

Abstract
Background
Cervical cancer (CC), a leading cause of cancer-related deaths in women worldwide, has been causally linked to genital human papillomavirus (HPV) infection. Although a host of genetic alterations have been identified, molecular basis of CC development is still poorly understood.
Results
We examined the role of promoter hypermethylation, an epigenetic alteration that is associated with the silencing tumor suppressor genes in human cancer, by studying 16 gene promoters in 90 CC cases. We found a high frequency of promoter methylation in CDH1, DAPK, RARB, and HIC1 genes. Correlation of promoter methylation with clinical characteristics and other genetic changes revealed the following: a) overall promoter methylation was higher in more advanced stage of the disease, b) promoter methylation of RARB and BRCA1 predicted worse prognosis, and c) the HIC1 promoter methylation was frequently seen in association with microsatellite instability. Promoter methylation was associated with gene silencing in CC cell lines. Treatment with methylation or histone deacetylation-inhibiting agents resulted in profound reactivation of gene expression.
Conclusions
These results may have implications in understanding the underlying epigenetic mechanisms in CC development, provide prognostic indicators, and identify important gene targets for treatment.
Keywords: cervical carcinoma, promoter hypermethylation, CDH1, DAPK, RARB, tumor suppressor gene, gene expression
Background
Carcinoma of cervix uteri (cervical cancer, CC) is a leading cause of cancer-related mortality in women worldwide [1,2]. CC arises by distinct morphologic changes from normal epithelium and progresses to carcinoma through a series of well-defined preinvasive lesions. Histologically, CC presents as either squamous cell carcinoma (SCC) or adenocarcinoma (AC) [3] with SCC predominating. Converging evidence from epidemiological and molecular studies suggests that infection of genital human papillomavirus (HPV) is causally linked to the development of CC [4]. Since only a small fraction of HPV-infected cervical intra-epithelial neoplastic (CIN) lesions progress to invasive cancer, these studies further suggest that in addition to HPV, other host genetic factors play a role in cervical carcinogenesis [5]. A number of molecular studies have identified genetic alterations in these two histologic types of CC and at various stages of precursor lesions [6-8]. Despite this molecular characterization, the genetic basis of CC initiation and progression is still very poorly understood. Therefore, identification of the underlying genetic changes may provide further insight into the molecular basis of CC.
Epigenetic hypermethylation in the promoter regions of a number of genes has been recognized as an important change in the development of human cancer [9]. A growing number of cancer-related genes have been identified to harbor methylation of cytosine residues in CpG-rich promoter sequences. The pattern of such promoter methylation has been noted to be non-random in various tumor types, while certain genes are commonly methylated in diverse tumor types [10,11]. The extent of aberrant promoter hypermethylation and its association with loss of gene function in cancer suggests that CpG island methylation is an important mechanism in inactivating tumor suppressor genes (TSGs).
The role of epigenetic gene inactivation in cervical tumorigenesis is poorly understood. A few previously published reports on CC and its precursor lesions showed promoter methylation of specific genes [12,13]. However, these studies were limited by the small number of genes and tumors examined. To investigate the role of promoter methylation in detail in cervical tumorigenesis, we evaluated CpG methylation of 16 genes in 90 CC specimens and cell lines. We found 86.6% of CC patients exhibiting promoter methylation. The CDH1, DAPK, RARB, and HIC1 gene promoters were frequently methylated. Methylation status was correlated with clinical and histologic characteristics, and microsatellite instability (MSI). We also found evidence that promoter methylation inactivates gene expression in CC and exposure to methylation and/or histone deacetylase (HDAC)-inhibiting agents reactivate the gene expression.
Results
CDH1, DAPK, RARB and HIC1 gene promoters are frequently methylated in CC
We examined the status of promoter hypermethylation of 16 genes (CDH1, DAPK, RARB, HIC1, FHIT, RASSF1A, APC, CDKN2A, MGMT, BRCA1, TP73, TIMP3, GSTP1, MLH1, p14ARF, and RB1) in eight specimens of normal cervical squamous epithelia and 90 CC specimens. Promoter hypermethylation was not found in the DNA isolated from normal cervical smears for any of the tested genes. However, hypermethylation was detected in one or more genes in 79 of 90 (87.8%) CC specimens. The frequency of promoter hypermethylation for individual genes was: CDH1, 51.1%; DAPK, 43.3%; RARB, 33.3%; HIC1, 22.2%; FHIT, 11.1%; RASSF1A, 10%; APC, 10%; CDKN2A, 8.9%; MGMT, 6.7%; BRCA1, 5.6%; TP73, 2.2%; TIMP3, 1.1%; GSTP1, 1.1%; and MLH1, 1.1% (Table 1). The remaining two genes (p14 ARF and RB1) did not show promoter methylation. Seventy-one of 82 (86.6%) primary tumors and 8 of 8 (100%) cell lines exhibited methylation. Although the patterns are similar, primary tumors had a higher frequency of methylation of CDH1 and DAPK genes as compared to cell lines, while RARB, HIC1, RASSF1A, MGMT, and TP73 genes had higher prevalence in cell lines as compared to primary tumors (Table 1). A high frequency of promoter methylation was detected in primary tumors for CDH1 (54.9%; 45 of 82 tumors) and DAPK (45.1%; 37 of 82 tumors). RARB (29.3%) and HIC1 (18.3%) genes were less frequently methylated in primary tumors. Other genes, FHIT (11%), APC (11%), CDKN2A (8.5%), RASSF1A (7.3%), BRCA1 (6.1%), MGMT (4.9%), and TIMP3 (1.2%), were rarely methylated in primary CC (Table 1, Fig. 1).
Table 1.
Frequency of promoter hypermethylation in cervical carcinoma
| Gene | ||||||||||||||||
| Specimen type | No. studied | Any gene | CDH1 | DAPK | RARB | HIC1 | FHIT | RASSF1A | APC | CDKN2A | MGMT | BRCA1 | TP73 | TIMP3 | GSTP1 | MLH1 |
| Controls | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CC cell lines | 8 | 8 (100) | 1 (12.5) | 2 (25) | 6 (75) | 5 (62.5) | 1 (12.5) | 3 (37.5) | 0 | 1 (12.5) | 2 (25) | 0 | 2 (25) | 0 | 1 (12.5) | 1 (12.5) |
| Primary tumors | 82 | 71 (86.6) | 45 (54.9) | 37 (45.1) | 24 (29.3) | 15 (18.3) | 9 (11) | 6 (7.3) | 9 (11) | 7 (8.5) | 4 (4.9) | 5 (6.1) | 0 | 1 (1.2) | 0 | 0 |
Figures in parenthesis indicate percentage
Figure 1.

Methylation-specific PCR of CDH1, DAPK, RARB, and HIC1 genes. U, unmethylated; M, methylated.
Analyses of HPV and microsatellite instability
Eighty-six of 90 (95.6%) tumors were positive for HPV DNA. The remaining four tumors were HPV negative. HPV 16 alone or in combination with other HPV types was found in 55 (61.1%). HPV 18 was found in eight (8.9%) tumors, while other HPV types (20 different types) in 23 (25.6%) tumors.
