

Abstract
Background
Actin is required for the gene expression and morphogenesis of respiratory syncytial virus (RSV), a clinically important Pneumovirus of the Paramyxoviridae family. In HEp-2 cells, RSV infection also induces actin stress fibers, which may be important in the immunopathology of the RSV disease. Profilin, a major regulator of actin polymerization, stimulates viral transcription in vitro. Thus, we tested the role of profilin in RSV growth and RSV-actin interactions in cultured cells (ex vivo).
Results
We tested three cell lines: HEp-2 (human), A549 (human), and L2 (rat). In all three, RSV grew well and produced fused cells (syncytium), and two RSV proteins, namely, the phosphoprotein P and the nucleocapsid protein N, associated with profilin. In contrast, induction of actin stress fibers by RSV occurred in HEp-2 and L2 cells, but not in A549. Knockdown of profilin by RNA interference had a small effect on viral macromolecule synthesis but strongly inhibited maturation of progeny virions, cell fusion, and induction of stress fibers.
Conclusions
Profilin plays a cardinal role in RSV-mediated cell fusion and viral maturation. In contrast, interaction of profilin with the viral transcriptional proteins P and N may only nominally activate viral RNA-dependent RNA polymerase. Stress fiber formation is a cell-specific response to infection, requiring profilin and perhaps other signaling molecules that are absent in certain cell lines. Stress fibers per se play no role in RSV replication in cell culture. Clearly, the cellular architecture controls multiple steps of host-RSV interaction, some of which are regulated by profilin.
Background
The actin cytoskeleton plays a central role in cell motility, movement and morphology. The ability of soluble monomeric G-actin to polymerize into filamentous F-actin in a reversible manner is central to the cellular function of actin [1,2]. Rearrangement of actin is promoted rapidly by extracellular stimuli, and is facilitated by sets of actin-associated proteins, one of which is profilin [2]. It is now appreciated that a large number of cellular parasites co-opt actin for their gene expression and replication in the host cell [3,4]. Due to its primary role in pediatric respiratory disease, and the enormous human and economic toll it exacts throughout the world, the respiratory syncytial virus (RSV) and its interaction with the host cell have received serious attention. RSV is a member of the Pneumovirus genus in the Paramyxoviridae family, and contains a nonsegmented negative-stranded RNA genome of about 15 kb [5]. The minimal viral RNA-dependent RNA polymerase (RdRP) consists of two virally encoded proteins, the large protein L and the phosphoprotein P [6-8], and a third viral protein, M2-1 (22K), contributes RNA antitermination function and optimal processivity [9]. In addition to these viral proteins, the RdRP also requires cellular actin. Biochemical reconstitution studies using purified recombinant actin in vitro and studies in RSV-infected cells (ex vivo) demonstrated that actin functions as a viral transcription factor in both monomeric (G) and filamentous (F) forms, whereas the filamentous form appeared to be particularly important for viral morphogenesis [10]. Subsequent studies showed that at saturating actin concentration, profilin stimulated viral transcription even further, whereas profilin by itself had no transcriptional activity in the absence of actin [11]. These results suggested that the interaction of profilin with actin may be important for optimal activity of the RSV transcriptase. It remained unknown whether profilin plays a similar role in vivo.
Fusion of the infected cells is a hallmark of all Paramyxoviruses [12]; the fused mass of cells is called "syncytium", from which RSV derives its middle name. Although multiple viral proteins may be required for optimal syncytium induction, the fusion protein F is the central mediator of the process [13]. Syncytium most likely provides an efficient mechanism of cell-to-cell spread of the virus [12,14]; however, its exact role in productive RSV infection or in the pathology of the disease has remained unclear. Interestingly, RSV infection of HEp-2 cells also led to the formation of actin stress fibers and activation of the small cellular GTPase RhoA, a modulator of actin filamentation [14-17]. Inhibition of RhoA or its interaction with F resulted in the inhibition of syncytium as well as stress fiber formation and the second cycle of infection [14]. Recombinant F also activated RhoA in the absence of any other viral protein [17]. Based on these results, it was inferred that stress fiber formation might be important in some aspects of RSV infection, such as virus spread, elaboration of cytokines from infected cells, and contraction of airway muscles that promotes asthma.
