibition of the HERG channels after the channels had reached full activation (Figure 3C, open triangles). Thus, the per cent block of the tail currents continually increased with increasing depolarization (Figure 3C). The lack of effects of RP58866 on the activation properties was unlikely a result of an accumulated block with pulses as a consequence of incomplete recovery from block upon repolarization because the same effects were observed with longer pulsing intervals (20 s) or with detrimental (−10 mV) pulses from +50 mV to −40 mV.
Figure 3.
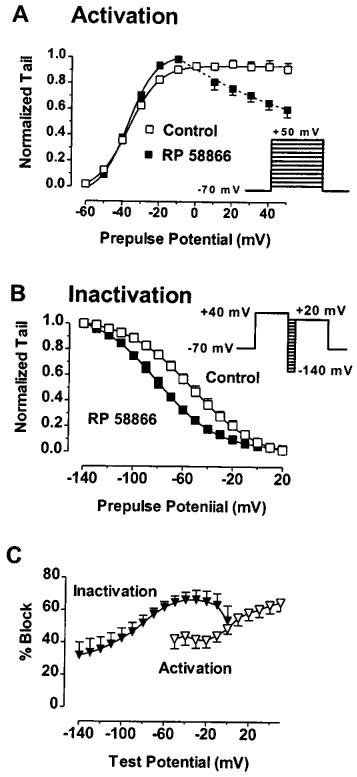
Effects of RP58866 on the voltage-dependent activation and inactivation properties of the HERG channels expressed in Xenopus oocytes. (A) Lack of effects of RP58866 (1 μM) on the activation conductance curves. Currents were elicited with the standard I–V protocols and the amplitude of the HERG tail currents at a repolarized test potential of −60 mV was normalized to the maximum values and plotted against the prepulse potentials. Symbols are experimental data and the curves represent the best fits to the Boltzmann distribution: I/Imax=1/{1+exp[V1/2−V)/k]}, where I is the HERG tail current amplitude at a prepulse potential V, V1/2 is the voltage for half-maximal activation, and k is a slope factor. (B) Negative shift of the inactivation curve produced by RP58866 (1 μM). To construct the inactivation curves, the voltage protocols shown in the inset were employed: an 2 s depolarizing pulse to +40 mV to inactivate the HERG channels, followed by varying repolarizing pulses to potentials between −140 to +20 mV for 20 ms followed by a test pulse to +20 mV. The amplitude of the HERG currents at the test potential was normalized and plotted against the prepulse potentials. Symbols are experimental data and the curves represent the best fits to the Boltzmann distribution. Here, V1/2 is the voltage for half-maximal inactivation. (C) Percentage of the HERG channel block determined from the tail currents (open symbols) elicited by the standard I–V protocol shown in the inset of (A) and from the currents (filled symbols) elicited by the inactivation protocols shown in the inset of (B).
On the contrary, the inactivation characteristics were markedly affected by RP58866 (Figure 3B). To construct the inactivation curves, the voltage protocols shown in the inset of Figure 3B were employed: a 2 s depolarizing pulse to +40 mV to inactivate the HERG channels followed by varying repolarizing pulses to potentials between −140 and +20 mV for a short period to allow full recovery of channels from inactivation at more negative potentials and rapid inactivation at less negative potentials. RP58866 (1 μM) caused an apparent shift of the inactivation curve to the negative direction. The V1/2 value (the half-maximal inactivation voltage) was changed from −53.3±4.6 mV under control conditions to −80.6±6.7 mV with the drug (P<0.01, n=6). The slope factor was not significantly altered (−26.7±3.4 mV for control vs −23.9±2.9 mV for RP58866, P>0.05, n=6). The blockade of the currents elicited at the TP of +20 mV was most prominent with prepulse potentials between −60 and 0 mV (see Figure 3C, filled triangles), which corresponded well with the voltage range where the HERG channel conductance rose from zero to the maximum.