Of the 82 primary tumors studied for promoter methylation, 48 tumors were also examined for microsatellite instability (MSI) using 49 di- or tetra-nucleotide markers [14-17]. We found 9 of 48 (18.8%) tumors had MSI-H, and 9 of 48 (18.8%) tumors exhibited MSI-L phenotype. The remaining tumors did not show MSI (Table 2).
Table 2.
Correlation of promoter methylation with clinical and histologic parametrs, HPV status and microsatellite instability in CC patients
| Methylated genes | |||||||||||
| Parameter | Overall | CDH1 | DAPK | RARB | HIC1 | FHIT | RASSF1A | APC | CDKN2A | MGMT | BRCA1 |
| Histology | |||||||||||
| SCC (N = 77) | 66 | 45 | 36 | 24 | 12 | 9 | 5 | 5 | 6 | 4 | 3 |
| AC (N = 5) | 5 | 0 | 1 | 0 | 3 | 0 | 1 | 4 | 1 | 0 | 2 |
| Significance | P = 1.00 | P = 0.02 | P = 0.372 | P = 0.315 | P = 0.040 | P = 1.00 | P = 0.323 | P = 0.0003 | P = 0.367 | P = 1.00 | P = 0.028 |
| Stage | |||||||||||
| I (N = 17) | 10 | 6 | 5 | 3 | 5 | 0 | 4 | 2 | 2 | 0 | 0 |
| II (N = 19) | 19 | 13 | 12 | 5 | 3 | 3 | 2 | 2 | 1 | 0 | 2 |
| III-IV (N = 46) | 42 | 16 | 20 | 16 | 7 | 6 | 0 | 5 | 4 | 4 | 3 |
| Significance | P = 0.0005 | P = 0.0005 | P = 0.129 | P = 0.394 | P = 0.460 | P = 0.279 | P = 0.003 | P = 1.00 | P = 0.759 | P = 0.395 | P = 0.397 |
| Size of tumor | |||||||||||
| 1–5 cm (N = 32) | 28 | 20 | 13 | 10 | 8 | 3 | 4 | 4 | 1 | 0 | 1 |
| ≥ 6 cm (N = 38) | 34 | 21 | 19 | 13 | 6 | 4 | 1 | 3 | 4 | 2 | 2 |
| Significance | P = 1.000 | P = 0.540 | P = 0.433 | P = 0.793 | P = 0.337 | P = 1.00 | P = 0.171 | P = 0.695 | P = 0.366 | P = 0.497 | P = 1.000 |
| Age (yrs) | |||||||||||
| ≤ 40 (N = 32) | 28 | 17 | 17 | 12 | 6 | 6 | 1 | 1 | 3 | 3 | 1 |
| 41–50 (N = 17) | 12 | 8 | 5 | 5 | 3 | 0 | 2 | 0 | 1 | 1 | 1 |
| 51–60 (N = 13) | 13 | 6 | 5 | 6 | 3 | 2 | 1 | 6 | 1 | 0 | 3 |
| > 60 (N = 20) | 18 | 14 | 10 | 1 | 3 | 1 | 2 | 2 | 2 | 0 | 0 |
| Significance | P = 0.113 | P = 0.439 | P = 0.400 | P = 0.021 | P = 0.956 | P = 0.169 | P = 0.600 | P = 0.0004 | P = 1.000 | P = 0.467 | P = 0.053 |
| Clinical Outcome | |||||||||||
| Alive (N= 39) | 30 | 18 | 19 | 8 | 7 | 3 | 5 | 4 | 2 | 0 | 0 |
| DOC (N= 43) | 41 | 27 | 18 | 16 | 8 | 6 | 1 | 5 | 5 | 4 | 5 |
| Significance | 0.015 | P = 0.131 | P = 0.533 | P = 0.097 | P = 0.939 | P = 0.487 | P = 0.097 | P = 1.000 | P = 0.436 | P = 0.118 | P = 0.056 |
| Treatment response¶ | |||||||||||
| CR (N= 21) | 19 | 11 | 15 | 3 | 4 | 1 | 2 | 3 | 2 | 0 | 0 |
| DOC/PR (N = 26) | 24 | 15 | 10 | 12 | 4 | 3 | 1 | 4 | 3 | 2 | 2 |
| Significance | 1.000 | 0.716 | 0.024 | 0.020 | 1.000 | 0.617 | 0.579 | 1.000 | 1.000 | 0.495 | 0.495 |
| HPV type | |||||||||||
| HPV 16 (N = 47) | 42 | 27 | 24 | 15 | 9 | 6 | 4 | 5 | 3 | 3 | 2 |
| HPV 18 (N = 6) | 5 | 2 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 0 | 1 |
| Multiple (N = 11) | 8 | 7 | 3 | 2 | 1 | 0 | 0 | 2 | 2 | 0 | 1 |
| Others (N = 16) | 12 | 9 | 8 | 5 | 2 | 3 | 0 | 0 | 1 | 1 | 1 |
| significance | P = 0.543 | P = 0.740 | P = 0.280 | P = 0.792 | P = 0.560 | P = 0.467 | P = 0.365 | P = 0.288 | P = 0.327 | P = 1.000 | P = 0.412 |
| MSI | |||||||||||
| MSS (N = 30) | 26 | 20 | 17 | 4 | 2 | 3 | 1 | 2 | 5 | 4 | 2 |
| MSI-L (N = 9) | 9 | 6 | 4 | 2 | 1 | 2 | 1 | 0 | 1 | 0 | 2 |
| MSI-H (N = 9) | 8 | 2 | 3 | 3 | 4 | 0 | 1 | 3 | 1 | 0 | 1 |
| Significance | P = 0.516 | P = 0.065 | P = 0.513 | P = 0.434 | P = 0.023 | P = 1.000 | P = 0.313 | P = 0.079 | P = 1.000 | P = 0.443 | P = 0.266 |
P value corresponds to the P value from the Fisher's Exact test for the first two rows and for the P value from the Analysis of Variance for the last 3 rows. ¶Only patients with at least 5 months follow-up and treatment were considered and patients with treatment unknown or no treatment were excluded. SCC, squamous cell carcinoma; AC, adenocarcinoma; MSS, microsatellite stable; MSI-L, microsatellite instability low; MSI-H, microsatellite instability high; DOC, died of cancer; PR, partial response
Promoter hypermethylation in relation to histological, clinical and genetic characteristics
To evaluate the relationship between promoter hypermethylation and clinico-pathologic features, as well as other genetic changes, methylation status was correlated with histology, age, stage and size of the tumor, clinical outcome, HPV type, and microsatellite instability. Table 2 shows the results of univariate analyses examining the associations between methylation and these characteristics. Although both squamous cell carcinoma (SCC; 85.7%) and adenocarcinoma (AC; 100%) exhibited an overall high rate of promoter methylation, we found statistically significant differences in various genes in these two histologic subsets (Table 2). Promoter methylation of the CDH1, RARB, FHIT, and MGMT genes was noted only in SCC, whereas the AC exhibited significantly higher frequency of promoter methylation of HIC1 (60% in AC vs. 15.6% in SCC), APC (80% in AC vs. 6.5% in SCC), and BRCA1 (40% in AC vs. 3.9% in SCC) (Table 2).