The epithelial A549 cells of alveolar carcinoma origin exhibit many properties characteristic of the type II cells, such as the ability to produce surfactants. In cell culture, A549 monolayer also faithfully exhibits various aspects of RSV infection and RSV-host interaction, including robust virus growth, formation of syncytia, activation of NF-kappa B (NF-κB), and elaboration of a battery of cytokines in response to virus infection [18-22]. In this paper we have, therefore, continued the use of A549 cells to elucidate the mechanism of RSV-actin interactions, and in particular, test the role of profilin in the pathways that are likely to involve such interactions.
Results
Viral partners of cellular profilin
The minimal constituents of a nonsegmented negative-strand RNA viral RdRP are the viral large protein L and the phosphoprotein P [23]. While the L protein is believed to possess the basic RNA polymerase function, the P protein serves as its transcription factor. Together these proteins transcribe the genomic RNA wrapped with nucleocapsid protein N. It is only the N-RNA complex, and not the naked RNA, that serves as the functional template for the viral RdRP. Because profilin, like P, also acts to stimulate viral transcription [11], we first tested if these two proteins interact directly in a purified system. Recombinant His-tagged P protein was immobilized on a Ni-NTA column and purified recombinant human profilin was passed through it. Results (Fig. 1) showed that profilin indeed bound to P, and the two proteins were identified by staining as well as immunoblot analysis. No profilin bound to a control column lacking P. When an extract of A549 cells was passed through the P column, a few additional bands of 30–34 kDa size range besides the 14 kDa profilin were found to associate to the column. The identity of these bands was not determined, but we have previously shown that the catalytic subunit of human PP2A, which has a similar size, binds to P [22]. The specific affinity of all of these proteins for the P protein in the column was ascertained by the demonstration that pre-incubation of the A549 extract with excess purified P abolished their binding to the column. The reciprocal affinity experiment was not performed because we did not have a suitable matrix to immobilize profilin.
Figure 1.
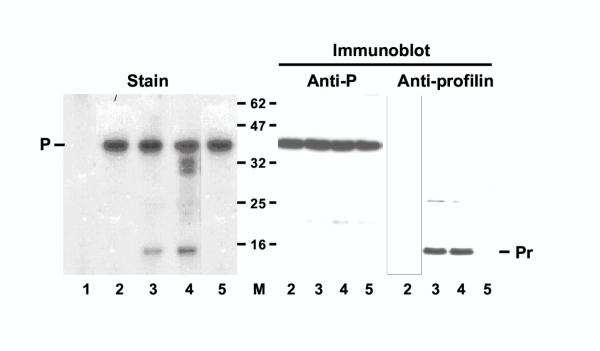
Interaction between RSV P protein and profilin in vitro. Ni+2-agarose matrix with bound His-tagged P protein was incubated with either of the following, and the bound proteins analyzed as described under Materials and Methods: none (lane 1), 2 μg profilin (lane 2), 40 μg S100 extract of A549 cells (see Materials and Methods) (lane 4), A549 extract as in lane 4 except that it was pre-incubated with 10 μg of purified P protein at room temperature for 5 min (lane 5). About 90% of the eluted material was analyzed by SDS-PAGE and staining with Coomassie Blue R250 (left); the rest divided into equal parts and analyzed in parallel Western blots using anti-P or anti-profilin antibody (right). Lane 1 represents eluate from a blank Ni+2-column to which no P protein was bound. Molecular weights of protein size markers are indicated in the middle (62 through 16 kDa). The P and the profilin bands are so indicated.
We then tested whether profilin interacts with viral proteins in the RSV-infected cell. An extract of the RSV-infected A549 cells was first subjected to immunoprecipitation by anti-P antibody and the precipitated proteins probed in immunoblot with profilin antibody. Results (Fig. 2) showed that profilin was indeed present in association with RSV P protein (lane 2). A reciprocal immunoprecipitation of the infected cell extract was carried out using anti-profilin; the precipitate was found to contain P protein (lane 6) as well as profilin (lane 5). Interestingly, when this precipitate was probed with a mixture of antibodies against all RSV proteins, in addition to P, the nucleocapsid protein N was detected (lane 7). To further test the specificity of the interaction of N and P with profilin, another antibody, specific for the viral fusion protein, F, was used to probe the profilin immunoprecipitate; however, no F was detected (lane 8).
Figure 2.