The data shown in Figure 3 suggest that RP58866 likely acts on the inactivated channels but not on the activated state. Interestingly, RP58866 (1 μM) caused about 40% inhibition of the currents with a prepulse to −100 mV (Figure 3C), in good agreement with the per cent block with the instantaneous I–V protocols (Figure 2C). This implies that the inhibition seen with the instantaneous I–V protocols was not due to open channel block but the block occurring at the prepulse to −100 mV or/and the residual block from the conditioning pulse to +20 mV which was not yet relieved at the subsequent hyperpolarizing prepulse (−100 mV) because of the short period (20 ms).
Time-dependence of HERG blockade by RP58866
State-dependent channel blockers often demonstrate characteristic effects on channel kinetics. The HERG channels once activated undergo complex processes from closed states to open/inactivation, inactivation, deactivation, and back to closed states again. We therefore analysed the effects of RP58866 on the kinetics of the HERG channels.
Activation
Open channel blockers often cause apparent acceleration of the activation time course. The activation time course was described by the double exponential function on the currents evoked at a TP of −20 mV from a HP of −70 mV. RP58866 did not produce any significant changes in the activation time constants (Figure 4A). The results suggest either that the drug does not affect the activation process or that it binds to the channels slowly.
Figure 4.

Effects of RP58866 (1 μM) on the kinetics of the HERG channels expressed in Xenopus oocytes. (A) Activation time constants were obtained by fitting the current traces elicited by a step to 0 mV from an HP of −70 mV to the double exponential function (fast and slow time constants). (B) Deactivation time constants were calculated by best fits to the triple exponential function of the currents elicited by a test pulse to −120 mV preceded by a 2 s prepulse to +20 mV. The fitted values for the rising phase (or reduced inward current) of the currents were taken as the time constants (fast and slow components) for deactivation. (C) For reactivation time course, the currents were elicited with voltage protocols the same as for deactivation in (B). Reactivation was best described with the single exponential fit. The time constants derived from the fits for the first phase (descending phase or increasing inward current) are used to indicate the reactivation kinetics. (D) Acceleration of inactivation time courses by RP58866. Currents were elicited with the instantaneous I–V protocols and the traces recorded at a TP of 0 mV were used to perform single exponential fits. Typical examples of such fit are shown (current amplitude was normalized for better comparison). (E) Mean data (n=7) on the inactivation kinetics of the HERG channels. **P<0.01, compared with control.
Deactivation
Open channel blockers often cause apparent deceleration of the deactivation time course because of re-opening of the channels upon drug unbinding. The deactivation time constants were obtained by fitting, to the triple exponential function, the tail currents elicited by a hyperpolarizing pulse to −120 mV following a 2 s prepulse at +20 mV. The descending phase represents the recovery time course from inactivation and the following rising phase represents the deactivation time course. The data are shown in Figure 4B and no appreciable effects were seen with RP58866 at all. The results indicated that the drugs did not affect the deactivation time course either because the drug unbinding occurred rapidly before the apparent deactivation started or because the drug dissociation was slower than the deactivation process.
Recovery
The recovery time constants were calculated in the way as described for deactivation. The initial descending phase of the tail current upon hyperpolarization to −140 mV was taken as an indication of reactivation process. The drug failed to cause any significant changes in the recovery time course (Figure 4C).
Inactivation
The inactivation process was analysed by fitting the currents elicited with the voltage protocols described for the voltage-dependent inactivation (shown in Figure 4D and E) to the single exponential function. A short hyperpolarizing pulse to −100 mV was sufficient for channel recovery from inactivation, but too short to allow significant deactivation. The following depolarization-mediated initial outward current reflects the open state of the HERG channels that then again inactivate rapidly. Therefore, the decaying tail currents represent the HERG channel inactivation but not deactivation. RP58866 considerably accelerated the inactivation time course. For example, the inactivation time constants were 14.2±2.1 ms before and 7.0±1.0 ms (P<0.01, n=8) after RP58866 (1 μM). Acceleration of inactivation time course can be interpreted as either an indication of drug binding to the inactivated channels or drug blockade of the open channels.