A significant correlation was observed between the overall frequency of methylation and the tumor stage (P = 0.001). Higher stage (stages II-IV) tumors exhibited an increased overall frequency of promoter methylation (Table 2). However, the differences were not statistically significant when individual genes examined. On the other hand, RASSF1A methylation was associated with low-grade tumors (P = 0.003). Tumor size has no effect on the overall frequency of methylation. Age also has no influence on overall frequency of promoter methylation. However, RARB gene was more frequently methylated in younger patients (34.7% in below 50 years and 21.2% in above 50 years age) (P = 0.02). Promoter methylation of APC (2.0% in patients below 50 years vs. 24.2% in patients above 50 years age) (P= 0.001) and BRCA1 (4.1% in below 50 years and 9.1% in patients above 50 years) (P = 0.05) genes were significantly higher in older patients.
In order to assess the prognostic role, we also correlated treatment response and clinical outcome with promoter methylation. The overall frequency of methylation was higher in patients who died of cancer (DOC) (95.3%) as compared to those who were alive with recurring disease or complete remission (76.9%) (Table 2) (P = 0.015). Although statistically not significant, all the patients whose tumors showed promoter methylation of MGMT (4 tumors) and BRCA1 (5 tumors) genes DOC within 1–21 months of follow-up after the initial diagnosis. Promoter methylation of the remaining genes did not show such differences in relation to the outcome. To assess the role of radiation and/or chemotherapy treatment on outcome, we correlated methylation with response in patients with at least 5 months follow up after treatment. No significant differences in the overall promoter methylation were found in patients with complete remission (CR) compared to patients who DOC or had only partial response (PR). In this group, all the patients that showed methylation of MGMT and BRCA1 genes failed to respond to the treatment suggesting promoter methylation of these genes may be a bad prognostic indicator (Table 2). Promoter methylation of RARB gene was also predicted a worse prognosis as 80% of the patients with methylation were either DOC or only partial responders compared to only 20% patients with CR exhibited methylation of this gene (Table 2).
Table 3 summarizes the results from multivariate Cox Survival models. Overall, after adjusting for age, treatment, tumor size, stage and HPV type, neither total number of methylated genes nor presence of methylation of each specific gene were statistically significantly related to survival. However, methylation of 3 genes did suggest a doubling (RASSF1A and MGMT) or tripling (BRCA1) of risk compared to those who did not have these genes methylated. These differences were not statistically significant due to small number of tumors analyzed and need to be confirmed using a larger sample size.
Table 3.
Promter Hypermethylation and cervical cancer survival
| Alive | Dead | Hazard Ratio§ | 95%CI | ||
| Total loci | 0 | 9 | 2 | 1.00 | – |
| 1 | 6 | 9 | 0.75 | 0.14–4.00 | |
| 2 | 13 | 19 | 1.04 | 0.20–5.50 | |
| ≥3 | 11 | 13 | 1.07 | 0.19–5.92 | |
| CDH1 | + | 18 | 27 | 0.69 | 0.29–1.63 |
| - | 21 | 16 | 1.00 | – | |
| DAPK | + | 19 | 18 | 0.53 | 0.23–1.19 |
| - | 20 | 25 | 1.00 | – | |
| RARB | + | 8 | 16 | 1.66 | 0.80–3.46 |
| - | 31 | 27 | 1.00 | – | |
| HIC1 | + | 7 | 8 | 1.67 | 0.69–4.04 |
| - | 32 | 35 | 1.00 | – | |
| FHIT | + | 3 | 6 | 1.18 | 0.38–3.66 |
| - | 36 | 37 | 1.00 | – | |
| RASSF1A | + | 5 | 1 | 2.02 | 0.16–25.08 |
| - | 34 | 42 | 1.00 | – | |
| APC | + | 4 | 5 | 0.70 | 0.21–2.41 |
| - | 35 | 38 | 1.00 | – | |
| CDKN2A | + | 2 | 5 | 1.64 | 0.51–5.26 |
| - | 37 | 38 | 1.00 | – | |
| MGMT | + | 0 | 4 | 2.30 | 0.46–11.45 |
| - | 39 | 39 | 1.00 | – | |
| BRCA1 | + | 0 | 5 | 3.05 | 0.72–12.93 |
| - | 39 | 38 | 1.00 | – |
Adjusted for age (continuous), treatment (3 groups), tumor size (binary), stage (3 groups) and HPV type (4 groups)
Type of HPV infection had no association with the status of promoter methylation (Table 2). Although the MSI phenotype also had no significant correlation with overall promoter methylation, the HIC1 (P = 0.02) and APC (P = 0.08) genes were frequently methylated in patients exhibiting MSI-H phenotype (Table 2).
CDH1 and RARB expression in CIN
To examine the role of CDH1 and RARB genes in CC progression, we carried out immunohistochemical analysis in 39 CIN specimens (Nine low-grade and 30 high-grade). None of the low-grade CINs showed down-regulated expression, while only 2 of 30 (6.7%) showed absent or reduced expression suggesting that CDH1 inactivation is a later event in CC development. The expression pattern of RARB in normal epithelium is complex since the staining intensities are highly variable between nuclear and cytoplasmic compartments. There was no normal epithelium to evaluate the expression in most CIN cases studied. However, all the cases that we evaluated for RARB immunoreactivity showed some positive mesenchymal cells. We evaluated the intensity of nuclear staining in 34 cases of paraffin-embedded tissues. One (11.1%) of the nine low-grade CINs showed decreased nuclear expression, whereas, 15 (60%) of 25 evaluable high-grade CIN lesions showed complete lack of nuclear expression (Fig. 2). If confirmed, the down-regulated RARB expression in high-grade CINs suggests that this occurs early in progression and plays a role in the development of CC.
Figure 2.

Immunohistochemical analysis of RARB gene expression in low- and high-grade CIN lesions from the same patient. A. Low-grade CIN showing intense nuclear staining. B. High-grade CIN showing lack of nuclear expression.
Gene expression is down regulated by promoter hypermethylation and reactivated by demethylation
To examine the biological role of promoter methylation in CC, we assessed the levels of mRNA by semi-quantitative RT-PCR in eight cell lines and compared with the respective control values obtained from the averages calculated from 2 normal cervix after normalization against ACTB. This analysis was performed on 7 genes (CDH1, DAPK, RARB, HIC1, MGMT, RASSF1A, and MLH1) that exhibited methylation in one or more cell lines and primary tumors (Fig. 3). RARB gene showed either complete absence of or down regulated expression in all cell lines (6 methylated and 2 unmethylated). Of the six cell lines with RARB gene methylated, three (HT-3, MS751, and ME-180) exhibited reactivation upon treatment with 5-Aza-2' deoxycytidine or n-butyrate alone or the combination of the two. Three cell lines (C4-I, CaSki, and SW756) that had promoter methylation of RARB did not reactivate expression after treatment with these drugs (Fig. 4). One of the two unmethylated cell lines also reactivated the expression of RARB.