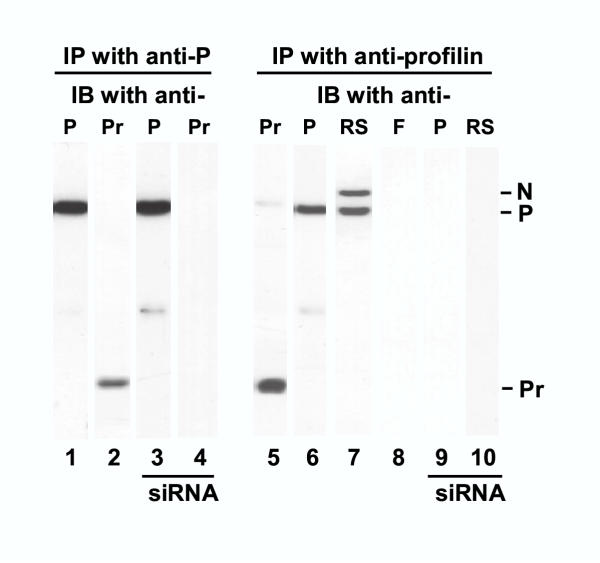
Interaction of RSV N and P with profilin in RSV-infected cell. RSV-infected cells at 18 h p.i. were lysed and subjected to immunoprecipitation (IP) by anti-P (left) or anti-profilin (right) antibodies as described under Materials and Methods. Western (immunoblot, IB) analysis of the precipitate was carried out using antibodies against the indicated proteins: P, F, profilin (Pr), and a mixture of antibodies against RSV N, P, F, G, and M. The N, P, and profilin bands are so marked. siRNA indicates samples from cells treated with anti-profilin siRNA.
As components of the functional viral RdRP, N and P bind each other [23]. It was, therefore, important to ask whether they need each other (or other viral proteins) in order to interact with profilin, or whether they are individually capable of binding profilin. To answer this question, we expressed recombinant N and P proteins individually by transient transfection of uninfected A549 cells [21], and studied their association with profilin by co-immunoprecipitation. Results (Fig. 3) clearly demonstrate that N and P can each bind profilin, independent of each other and in the absence of any other viral protein.
Figure 3.
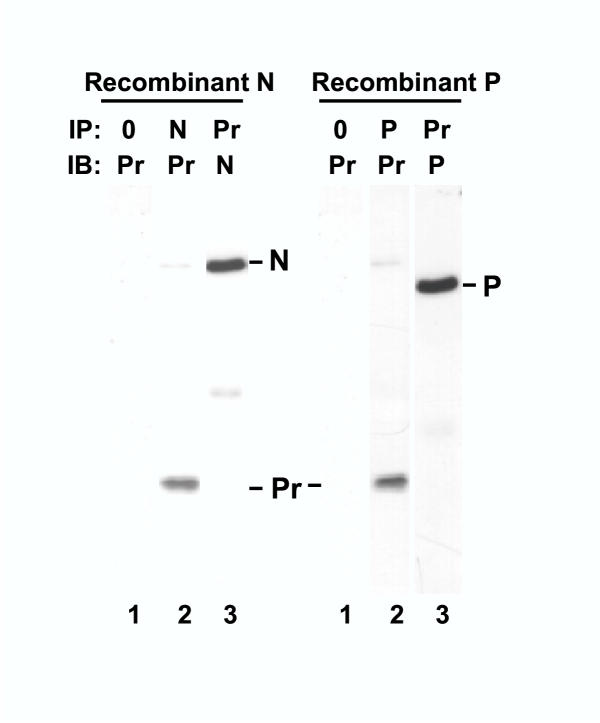
Interaction of recombinant N and P proteins with profilin ex vivo. A549 cells were transfected with varying amounts of pcDNA3 clone of N or P, and a representative set of results is shown. Lysis and immunoprecipitation (IP) of the transfected cells were carried out as described in Materials and Methods. The resultant precipitate was probed in immunoblot (IB). The antibodies were against N, P, or profilin (Pr), as indicated. Lane 0 indicates that IP was performed with protein A-sepharose only without any antibody. The N, P, and profilin bands are so marked.