Effects of RP58866 on the HERG channels under high [K+]o condition
It is known that the unusually rapid inactivation of the HERG channels is due to the rapid C-type inactivation which is highly sensitive to external K+ concentrations (Wang et al., 1996b). Elevated [K+]o markedly slowed the kinetics of and decreased the magnitude of the HERG channel inactivation. If RP58866 blocks the HERG channels in the inactivated state, then weakening of HERG inactivation should relieve the block. This was indeed confirmed by the following experiments. The HERG currents were recorded with 20 mM [K+]o and the drug effects were evaluated with the three different I–V voltage protocols (Figure 5) as already described above. The degree of the inward rectification of the I–V relationship was weakened relative to that obtained with 5 mM [K+]o. The extent of the channel block by RP58866 (1 μM) also became smaller with higher [K+]o. Comparison of the per cent inhibition of the HERG channels with different [K+]o is presented in Figure 6. Inhibitory effects of RP58866 on the HERG channels were largely relieved after exposure to 20 mM [K+]o with the standard I–V protocols and virtually abolished with the other two I–V protocols (Figure 6A). To attain the same degree of block seen with 1 μM RP58866 under normal [K+]o (5 mM), the drug concentration must be raised to 100 μM or higher.
Figure 5.
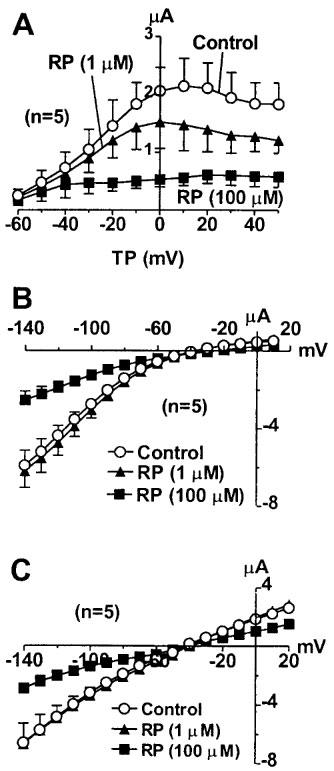
Effects of RP58866 on the HERG channels expressed in Xenopus oocytes under a high external K+ concentration (20 mM). (A) I–V relationships of the HERG channels with the standard I–V protocols shown in the inset before and after RP58866. (B) I–V relationships of the HERG channels with the fully activated I–V protocols shown in the inset before or after RP58866. (C) I–V relationships of the HERG channels obtained with the instantaneous I–V protocols shown in the inset before and after RP58866.
Figure 6.

(A) Comparison of the HERG channel blockade by RP58866 (1 μM) determined under normal [K+]o (5 mM) and under high [K+]o conditions (20 mM). Values were obtained from the measurements made at a test potential of 0 mV with the standard I–V protocols. *P<0.05 and **P<0.01 comparisons of groups between 5 mM [K+]o and 20 mM [K+]o. (B) Comparison of drug effects at different holding potentials with different extracellular K+ concentrations (n=7). Experiments with 5 and 20 mM [K+]o were performed in the same oocytes.
Elevation of [K+]o from 5–20 mM is expected to cause ∼40 mV depolarization of the cell membrane according to the Nernst equation. Supposed that the membrane potential is −80 mV with 5 mM [K+]o, then it will be shifted to −40 mV with elevating [K+]o to 20 mM. The effects of RP59966 (1 μM) at a holding potential of −80 mV with 5 mM [K+]o and at a holding potential of −40 mV with 20 mM [K+]o were evaluated. The results are illustrated in Figure 6B. Clearly, the inhibitory effects of RP58866 were greater at a holding potential of −40 mV with 20 mM [K+]o than at −80 mV with 5 mM [K+]o, although the differences did not reach statistical significance. This implies that the voltage-dependent blockade outweighed the [K+]o-dependent relief of block. It is possible that blockade at a HP of −40 mV was a consequence of mixed effects of RP58866 on the inactivated and open channels because the HERG channels were also activated by holding at −40 mV. It is extremely hard, if not impossible, to separate directly the inactivation block from open channel block at a HP of −40 mV, because of a significant ‘window current' due to overlapping of channel activation and inactivation at this potential. One way of surmounting this problem is to compare the blockade at −40 mV with 20 mM [K+]o with that at −40 mV with 5 mM [K+]o. Under the two different conditions, the activation of the HERG channels at −40 mV is the same (because [K+]o does not alter the voltage-dependence of activation) but the inactivation is different, being stronger with 5 mM [K+]o relative to 20 mM [K+]o. Thus, any differences in the block under the two different conditions are more likely due to inactivation-dependent alterations. In our case, the per cent block (at −40 mV) was 67.3±7.5% with 5 mM [K+]o and 54.0±6.0% (P<0.05, n=6) with 20 mM [K+]o (Figure 6B), indicating that when activation was kept the same, the stronger inactivation favoured HERG blockade by RP58866.