Figure 3.

Semi-quantitative multiplex RT-PCR analysis of gene expression in CC cell lines. Genes are shown on left; cell lines are indicated on top. Empty arrows indicate the PCR product of actin gene used as control and filled arrowheads represent the indicated gene.
Figure 4.
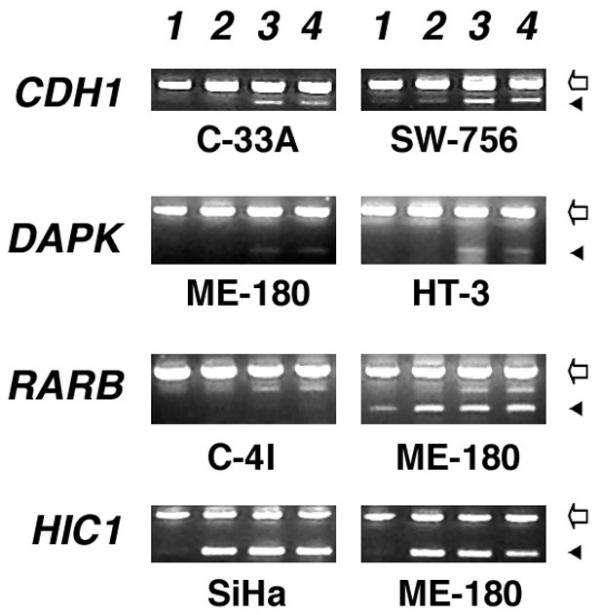
Effect of demethylation and acetylation on gene expression. Note the lack of reactivation of gene expression in C-4I in RARB gene. Lane 1, untreated; Lane 2, butyrate-treated; Lane 3, 5-Aza-2' Deoxycytidine + butyrate treated, and Lane 4, 5-Aza-2' Deoxycytidine-treated. Genes are shown on left; cell lines are indicated below; Empty arrows indicate the PCR product of actin gene used as control and filled arrowheads represent the indicated gene.
The CDH gene exhibited a complete absence of expression in one cell line (SW756) that showed promoter hypermethylation and two other cell lines (C-33A and SiHa) without evidence of methylation (Fig. 3). The SW756 cell line with methylation and one of the two unmethylated cell lines showed reactivated expression upon treatment with 5-Aza-2' deoxycytidine or in combination with n-butyrate, but not with n-butyrate alone. The remaining five cell lines neither showed evidence of decreased mRNA nor any effect of demethylating or HDAC-inhibiting agents on expression levels.
The DAPK gene showed a complete absence of expression in five and down regulated expression in one of the 8 CC cell lines studied (Fig. 3). Of the six cell lines with down regulated expression, 2 had promoter methylation and 4 had unmethylated promoter of the DAPK gene. Both methylated cell lines (HT-3 and SiHa) and three (C-33A, MS751, and ME-180) of the four unmethylated cell lines showed reactivation upon exposure to 5-Aza-2' deoxycytidine or in combination with n-butyrate. One of the unmethylated cell lines (SW756) that showed a complete absence of expression did not reactivate after treatment.
The HIC1 gene was methylated in 5 of 8 cell lines. Three of the methylated cell lines (C-4I, SiHa, and ME-180) and one of the three unmethylated cell lines (MS751) showed down regulated expression. Treatment of 5-Aza-2' deoxycytidine or n-butyrate alone or the combination of both activated gene expression in all the cell lines, whether or not promoter had detectable methylation (Fig. 4).
The RASSF1A gene was methylated in 3 cell lines and all three showed a complete lack of expression. None of the unmethylated cell lines showed evidence of down-regulated expression of RASSF1A mRNA. Two of three cell lines with RASSF1A methylation and complete lack of expression reactivated upon treatment with 5-Aza-2' deoxycytidine alone or in combination with n-butyrate. One of the methylated cell lines (HT-3), however, did not reactivate gene expression after treatment. The unmethylated cell lines showed normal expression and the treatment had no effect on expression.
MGMT was methylated in two cell lines. One of the two methylated cell lines (CaSki) showed a complete absence of expression while the other methylated cell line (SW756) showed normal levels of expression. Treatment with demethylating and deacetylating inhibitor did not activate the expression in any cell line regardless of promoter methylation. MLH1 gene was methylated in one cell line (SW756) but we found no evidence of down-regulated expression in this cell line (data not shown).
Discussion
Genetic alterations play an important role in the development of CC [4,7,8]. Our extensive molecular analyses identified a number of specific genetic changes in invasive CC and its precursor lesions [6,14-19]. However, epigenetic alteration in cervical tumorigenesis is not well characterized [12,13]. In the present study, we identified an overall methylation in 87.8% CC cases. Previous studies by Dong et al (79%) and Virmani et al (72.4%) have also reported a similar overall high frequency of promoter methylation [12,13]. We identified promoter methylation in >25% cases in CDH1, DAPK, and RARB genes, while it was less frequent (10–25% cases) in HIC1, FHIT, RASSF1A, and APC genes in CC patients. The promoters of CDKN2A, MGMT, BRCA1, TP73, TIMP3, GSTP1, and MLH1 genes were rarely (<10% cases) methylated.
CDH1 is the most commonly methylated gene in the present study showing promoter methylation in 51.1% of CC cases. Dong and co-workers have shown earlier that the CDH1 promoter is methylated in 28% of invasive CC [13]. E-cadherin, CDH1, is a major adhesion component of epithelial cells, which plays an important role as an invasion suppressor gene. Loss of function of CDH1 gene has been shown to occur in widespread of epithelial tumor types by mutational or promoter hypermethylation mechanisms [11,20]. We have also identified loss of expression of CDH1 gene in 37.5% of CC cell lines analyzed. Thus, these data suggest that inactivation of the CDH1 gene by promoter methylation plays a major role in CC tumorigenesis. However, our immunohistochemical analysis of E-cadherin protein showed no evidence of decreased expression in precursor CIN lesions. Therefore, these data indicate that the methylation of CDH1 gene is a late event in the development of CC.
DAPK gene is methylated in 43.3% cases in the present study. Dong and co-workers also previously showed promoter methylation in 51% of CC [13]. DAPK is a positive mediator of the programmed cell death induced by gamma-interferon [21]. Loss of DAPK expression has been shown to occur in a number of human malignancies, primarily by promoter hypermethylation [11]. It has also been shown that loss of DAPK gene expression was associated with aggressive and metastatic phenotype in many tumor types [22]. In the present study, in addition to showing high frequency of promoter methylation of DAPK gene, we found a complete lack of expression in two of the methylated cell lines and four of 6 unmethylated cell lines. Thus, our results suggest that loss of expression occurs in CC cell lines by mechanism other than complete promoter methylation. Therefore, our data further suggest a role for DAPK in CC tumorigenesis.