To investigate the role of profilin in host-virus interaction in intact cells, we sought to develop a profilin knock-down cell by using RNA interference targeted against profilin mRNA as described under Materials and Methods. RNA interference is now an established tool in knocking down specific intracellular mRNA in mammalian cell culture [24], and our laboratory has pioneered its use in virus-infected cells [25]. Results presented later (Fig. 4, panel A) show that cells were substantially depleted of profilin at 200 nM siRNA. In our routine experiments, therefore, we have used 400 nM siRNA to achieve an essentially profilin-null cell. These cells were infected with RSV, and the infected cell lysates subjected to immunoprecipitation studies. As shown in Fig. 2, the anti-P immunoprecipitate obtained from these cells, while containing appreciable quantities of the P protein (lane 3), did not contain any profilin (lane 4).
Figure 4.
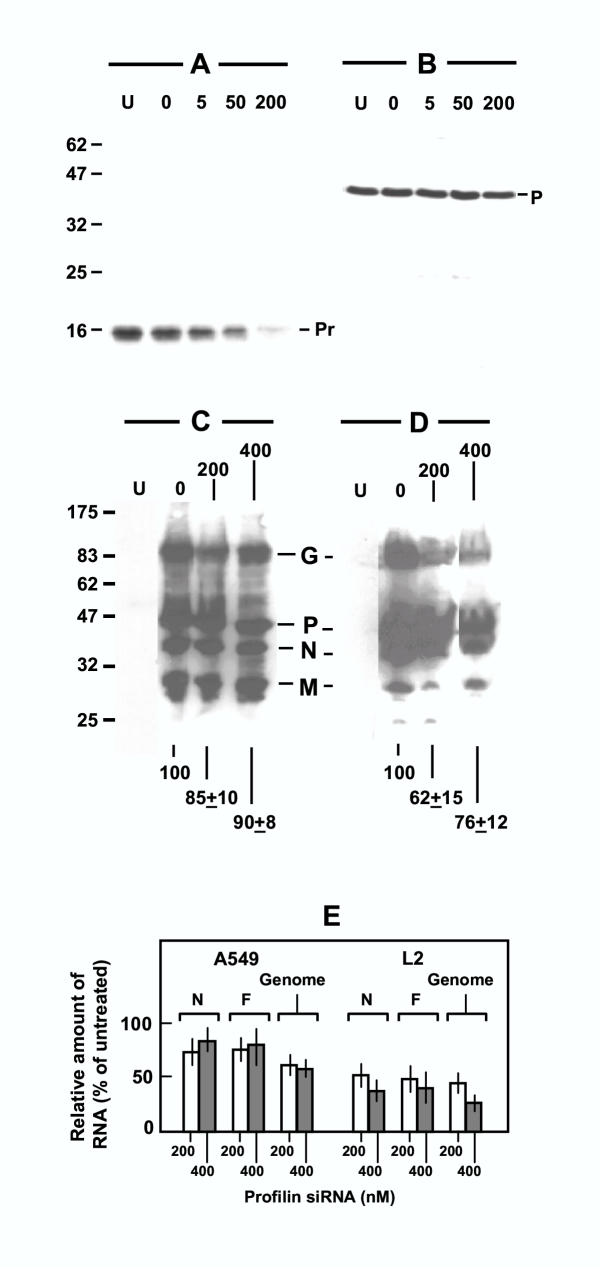
RSV gene expression in profilin-depleted cells. Panels A, B: siRNA-mediated knock down of profilin. A549 monolayer was transfected with 0, 5, 50, or 200 nM profilin siRNA, and then infected with RSV. Lane U indicates control cells that were neither transfected nor infected. About 40 μg of total cellular protein was subjected to immunoblot in each lane using antibody against profilin (Pr) (panel A) or RSV P (panel B). The chemiluminographs are presented. Note that profilin siRNA did not appreciably affect the P protein. Panel C, D: Immunoblot of RSV proteins. A549 (panel C) or L2 (panel D) cells were pre-treated with 0, 200, or 400 nM profilin siRNA and then infected with RSV as in the previous panels. A total of 40 μg of total protein from the infected cells were subjected to immunoblot using an antibody mixture against RSV G, N, P, and M proteins. Lane U represents identical amount of protein from the respective uninfected cells. All bands were digitally scanned by a Fujifilm LAS-1000 Intelligent Dark Box II Image Reader (Fujifilm, Edison, NJ), and the intensities integrated and added using the Image Gauge v4.0 software. Total intensities of the G, P, N, and M bands of the siRNA-treated lanes, expressed as percent of the untreated (0 nM) lane, are presented at the bottom. Values represent mean of three experiments with the range in ±. In panels A-D, the protein size markers are indicated on the left in kDa. Panel E: RT-PCR quantitation of N and F mRNA and genomic RNA of RSV. Design of primers and the procedure of RT-PCR have been described [25]. All values were mean of three experiments with the range indicated by bars. Results were normalized against control actin RNA amplified by RT-PCR using primers described previously [25].