Effects of RP58866 on the mutated HERG channels (S631A)
To further investigate the possibility of inactivated state block of the HERG channels by RP58866, we evaluated the effects of this compound on S631A mutant of the HERG channels, in which the rapid C-type inactivation is substantially weakened due to a positive shift of the voltage-dependence of inactivation by >100 mV (Ficker et al., 1998; Herzberg et al., 1998; SchÖnherr & Heinemann, 1996; Suessbrich et al., 1997; Zou et al., 1998). Expression of S631A in oocytes gave rise to delayed rectifier-like currents with rapid activation, and the inward rectification, seen with the wild-type HERG channels, was absent within the voltage range between −60 and +50 mV (Ficker et al., 1998; SchÖnherr & Heinemann, 1996; Suessbrich et al., 1997). For comparison, three different I–V protocols were again used to assess the drug effects. The results are illustrated in Figure 7. The potency of RP58866 was apparently diminished in the mutant. RP58866 at 1 μM, which suppressed the wild-type HERG channels by ∼50%, did not affect the currents expressed by the mutant HERG channels. Elevation of the drug concentration up to 100 μM produced significant block of the mutated channels to the extents comparable with the blockade of the wild-type HERG channels by 1 μM RP58866. The per cent inhibition by 100 μM RP58866 was in the same range irrespective of voltage protocols (Figure 7C) and the block was not dependent on voltage, thus the I–V relationships were linear before and after the drug (Figure 7B). To see whether RP58866 can cause time-dependent block of S631A mutant due to slow binding to the open channels, prolonged pulse duration was used to assess the drug effects. The per cent block of S631A with 10 s pulses slightly increased (68.4±16.6% reduction at +50 mV, n=4 cells) when compared with the effects with 2 s pulses (62.5±12.7% decrease, n=7, P>0.05). Therefore, there was no apparent time-dependent component of S631A block by RP58866.
Figure 7.
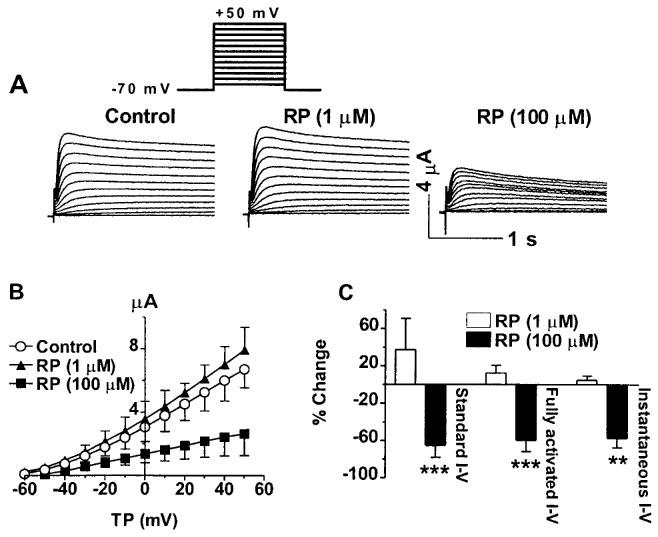
Effects of RP58866 on the mutant HERG channels (S631A) expressed in Xenopus oocytes. (A) Analogue data showing the effects of RP58866 with the standard I–V protocols. (B) I–V relationships of the HERG channels before and after RP58866, with the standard I–V protocols shown in the inset. (C) Per cent block of the mutated HERG channels by RP58866 as assessed by the three different I–V protocols indicated. **P<0.01 and ***P<0.001 vs control values.