The third most commonly methylated gene in the present study is RARB. A previous study also has reported promoter methylation in 33.3% of invasive CC, and 11% and 29% of low- and high-grade CIN lesions, respectively [12]. These authors suggest that RARB methylation is an early event in multistage cervical carcinogenesis. Our semi-quantitative analysis of RARB gene expression by RT-PCR showed down-regulated expression in all the eight cell lines analyzed whether or not the promoter methylated (Fig. 3). In addition, our immunohistochemical analysis of RARB showed lack of expression in 60% of high-grade CINs. It has been reported that RARB mRNA is down regulated in a number of tumor types [23], including cervical cancer [24]. Taken together, these data support the concept that RARB play an important role early in cervical cancer progression. The direct role of RARB in regulating gene expression and its retinoid-mediated antiproliferative, differentiative, immuno-modulatory, and apoptotic-inducing properties may offer a therapeutic target in the future for CC treatment [25].
As reported in a previous study, we also found promoter methylation of HIC1 in 22.2% of CC [13]. HIC1 is a zinc finger transcription factor that is transcriptionally silenced by promoter methylation in several types of human cancer [26]. Heterozygous Hic1 mouse with promoter methylation of wild type allele develop carcinomas, sarcomas and lymphomas, suggesting that this gene acts as a tumor suppressor gene [27]. HIC1 gene was down regulated in majority of the CC cell lines examined by us and this down-regulated expression was reactivated upon treatment with demethylating and HDAC-inhibiting agents. These results support the tumor suppressor role of HIC1 and its inactivation by promoter methylation in CC may be a critical epigenetic change in tumor development.
The present methylation profile of CC identified significant differences in the methylated genes between SCC and AC. We found that methylation of HIC1, APC, and BRACA1 genes was more common in AC while CDH1 promoter methylation was seen only in SCC. Such differences in the two major histologies have also been reported previously for DAPK, HIC1, and APC genes [13]. However, we found CDH1 promoter methylation restricted to SCC in the present study, while Dong et al found methylation in both histological types [13]. This difference may be due to small number of AC patients we studied. Distinct patterns of promoter methylation between SCC and AC, however, suggest that epigenetic pathways of tumor suppression may be different in these histologic subtypes. Contrary to the previous reports, we found a low frequency of p16/CDKN2A gene promoter methylation [12,13,28,29]. The reason for these differences is currently unclear.
Our statistical correlative analysis of promoter methylation with clinicopathologic parameters, HPV type, and microsatellite instability identified significant associations. A most significant finding was that overall promoter hypermethylation is associated with the progression and predicts significantly poor disease-free survival. This was most evident in BRCA1 and MGMT genes, where all the patients with promoter methylation were dead (Table 2). The multivariate Cox regression analysis showed that promoter methylation is an independent predictor of outcome for BRCA1 (3-fold risk) and MGMT (2.3 fold risk) genes. Promoter methylation of cancer-related genes has been reported to predict patient prognosis for a number of genes [30,31]. BRCA1 plays a critical role in DNA repair and recombination, cell cycle checkpoint control, and transcription. This gene has been shown to be hypermethylated in tumors arising from breast-ovarian cancer syndrome [32]. Although we detected complete promoter methylation of BRCA1 gene in only 6% of primary tumors, other mechanisms that inactivate this gene might exist. The present MSP data and its relation to poor clinical outcome warrants a further study of BRCA1 role in CC. MGMT gene encodes O(6)-methylguanine-DNA methyltransferase and this enzyme effectively removes DNA adducts formed by alkylating agents [33]. Epigenetic gene silencing of MGMT confers enhanced sensitivity to alkylating-based chemotherapy, and lack of methylation associates with failure to respond to chemotherapy [34,35]. On the contrary, all patients that had MGMT promoter methylation were dead within 21 months of initial diagnosis, which predicts a poor prognosis. A possible explanation for this failure to respond may be that all the four patients in the present study were treated only with radiotherapy with or without combination of surgery. The standard treatment for advanced-stage cervix cancer has been the combination of radical surgery and radiotherapy [36]. The utility of cytotoxic chemotherapy in patients with advanced CC has been recognized recently [37]. Thus, the epigenetic gene silencing of MGMT, although in a small proportion, may serve as indicator of responsiveness to treatment with chemotherapeutic drugs that cause DNA adducts in CC.
Most patients in the present study population were treated with traditional radiation and surgery treatment protocols. To assess the impact of promoter methylation on treatment, we compared the patients followed for more than 5 months after treatment. This analysis suggests that RARB methylation predict a worse prognosis (P = 0.02). Twelve of the 15 (80%) patients with RARB methylation were DOC or did not respond to the current treatment, while only 3 of the 15 (20%) patients with RARB methylation exhibited complete response. Since RARB functions as a tumor suppressor gene and its chemopreventive effects are purely based on retinoid induced reactivation, the patients that received radiation therapy will have no effect on these tumors. Our data, therefore, suggest that the CC patients showing RARB promoter methylation may benefit from targeted chemopreventive treatment of combination of retinoic acid, demethylating, and chromatin modifying agents that reactivate the gene expression. An understanding of the exact mechanisms of RARB loss of function in CC is essential for such a strategy.
Microsatellite instability (MSI) is hallmark of mismatch repair-deficient cancers, which has been observed in all cancers arising from Hereditary Non-polyposis Colorectal Cancer (HNPCC) syndrome, some sporadic colorectal and other cancers [38]. MSI is generally associated with germ-line mutations in one of two MMR genes, hMLH1 and hMSH2. Epigenetic inactivation of promoter hypermethylation of APC and HIC1 also results in MSI phenotype in sporadic colorectal cancers [39,40]. Accumulating evidence supports the view that promoter methylation of mismatch repair genes may be related to MSI phenotype [41,42]. A number of previous studies have shown the existence of MSI phenotype in CC [43,44]. In the present study, we found that MSI-H occurs at an increased frequency in tumors with HIC1 and APC promoter hypermethylation. Association of APC promoter methylation with MSI phonotype has been previously reported in endometrial cancer [45]. Although the significance of these results is currently unclear, we believe that inactivation of these genes by promoter methylation causes microsatellite instability and may contribute to the development of CC.
Aberrant promoter methylation changes that occur in cancer are associated with transcriptional repression and loss of function of the methylated gene [9]. Since we found evidence of down-regulated expression of several methylated genes in CC cell lines, we tested to see if this down-regulation could be reactivated by methylation and HDAC-inhibiting agents. Transcriptional silencing of genes resulting from DNA hypermethylation of CpG islands is reversed by treatment with the hypo-methylating agent 5-aza-2'-deoxycytidine and HDAC-inhibiting agent n-butyrate in a dose and duration-dependent manner. We found that 5-Aza-2'-deoxycytidine treatment alone or in combination with n-butyrate resulted in reactivation of gene expression in most cell lines that showed promoter methylation. In some cell lines, exposure to n-butyrate alone has resulted in reactivation of the gene expression, for example in HIC1 gene (Fig. 4). These data, therefore, suggest that other epigenetic mechanisms involving defects in chromatin modification factors such as acetylation and methylation of histone proteins may also play role in gene silencing. Thus, our data also suggest that epigenetic changes that accompany methylation are common in CC and play critical role in CC development.