We note here that no stress fiber or syncytium was observed in cells expressing N and P (data not shown), which is consistent with the notion that these proteins are primarily involved in viral RNA synthesis.
Profilin plays a minor role in viral gene expression
Because profilin stimulated RSV transcription in vitro [11] and associated with viral transcriptional proteins, N and P, the obvious next question was to ask how important profilin was for viral gene expression in the intact RSV-infected cell. Our ability to knock down profilin now provided us with an opportunity to answer this question. The profilin-depleted cells were, therefore, infected with RSV, and virus-specific RNA and proteins were quantified by appropriate techniques. In the very first experiment of this series, we sought to optimize the amount of siRNA, and tested their effects on profilin and viral P protein in infected A549 cells. As shown in Fig. 4, a progressive loss of profilin (panel A) was not accompanied by any pronounced effect on P (panel B), suggesting that profilin may not have a major role in viral gene expression. We then extended these studies to include two different cell lines and other viral proteins. Use of an antibody mixture allowed us to detect viral G, N, P, and M proteins (panels C, D). In A549 human cells (panel C) there was a small reduction in these proteins (ranging from 15% to 10% reduction at 200–400 nM siRNA); in rat L2 cells (panel D) a somewhat greater reduction (about 30–40%) was observed. To test if profilin had a role in viral RNA synthesis, representative N and F mRNAs as well as the negative-stranded viral genomic RNA were quantitated by real-time RT-PCR as described under Materials and Methods. Results presented graphically (Fig. 4, panel E) show that even at the highest siRNA concentration (400 nM), at which essentially all the profilin was knocked down (data not shown), 60–80% of viral RNA synthesis continued in A549 cells, whereas in L2 cells the effect was slightly more severe, with 30–50% RNA synthesis remaining. We conclude that profilin has only a minor role, if any, in the expression of RSV macromolecules in the infected cell, although it is impossible to rule out that small amounts of profilin survived the knock-down and sufficed for viral transcription.
Induction of actin stress fibers by RSV is cell-specific and profilin-dependent
As discussed earlier, RSV-infection of HEp-2 cells was shown to rapidly induce the formation of actin stress fibers, and this process required an interaction between viral fusion protein, F and cellular G-protein, RhoA [17]. To initiate studies of its mechanism, we first tested the universality of this finding. Three kinds of established epithelial cells of lung origin – namely HEp-2 (human), A549 (human), and L2 (rat) – were infected with RSV at various multiplicities of infection (m.o.i.), and stress fibers were probed for at various times by staining as described under Materials and Methods. Representative pictures clearly show that RSV-infection evoked stress fiber response in HEp-2 cells (Fig. 5; compare uninfected cells in panel A with infected cells in panel B), confirming previous findings [17]. A particularly robust induction of stress fiber was also observed in the rat L2 cells (Fig. 6, panel B). In contrast, stress fibers were completely absent in RSV-infected A549 cells (Fig. 7), suggesting that the signaling pathway that leads to stress fiber formation is missing or nonfunctional in A549 cells.
Figure 5.