Discussion
RP58866 has been demonstrated to be highly effective against a variety of arrhythmias in various animal models. Most of IKr blockers loss their efficacy with depolarized membrane potential or elevated external K+ concentration (Rees & Curtis, 1996). A major distinction of RP58866 from most of other IKr blockers is its ability to prolong ERP and suppress arrhythmias under conditions that can manifest during ischaemia such as membrane depolarization due to elevated [K+]o (Rees & Curtis, 1996). The present study is the first to characterize in detail the interactions between RP58866 and the HERG channels. The major finding of the present study is that the HERG blockade by RP58866 is mainly associated with the binding of the drug to the inactivated channels but not the open channels. This finding provides one explanation for the unique pharmacological properties of RP58866.
Our finding that RP58866 preferentially binds to the inactivated channels is supported by several lines of evidence. First, the voltage protocols which favour channel inactivation enhanced drug block of the HERG channels. For example, RP58866 blocked the HERG channels to a greater extent at more depolarized membrane potentials (prepulse potentials or holding potentials). RP58866 inhibited HERG by ∼20% at a prepulse potential of −140 mV and by 65% at 0 mV. Second, RP58866 produced a negative shift of the inactivation curve and accelerated the inactivation time course of the HERG channels. These data provide suggestive evidence for the inactivation block of the HERG channels by RP58866. Third, the effects of RP58866 were largely diminished by increasing [K+]o or by using the instantaneous I–V protocols, manoeuvres which attenuated the channel inactivation (Smith et al., 1996; Wang et al., 1996b). And finally, more direct data in favour of inactivated channel block were acquired from the experiments on the HERG mutant (S631A) with weakened inactivation. The inhibitory effects of RP58866 on this construct were remarkably weakened. Like in the case of high [K+]o, much higher concentrations (∼100 fold higher) of the drugs were required to achieve the comparable degrees of blockade seen with the wild type HERG channels under normal [K+]o.
Nonetheless, our experiments do not exclude the possibility of open channel and/or closed state-dependent block by the drug. As shown in Figure 2, with the instantaneous I–V protocols devoid of channel inactivation, RP58866 was still able to cause reduction of the currents despite the fact that the degree of blockade was largely decreased. The results could also result from block of the HERG channels in the closed states. On the other hand, these observations could also be explained by inactivated state block of the HERG channels at −100 mV where steady-state channel inactivation has already occurred, as shown in the inactivation curves in Figure 3. Therefore, the inhibitory effects of RP58866 are probably the consequence of blocking the channels in different states, but with higher affinity for association to the inactivated channels. It is also likely that like Ito (the cardiac transient outward K+ current) blockade by flecainide, quinidine and 4-aminoprydine from our previous study (Wang et al., 1995), although RP58866 preferentially binds to the inactivated channels, the blockade (viewed as an occlusion of the channels by the drug molecules) occurs only when the channels open.