Conclusions
The present study identified promoter methylation in a number of genes in CC. The most frequently methylated genes were CDH1, DAPK, RARB, and HIC1. We have also shown that promoter methylation of these genes result in transcriptional repression. Furthermore, RARB gene expression is down regulated in high-grade CINs. These data suggest that some of the methylated genes may play a role early in the progression of cancer. Here we showed that promoter methylation is associated with advanced stage of the disease, RARB and BRCA1 promoter methylation predict a worse prognosis, and the HIC1 and APC promoter methylation is frequently associated with MSI. This knowledge of epigenetic changes in CC may have implications in our understanding of the role they play in CC development. Particularly, if the present results are replicated and identified in precancerous lesions, this may be useful in predicting the high-risk lesions to progression and serve as targets of treatment.
Methods
Patients, Tumor tissues, and cell lines
A total of 82 tumor biopsies derived from previously untreated primary invasive CCs and eight cell lines were used. The tumor biopsies were ascertained from patients evaluated at the Instituto Nacional de Cancerologia (Santa Fe de Bogota, Colombia) and from the Department of the Obstetrics and Gynecology of Friedrich Schiller University, Jena, Germany after appropriate informed consent and approval of protocols by institutional review boards. The primary tumors were clinically classified as FIGO stage IB (17 tumors), IIB (19 tumors), IIIB (43 tumors), and IV (3 tumors). Histologically, 77 tumors were classified as squamous cell carcinoma (SCC) and five as adenocarcinoma (AC). Clinical information such as age, stage and size of the tumor, follow-up after treatment was collected for all patients. Patient follow-up data was collected from the review of institutional medical records, and by contacting outside physicians and institutions. The follow up ranged from 1–72 months. Thirty-nine cervical intraepithelial neoplastic lesions derived from 37 patients were also obtained as formalin-fixed and paraffin-embedded archival tissues from Columbia University and Instituto Nacional de Cancerologia (Santa Fe de Bogota, Colombia). Eight normal cervical swabs, diagnosed on Pap smear as normal squamous epithelium, were collected in phosphate buffer saline from the patients attending Gynecology Clinic at the Columbia Presbyterian Medical Center, New York after appropriate informed consent. Cell pellets obtained from the swabs were used to isolate DNA. CC cell lines SiHa, SW756, C-4I, Ca Ski, C-33A, HT-3, MS751 and ME-180 were obtained from the American Type Culture Collection (Manassas, VA), and grown in tissue culture according to the supplier's recommendations. Frozen tumor tissues or cell pellets were utilized for isolating DNA and/or RNA by standard methods.
Methylation Specific PCR (MSP)
Genomic DNA was treated with sodium bisulphite as previously described [46]. Placental DNA treated in vitro with SssI methyltransferase (New England Biolabs, Beverly, MA) and similarly treated normal lymphocyte DNA were used as controls for methylated and unmethylated templates, respectively. The primers used for amplification of methylated and unmethylated promoters of genes RARB, TIMP3, CDKN2A, p14ARF, MGMT, DAPK, CDH1, GSTP1, APC promoter 1A, RB1, MLH1, TP73, BRCA1, FHIT, and HIC1 have been described previously http://pathology2.jhu.edu/pancreas/prim0425.htm#MSP; [32,47-49]. Methylated (MF and MR) and unmethylated (UF and UR) primers for RASSF1A promoter were designed according to Herman et al [46] and as described earlier [50]. PCR products were run on 2% agarose gels and visualized after ethidium bromide staining. Purified MSP products were sequenced in representative specimens by direct sequencing to confirm the methylation identified on agarose gels.
5-Aza-2' deoxycytidine and n-butyrate treatment, and analysis of gene expression
Cell lines were treated with demethylating agent 5-Aza-2' deoxycytidine (Sigma) for five days at a concentration of 2 to 5 μM, HDAC-inhibiting agent n-butyrate at a final concentration of 5 μM for the last 24 hours or a combination of both. Total RNA isolated from treated and untreated cell lines, and the total RNA and poly A+ RNA from normal cervix obtained from Ambion (Austin, TX) was reverse transcribed using random primers and the Pro-STAR first strand RT-PCR kit (Stratagene, La Jolla, CA). A semi-quantitative analysis of gene expression was performed in replicate experiments using 26–28 cycles of multiplex RT-PCR with β-actin (ACTB) as control and gene specific primers spanning at least 2 exons. The primers used for CDH1, MGMT, MLH1, and RARB have been described earlier [50]. The gene primers used for DAPK and RASSF1A and their positions in respective cDNAs were:
DAPK-F 5'-CAGTTTGCGGTTGTGAAGAA-3' (403–422 bp)
DAPK-R 5'-CCTGCAACGAGTTCCAAGAT-3' (610–629 bp)
RASSF1A-F 5'- GACCTCTGTGGCGACTTCAT-3' (232–251 bp, exon 1A)
RASSF1A-R 5'-GCTGTTGATCTGGGCATTGT-3' (461–480 bp)
The PCR products were run on 1.5% agarose gels, visualized by ethidium bromide staining and quantitated using the Kodak Digital Image Analysis System (Kodak, New Haven, CT).
Microsatellite analysis
High-molecular weight DNA from frozen tumor and the corresponding peripheral blood specimens was isolated as previously described [14]. A panel of 49 sequence tagged repeat polymorphic (STRP) (24 markers mapped to chromosome 2q, nine on 5p, 11 on 6p, and five on 11q) markers, previously analyzed for LOH, was chosen for scoring MSI [14-17]. The criterion for analysis and classification of MSI has been described earlier [51,52].
Detection of HPV
PCR amplification with PGMY09/11 L1 consensus HPV L1 primers followed by reverse line blot hybridization was used to detect 38 genital HPV types as described earlier [53,54]. Appropriate control experiments were set up using bulk master mix components to eliminate potential contamination. Individual assay sensitivity was analyzed by the use of serial dilutions of SiHa cell line crude cell digest, targeting 104, 103, 102, 101, and 100 input copies of HPV-16. A no template negative control was set up in all experiments.
Immunohistochemistry
Four μ-thick sections were cut from paraffin-embedded blocks and immuno-stained following deparaffinization and antigen retrieval using citrate buffer at pH 6.0. Following incubation with antibodies against E-cadherin (Dako, Carpinteria, CA) and RARB (NeoMarkers, Fremont, CA), signal was detected with the Envision plus (DAKO, Carpenteria, CA) system, using diaminobenzidine as a chromogen. Tumors were considered positive for E-cadherin when cells showed membrane staining. For RARB, staining in normal epithelium either nuclear or cytoplasmic or both were considered positive. Normal epithelium and mesenchymal cell expression were used as internal controls.
Statistical Analysis
We first assessed associations between presence of promoter methylation for each gene and various clinical, demographic, and genetic characteristics using Chi-Square test [55] and Fisher's Exact test (for small cell counts) [56]. Characteristics that we examined included: histology, stage, tumor size, patient's age, clinical outcome, treatment response, HPV type, and microsatellite instability. Cox Proportional Hazard Models [57] were further applied to assess the relative hazard (risk) of dying associated with each methylated gene after adjusting for age, treatment status, tumor size, stage, and HPV type.