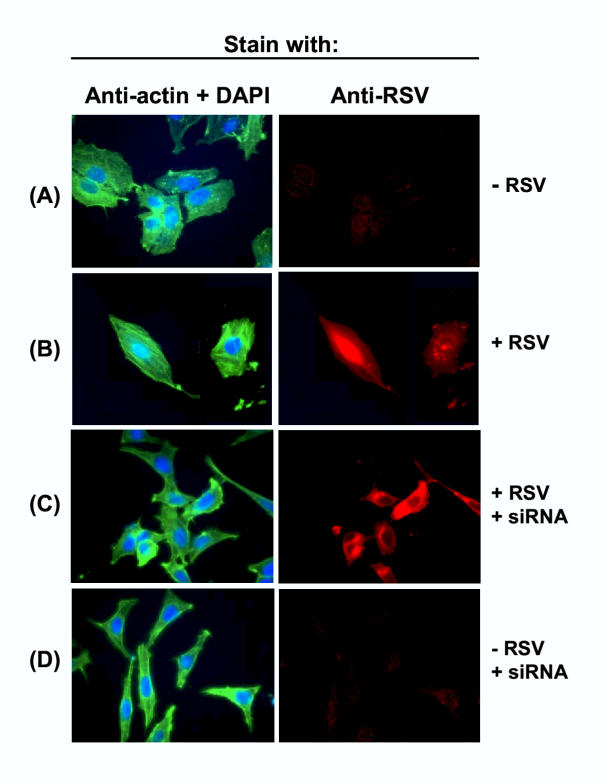
Profilin-dependent stress fiber formation in HEp-2 cells in response to RSV infection. HEp-2 monolayers were transfected with siRNA against profilin and infected with RSV as and where indicated. Actin (green), RSV antigens (red), and nuclei (blue) were stained as described in Materials and Methods. Note the long green stress fibers in panel B, and their abrogation in siRNA-treated cells in panel C.
Figure 6.
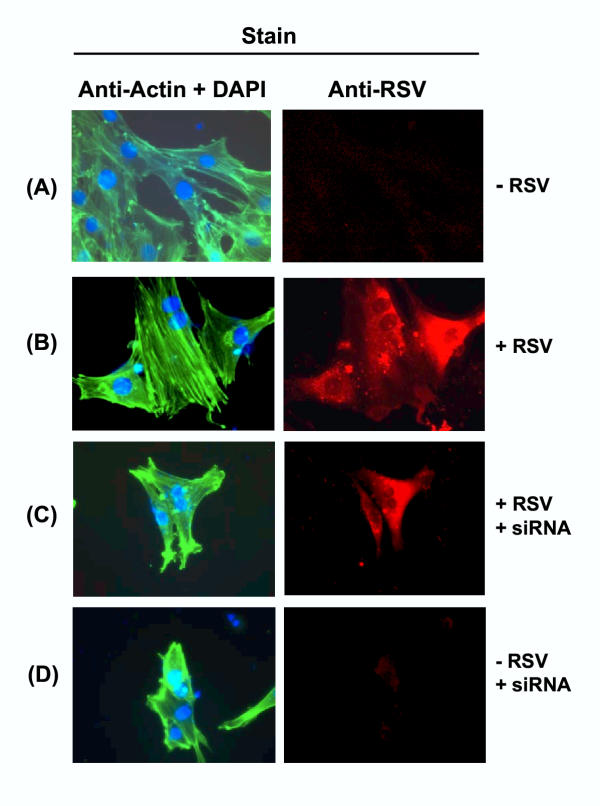
Profilin-dependent stress fiber formation in L2 cells in response to RSV infection. The legend is essentially as in Fig. 5.
Figure 7.

Absence of stress fiber in RSV-infected A549 cells. RSV-infected (and uninfected control) A549 cells were stained for nuclei, actin filament, and RSV as described for Fig. 5.
In order to investigate whether the formation of actin stress fibers requires profilin, we knocked down profilin in the two cells where stress fibers were seen, namely, HEp-2 and L2. Upon RSV infection, the profilin-depleted cells failed to produce stress fibers (Panel C in Figs. 5, 6), suggesting that stress fiber formation is a profilin-dependent process. However, the loss of profilin had no detectable effect on intracellular RSV replication, as determined by immunostaining of the cells with anti-RSV antibody. It also did not have any detrimental effect on growth or morphology of the host cells e.g., (Panel D in Figs. 5, 6), at least up to 70 hrs of cell culture.
As stress fiber induction is a relatively early event in virus infection, one might wonder whether it plays an important role in intracellular virus growth, especially in the late phases. However, the results of Western blot, real-time PCR, and immunostaining clearly revealed that RSV replicated equally well inside all three cells, despite the fact that two of these cells (HEp-2, L2) evoked stress fibers whereas the third (A549) failed to do so. Moreover, all three cell lines elaborated very similar titers of infectious progeny virus in the range of 1–3 × 107 pfu /ml (e.g., Table 1). Combining all these results with the previous ones [14-17], we conclude that profilin is important for stress fiber formation, which is triggered by an interaction between viral F protein and cellular RhoA. However, neither profilin nor stress fibers may be essential for the intracellular replication of RSV.