One of the arguments in favour of IKr block not being likely to contribute to the RP58866-induced increase in QT is that there is little change of activity when [K+]o is raised. This is in contrast to IKr blockers, which show a marked loss of effects with elevated [K+]o (Rees & Curtis, 1996). However, considering that elevating [K+]o would depolarize the membrane which in turn would render more IKr channels in the inactivated state, it is expected that high [K+]o would enhance the inhibition of IKr by RP58866 or any other drugs which block IKr in the inactivated states, like dofetilide (Andersen et al., 1994; Black et al., 1991; Mortensen et al., 1992). Our study actually provided data in favour of this notion. With 20 mM [K+]o, the blockade of the HERG channels by RP58866 at a given voltage were indeed relieved relative to the effects with 5 mM [K+]o. This property would result in reduced potency of RP58866 at a given potential. However, considering that changing [K+]o from 5 to 20 mM is expected to cause a positive shift of the potassium equilibrium potential by about 40 mV according to the Nernst equation, it is not difficult to expect that the effects of RP58866 would be actually greater at more depolarized potential with higher [K+]o than at less depolarized potentials with lower [K+]o. For example, in our study RP58866 caused 45.1±6.0% decrease (test potential 0 mV) in the HERG amplitude at −80 mV (holding potential) with 5 mM [K+]o and 54.0±6.0% reduction at −40 mV with 20 mM [K+]o. These data explain why the effects of RP58866 on APD/ERP had little change with elevated [K+]o relative to the normal [K+]o and in part why it is effective against ischaemia-induced arrhythmias. It is also speculated that the inactivated state blockade of the HERG channels is a desirable property of drugs for suppression of arrhythmias, particularly those occurring during ischaemia (Andersen et al., 1994; Black et al., 1991; Mortensen et al., 1992). Certainly, this prospect needs to be put into appropriate pathological models for testing.
There are several limitations in our study. First, we are unable to provide information regarding the detailed structural determinants of drug binding and blocking. Extensive site-directed mutagenesis is required to answer this question. Nevertheless, our data do suggest that the binding site of RP58866 is likely within the pore region close to the residues determining the C-type of inactivation of the HERG channels. Second, our data also failed to answer why RP58866 has rather similar mode of actions on the HERG channels as other drugs like dofetilide and E-4031 (Ficker et al., 1998; Herzberg et al., 1998) considering that RP58866 is structurally unrelated to these two compounds. Although these topics are beyond the scope of the present work, it is of great interest and importance to make further investigation in order to have better understanding of mechanisms of the drug actions.
Acknowledgments
This work was supported in part by the Medical Research Council of Canada, the Heart and Stroke Foundation of Quebec, and Establishment Grant for young investigators from the Fonds de Recherche en Sante de Quebec awarded to Dr Wang, and the Fonds de la Recherche de l'Institut de Cardiologie de Montreal. Dr Wang is a research scholar of the Heart and Stroke Foundation of Canada. The authors wish to thank XiaoFan Yang for her excellent technical assistance and Dr Stanley Nattel for discussion on the manuscript. We also thank Drs Mar Keating and Terry Hebert for kindly providing the clones used in this study.
Abbreviations
- APD
action potential duration
- ERP
effective refractory period
- HERG
the human ether-a-go-go related gene
- HP
holding potential
- IC50
concentrations of drug needed to achieve half-maximal inhibitory effects
- RP58866
1-[2-(3,4-dihydro-2H-1-benzopyran-4-yl)ethyl]-4-(3,4-dimethoxyphenyl)piperidine)
- TP
test potential
- V1/2
half-maximal activation (or inactivation) voltage
References
- ANDERSEN H.R., WIGGERS H.S., KNUDSEN L., SIMONSEN I., THOMSEN P.E.B., CHRISTIANSEN N. Dofetilide reduces the incidence of ventricular fibrillation during acute myocardial ischemia. A randomized study in pigs. Cardiovasc. Res. 1994;28:1635–1640. doi: 10.1093/cvr/28.11.1635. [DOI] [PubMed] [Google Scholar]