Authors ' contributions
Author 1 (GN) performed cell culture, MSP and gene expression analysis. Author 2 (HAP) participated in the collection of tumors, clinical follow-up data, and performed HPV typing. Author 3 (SK) participated in MSP and MSI analyses. Author 4 (HV) participated in collection of tumor material and clinical information. Author 5 (FFZ) participated in statistical analysis. Author 6 (JV) participated in collection of normal cervical specimens. Author 7 (AS) participated in collection of cervical cancer specimens and clinical information. Author 8 (MBT) participated in statistical analysis and interpretation of the data. Author 9 (MM) participated in histologic diagnosis and immuno-histochemical analysis. Author 10 (VVM) has conceived and coordinated the study. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by the Elaine B. Lesser Fund, EIF pilot award from the Herbert Irving Comprehensive Cancer Center, Columbia University to VVM. HAP was supported by a grant from Colciencias, Colombia.
Contributor Information
Gopeshwar Narayan, Email: gn110@columbia.edu.
Hugo Arias-Pulido, Email: HArias@salud.unm.edu.
Sanjay Koul, Email: sk1276@columbia.edu.
Hernan Vargas, Email: hvargas76@yahoo.es.
Fang F Zhang, Email: fz2004@columbia.edu.
Jeannine Villella, Email: jav2004@columbia.edu.
Achim Schneider, Email: Achim.Schneider@med.uni-jena.de.
Mary B Terry, Email: mt146@columbia.edu.
Mahesh Mansukhani, Email: mm322@columbia.edu.
Vundavalli V Murty, Email: vvm2@columbia.edu.
References
- Jemal A, Thomas A, Murray T, Thun M. Cancer Statistics, 2002. CA Cancer J Clin. 2002;52:23–47. doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- Vizcaino AP, Moreno V, Bosch FX, Munoz N, Barros-Dios XM, Borras J, Parkin DM. International trends in incidence of cervical cancer: II. Squamous-cell carcinoma. Int J Cancer. 2000;86:429–435. doi: 10.1002/(SICI)1097-0215(20000501)86:3<429::AID-IJC20>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wright TC, Ferenczy A, Kurman R. Carcinoma and other tumors of the cervix. In: Kurman RJ, editor. "Pathology of the female genital tract ". Fourth. New Yori, Springer-Verlag; 1994. pp. 279–326. [Google Scholar]
- Zur Hausen H, Rosl F. Pathogenesis of cancer of the cervix. Cold Spring Harb Symp Quant Biol. 1994;9:623–628. doi: 10.1101/sqb.1994.059.01.071. [DOI] [PubMed] [Google Scholar]
- Ostor AG. Natural history of cervical intraepithelial neoplasia: a critical review. Int J Gynecol Pathol. 1993;12:186–192. [PubMed] [Google Scholar]
- Mitra AB, Murty VV, Li RG, Pratap M, Luthra UK, Chaganti RS. Allelotype analysis of cervical carcinoma. Cancer Res. 1994;54:4481–4487. [PubMed] [Google Scholar]
- Lazo PA. The molecular genetics of cervical carcinoma. Br J Cancer. 1999;80:2008–2018. doi: 10.1038/sj.bjc.6690635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KR. Cervical Cancer. In: Vogelstein B, Kinzler KW, editor. In "The genetic basis of human cancer". 2. New York, McGraw-Hill; 2002. pp. 689–696. [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg962. [DOI] [PubMed] [Google Scholar]
- Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O'Dorisio MS, Held WA, Cavenee WK, Plass C. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- Virmani AK, Muller C, Rathi A, Zoechbauer-Mueller S, Mathis M, Gazdar AF. Aberrant methylation during cervical carcinogenesis. Clin Cancer Res. 2001;7:584–589. [PubMed] [Google Scholar]
- Dong SM, Kim HS, Rha SH, Sidransky D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001;7:1982–1986. [PubMed] [Google Scholar]
- Pulido HA, Fakruddin MJ, Chatterjee A, Esplin ED, Beleño N, Martínez G, Posso H, Evans GA, Murty VVVS. Identification of a 6 cM minimal deletion at 11q23.1-23.2 and exclusion of PPP2R1B gene as a deletion target in cervical cancer. Cancer Res. 2000;60:6677–6682. [PubMed] [Google Scholar]
- Chatterjee A, Pulido HA, Koul S, Beleño N, Perilla A, Posso H, Manusukhani M, Murty VVVS. Mapping the Sites of Putative Tumor Suppressor Genes at 6p25 and 6p21.3 in Cervical Carcinoma: Occurrence of Allelic Deletions in Precancerous Lesions. Cancer Res. 2001;61:2119–2123. [PubMed] [Google Scholar]
- Arias-Pulido H, Narayan G, Vargas H, Mansukhani M, Murty VV. Mapping common deleted regions on 5p15 in cervical carcinoma and their occurrence in precancerous lesions. Mol Cancer. 2002;1:3. doi: 10.1186/1476-4598-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan G, Pulido HA, Koul S, Lu X-Y, Harris CP, Yeh YA, Vargas H, Posso H, Terry MB, Gissmann L, Schneider A, Mansukhani M, Rao PH, Murty VV. Genetic analysis identifies putative tumor suppressor sites at 2q35-3q36.1 and 2q36.3-q37.1 involved in cervical cancer progression. Oncogene. 2003. [DOI] [PubMed]
- Mitra AB, Murty VV, Pratap M, Sodhani P, Chaganti RSK. ERBB2 (HER2/neu) oncogene is frequently amplified in squamous cell carcinoma of the uterine cervix. Cancer Res. 1994;54:637–639. [PubMed] [Google Scholar]
- Mitra AB, Murty VV, Singh V, Li R-G, Pratap M, Sodhani P, Luthra UK, Chaganti RSK. Genetic alterations at 5p15: A potential marker for progression of precancerous uterine cervix lesions. J Natl Cancer Inst. 1995;87:742–745. doi: 10.1093/jnci/87.10.742. [DOI] [PubMed] [Google Scholar]
- Strathdee G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol. 2002;12:373–379. doi: 10.1016/S1044-579X(02)00057-3. [DOI] [PubMed] [Google Scholar]
- Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995;9:15–30. doi: 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- Simpson DJ, Clayton RN, Farrell WE. Preferential loss of Death Associated Protein kinase expression in invasive pituitary tumours is associated with either CpG island methylation or homozygous deletion. Oncogene. 2002;21:1217–1224. doi: 10.1038/sj.onc.1205195. [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Berger J, Hermann M, Muller HM, Amberger A, Zeschnigk M, Widschwendter A, Abendstein B, Zeimet AG, Daxenbichler G, Marth C. Methylation and silencing of the retinoic acid receptor-beta2 gene in breast cancer. J Natl Cancer Inst. 2000;92:826–832. doi: 10.1093/jnci/92.10.826. [DOI] [PubMed] [Google Scholar]