Table 1.
Requirement of profilin for viral maturation
| Host cell | Extracellular viral titer (Pfu/ml) | ||
| Untreated | Profilin siRNA | Luciferase siRNA | |
| HEp-2 | 3 × 107 | 5 × 104 | 2 × 107 |
| A549 | 2 × 107 | 6 × 104 | 1.5 × 107 |
Monolayers of HEp-2 or A549 at low cell density (about 25% confluency) were infected with RSV Long at an m.o.i. of 3, in the presence or absence of the indicated siRNA (400 nM). The liberated virus was titered on HEp-2 cells using standard procedures. The coefficients have been rounded to the nearest integer and variations have been omitted for the sake of simplicity. Each number is an average of at least three experiments whereby variations never exceeded 20%.
Profilin is essential for syncytium formation and virion maturation
As stated earlier, the syncytium has been considered important for virus spread and pathology [17]. Because profilin was obviously important in bringing about cytoskeletal changes, it was logical to ask whether profilin also contributes to the fusion of cell membranes. To answer this question, we knocked down profilin by siRNA as before, infected the cells with RSV, and looked for syncytia at various time points. Representative results at 48 hr post-infection are presented in Fig. 8, showing that while syncytia occurred in all three control cells untreated with the siRNA, there were no syncytia in the profilin-depleted cells. However, the infected cells still exhibited a general cytopathic effect because, as we had shown earlier (e.g., Fig. 4), the intracellular growth of the virus occurred at about 85–90% efficiency in the absence of profilin.
Figure 8.
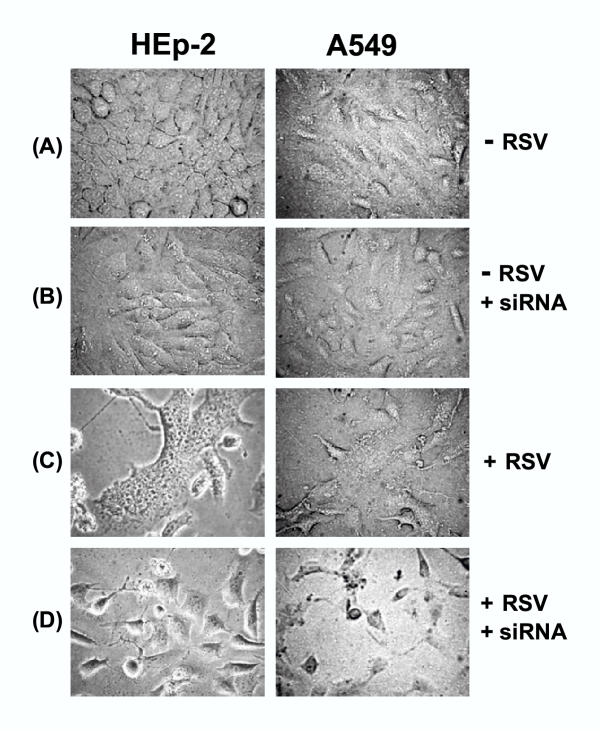
Requirement of profilin in RSV-induced cell fusion. Photomicrographs of HEp-2 and A549 monolayers were taken at 48 h post-infection as described under Materials and Methods. siRNA indicates pre-treatment with anti-profilin siRNA. Note the fused cells (syncytia) in panel C, and cytopathic effect (sick, rounded cells) but no syncytia in panel D. Also note that the siRNA alone had no discernible effect on cell morphology.
The next obvious question was whether profilin is required for the final steps of the viral life cycle, namely virus maturation and egress. To answer this, we measured the titer of infectious virions liberated from the three kinds of profilin-depleted cells infected after 10 hr of siRNA-treatment. A representative set of results (Table 1) show that such cells produced nearly a thousand-fold less infectious virion. Longer pre-treatment with siRNA prior to the addition of the virus did not lead to further reduction of progeny yield, suggesting that 10 hr of siRNA-treatment was sufficient to achieve maximal reduction. Virus titers were not affected in control cells treated with siRNA against luciferase mRNA [24].