- BLACK S.C., CHI L., MU D., LUCCHESI B.R. The antifibrillatory action of UK-69, 798, a class III antiarrhythmic agent. J. Pharmacol. Exp. Ther. 1991;258:416–423. [PubMed] [Google Scholar]
- DE BIASI M., WANG Z., ACCILI E., WIBLE B., FEDIDA D. Open channel block of human heart hKv1.5 by the β-subunit hKvβ1.2. Am. J. Physiol. 1997;272:H2932–H2941. doi: 10.1152/ajpheart.1997.272.6.H2932. [DOI] [PubMed] [Google Scholar]
- ESCANDE D., MESTRE M., CAVERO I., BRUGADA J., KIRCHHOF C. RP58866 and its active enantiomer RP62719 (terikalant): blockers of the inward rectifier K+ current acting as pure class III antiarrhythmic agents. J. Cardiovasc. Pharmacol. 1992;20 Suppl. 2:S106–S113. [PubMed] [Google Scholar]
- FICKER E., JAROLIMEK W., KIEHN J., BAUMANN A., BROWN A.M. Molecular determinants of dofetilide block of HERG K+ channels. Circ. Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- HERZBERG I.M., TRUDEAU M.C., ROBERTSON G.A. Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J. Physiol. 1998;511:3–14. doi: 10.1111/j.1469-7793.1998.003bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JURKIEWICZ N.K., WANG J., FERMINI B., SANGUINETTI M.C., SALATA J.J. Mechanism of action potential prolongation by RP58866 and its active enantiomer, terikalant: Block of the rapidly activating delayed rectifier K+ current, IKr. Circulation. 1996;94:2938–2946. doi: 10.1161/01.cir.94.11.2938. [DOI] [PubMed] [Google Scholar]
- MORTENSEN E., YANG T., BJORNSTAD H., REFSUM H. Class III antiarrhythmic action and inotropy: effects of dofetilide in acute ischemic heart failure in dogs. J. Cardiovasc. Pharmacol. 1992;19:216–221. doi: 10.1097/00005344-199202000-00010. [DOI] [PubMed] [Google Scholar]
- REES S., CURTIS M.J. Specific IK1 blockade: a new antiarrhythmic mechanism? Effect of RP58866 on ventricular arrhythmias in rat, rabbit, and primate. Circulation. 1993;87:1979–1989. doi: 10.1161/01.cir.87.6.1979. [DOI] [PubMed] [Google Scholar]
- REES S., CURTIS M.J. Which cardiac potassium channel subtype is the preferable target for suppression of ventricular arrhythmias. Pharmacol. Ther. 1996;69:199–217. doi: 10.1016/0163-7258(95)02063-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SCHÖNHERR R., HEINEMANN S.H. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J. Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH P.L., BAUKROWITZ T., YELLEN G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- SPECTOR P.S., CURRAN M.E., ZOU A., KEATING M.T., SANGUINETTI M.C. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUESSBRICH H., SCHÖNHERR R., HEINEMANN S.H., ATTALI B., LANG F., BUSH A.E. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br. J. Pharmacol. 1997;120:968–974. doi: 10.1038/sj.bjp.0700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Z. Ionic mechanisms and molecular basis of human cardiac repolarization: Implication for drug development. Curr. Topics Pharmacol. Res. Trends. 1998;4:19–35. [Google Scholar]
- WANG Z., FERMINI B., NATTEL S. Effects of flecainide, quinidine, and 4-aminopyridine on transient and sustained outward current in human atrial myocytes. J. Pharmacol. Exp. Ther. 1995;272:184–196. [PubMed] [Google Scholar]
- WANG Z., KIEHN J., YANG Q., BROWN A., WIBLE B. Comparison of K+ channel binding and block produced by alternatively spliced Kvβ1.2 subunits. J. Biol. Chem. 1996a;271:28311–28317. doi: 10.1074/jbc.271.45.28311. [DOI] [PubMed] [Google Scholar]
- WANG S., LIU S., MORALES M.J., STRAUSS H.C., RASMUSSON R.L. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J. Physiol. 1998a;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S., MORALES M.J., LIU S., STRAUSS H.C., RASMUSSON R.L. Time-, voltage and ionic-concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Lett. 1996b;389:167–173. doi: 10.1016/0014-5793(96)00570-4. [DOI] [PubMed] [Google Scholar]
- WANG Z., YUE L., WHITE M., PELLETIER G., NATTEL S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998b;98:2422–2428. doi: 10.1161/01.cir.98.22.2422. [DOI] [PubMed] [Google Scholar]
- ZOU A., XU Q.P., SANGUINETTI M.C. A mutation in the pore region of HERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of in activation. J. Physiol. 1998;509:129–137. doi: 10.1111/j.1469-7793.1998.129bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]