- Ivanova T, Petrenko A, Gritsko T, Vinokourova S, Eshilev E, Kobzeva V, Kisseljov F, Kisseljova N. Methylation and silencing of the retinoic acid receptor-beta 2 gene in cervical cancer. BMC Cancer. 2002;2:4. doi: 10.1186/1471-2407-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY, Lotan R. Retinoids and their receptors in cancer development and chemoprevention. Crit Rev Oncol Hematol. 2002;41:41–55. doi: 10.1016/S1040-8428(01)00144-5. [DOI] [PubMed] [Google Scholar]
- Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, Cavenee WK, Kuerbitz SJ, Baylin SB. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–577. doi: 10.1038/nm0695-570. [DOI] [PubMed] [Google Scholar]
- Chen WY, Zeng X, Carter MG, Morrell CN, Chiu Yen RW, Esteller M, Watkins DN, Herman JG, Mankowski JL, Baylin SB. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33:197–202. doi: 10.1038/ng1077. [DOI] [PubMed] [Google Scholar]
- Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB, Herman JG. In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci U S A. 1999;96:12754–12759. doi: 10.1073/pnas.96.22.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YF, Chung TK, Cheung TH, Nobori T, Yu AL, Yu J, Batova A, Lai KW, Chang AM. Methylation of p16INK4A in primary gynecologic malignancy. Cancer Lett. 1999;136:231–235. doi: 10.1016/S0304-3835(98)00327-9. [DOI] [PubMed] [Google Scholar]
- Tang X, Khuri FR, Lee JJ, Kemp BL, Liu D, Hong WK, Mao L. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. J Natl Cancer Inst. 2000;92:1511–1516. doi: 10.1093/jnci/92.18.1511. [DOI] [PubMed] [Google Scholar]
- Maruyama R, Toyooka S, Toyooka KO, Harada K, Virmani AK, Zochbauer-Muller S, Farinas AJ, Vakar-Lopez F, Minna JD, Sagalowsky A, Czerniak B, Gazdar AF. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001;61:8659–8663. [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- Teicher BA. Antitumor alkylating agents. In: De Vita VT jr., Hellman S, Rosenberg SA, editor. Cancer: Principles and practive of oncology. Vol. 1. Philadelphia: Lippincott-Raven; 1997. pp. 405–418. [Google Scholar]
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- Esteller M, Gaidano G, Goodman SN, Zagonel V, Capello D, Botto B, Rossi D, Gloghini A, Vitolo U, Carbone A, Baylin SB, Herman JG. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J Natl Cancer Inst. 2002;94:26–32. doi: 10.1093/jnci/94.1.26. [DOI] [PubMed] [Google Scholar]
- NIH Consensus statement. Cervical cancer. 1996;14:1–38. [PubMed] [Google Scholar]
- ACOG practice bulletin. Diagnosis and treatment of cervical carcinomas. Number 35, May. American College of Obstetricians and Gynecologists. Int J Gynaecol Obstet. 2002;78:79–91. doi: 10.1016/S0020-7292(02)00049-8. [DOI] [PubMed] [Google Scholar]
- Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Sugano K, Ushiama M, Fukayama N, Nomoto K, Kashiwabara H, Fujita S, Kakizoe T. Heterogeneity of DNA methylation status analyzed by bisulfite-PCR-SSCP and correlation with clinico-pathological characteristics in colorectal cancer. Clin Chem Lab Med. 2001;39:121–128. doi: 10.1515/CCLM.2001.021. [DOI] [PubMed] [Google Scholar]
- Ahuja N, Mohan AL, Li Q, Stolker JM, Herman JG, Hamilton SR, Baylin SB, Issa JP. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 1997;57:3370–3374. [PubMed] [Google Scholar]
- Yuen ST, Chan TL, Ho JW, Chan AS, Chung LP, Lam PW, Tse CW, Wyllie AH, Leung SY. Germline, somatic and epigenetic events underlying mismatch repair deficiency in colorectal and HNPCC-related cancers. Oncogene. 2002;21:7585–7592. doi: 10.1038/sj.onc.1205968. [DOI] [PubMed] [Google Scholar]
- Larson AA, Kern S, Sommers RL, Yokota J, Cavenee WK, Hampton GM. Analysis of replication error (RER+) phenotypes in cervical carcinoma. Cancer Res. 1996;56:1426–1431. [PubMed] [Google Scholar]
- Helland A, Borresen-Dale AL, Peltomaki P, Hektoen M, Kristensen GB, Nesland JM, de la Chapelle A, Lothe RA. Microsatellite instability in cervical and endometrial carcinomas. Int J Cancer. 1997;70:499–501. doi: 10.1002/(SICI)1097-0215(19970304)70:5<499::AID-IJC1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Zysman M, Saka A, Millar A, Knight J, Chapman W, Bapat B. Methylation of adenomatous polyposis coli in endometrial cancer occurs more frequently in tumors with microsatellite instability phenotype. Cancer Res. 2002;62:3663–3666. [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DJ, Hibberts NA, McNicol AM, Clayton RN, Farrell WE. Loss of pRb Expression in Pituitary Adenomas Is Associated with Methylation of the RB1 CpG Island. Cancer Res. 2000;60:1211–1216. [PubMed] [Google Scholar]
- Corn PG, Kuerbitz SJ, van Noesel MM, Esteller M, Compitello N, Baylin SB, Herman JG. Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt's lymphoma is associated with 5' CpG island methylation. Cancer Res. 1999;59:3352–3356. [PubMed] [Google Scholar]
- Zochbauer-Muller S, Fong KM, Maitra A, Lam S, Geradts J, Ashfaq R, Virmani AK, Milchgrub S, Gazdar AF, Minna JD. 5' CpG island methylation of the FHIT gene is correlated with loss of gene expression in lung and breast cancer. Cancer Res. 2001;61:3581–3585. [PubMed] [Google Scholar]
- Koul S, Houldsworth J, Mansukhani MM, Donadio A, McKiernan JM, Reuter VE, Bosl GJ, Chaganti RS, Murty VV. Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol Cancer. 2002;1:8. doi: 10.1186/1476-4598-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murty VV, Li R-G, Mathew S, Reuter VE, Bronson DL, Bosl GJ, Chaganti RSK. RER-type instability at 1q42-43 in human male germ cell tumors. Cancer Res. 1994;54:3983–3985. [PubMed] [Google Scholar]
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- Gravitt PE, Peyton CL, Apple RJ, Wheeler CM. Genotyping of 27 human papillomavirus types by using L1 consensus PCR products by a single-hybridization, reverse line blot detection method. J Clin Microbiol. 1998;36:3020–3027. doi: 10.1128/jcm.36.10.3020-3027.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton CL, Gravitt PE, Hunt WC, Hundley RS, Zhao M, Apple RJ, Wheeler CM. Determinants of genital human papillomavirus detection in a US population. J Infect Dis. 2001;183:1554–1564. doi: 10.1086/320696. [DOI] [PubMed] [Google Scholar]
- Daniel WW. Biostatistics: A foundation for Analysis in the Health Sciences. New York: John Wiley & Sons. 1991.
- Fleiss JL. Statistical Methods for Rates and Proportions. New York: John Wiley & Sons. 1981.
- Hosmer DW, Lemeshow S. Applied Survival Analysis: Regression Modeling of Time to Event Data. New York: John Wiley & Sons. 1998.