To examine the protein profile of the extracellular virions released from profilin-depleted and undepleted cells, the culture media of the two cells were collected and the virions were concentrated by sedimentation at 110,000 × g. As expected, a much larger volume of the profilin-depleted cell media was needed to produce a comparable amount of virus pellet. However, when equal amounts (by protein assay) of virus were analyzed by Western blot, the two kinds of virions exhibited an identical profile of viral proteins, including F (data not shown). In contrast, while the virions from untreated cells contained substantial amounts of profilin, confirming our previous finding [11], those from profilin-depleted cells were found to be essentially devoid of profilin (data not shown). Thus, it appeared that the small number of profilin-free virus particles that were released from profilin-depleted cells retained infectivity. Clearly, a more detailed and quantitative analysis will be needed to determine the relationship between the profilin content and infectivity of a virus particle. Nevertheless, we can conclude that profilin plays an essential role in the optimal yield of mature virions from all cells tested.
Discussion
Results in this communication point to the existence of a number of potential cellular and viral interactive pathways in which profilin may play a cardinal role. In fact, profilin was found to be essential for all three effects tested: viral maturation, cell fusion, and stress fiber formation. Based on our findings and relevant published data, we present a working model in Fig. 9. For the sake of simplicity, we have depicted profilin as functioning through actin in all these pathways. This is certainly true for stress fiber induction, as the stress fibers are physically made of filamentous actin. For syncytium formation and viral maturation, however, we could not rule out the possibility that profilin might also have a direct role. Knock-down of actin completely destroyed the cell's architecture [26], and therefore, was not a meaningful approach in these studies. Similarly, although the viral F protein is depicted as an important trigger for many pathways in this model (Fig. 9), other viral proteins and many cellular entities may act as important mediators at various steps. In the following sections we discuss these biochemical pathways in some detail.
Figure 9.
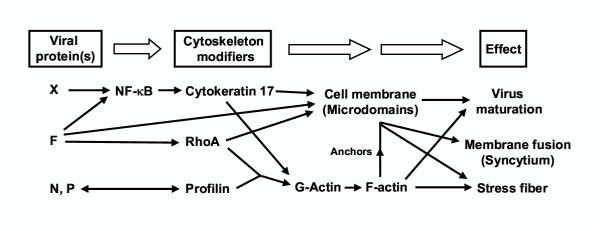
Model for host-RSV interacting pathways involving profilin (see Results and Discussion for details). F is postulated to activate cellular NF-κB, leading to the activation of a large family of NF-κB-responsive genes, including cytokeratin-17 [44]. NF-κB may also be activated by other RSV proteins, such as P [21]. F also activates RhoA, and this may lead to long actin stress fibers in selected cells, and perhaps shorter actin filaments in all cells, which may be important for cell fusion. The double-headed arrow between N/P and profilin means that we simply know that they interact, but not know the directionality of signaling between them. Membrane microdomains may serve as sites of assembly for virion particles, and anchoring of all forms of F-actin, RhoA, cytokeratin-17, and possibly other cytoskeletal components such as caveolin-1. Together, profilin is responsible for at least three morphologically visible "effects" shown on far right. Although profilin is depicted to work through actin, the possibility that profilin may directly participate in these effects is not excluded. Other actin-regulatory proteins may also play additional roles, but are not shown.
The membrane microdomains shown in Fig. 9 have recently received much attention as gateways of trafficking and signaling [27,28]. Also called membrane rafts or lipid rafts, they are specific membrane sites, rich in cholesterol and sphingolipids. These microdomains recruit an impressively large number of cellular proteins and regulatory molecules such as Rho, profilin, actin, caveolin-1, CD44, and phosphotidylinositol 4,5-biphosphate (PIP2), and have been shown to be important in a variety of cellular functions in health and disease [27-29]. Recently, a number of pathogens have been shown to hijack these sites for their entry, exit, and assembly; such agents include many RNA viruses such as HIV, measles virus, influenza virus, and RSV [29-33]. In a very recent study [34], filamentous RSV was confirmed to exit through membrane microdomains, and immunological analysis placed RSV proteins F, N, and P at these sites. Combining these findings, we propose that the activated RhoA utilizes profilin to bring about a net polymerization of actin in the RSV-infected cells (Fig. 9). The actin filaments may anchor to specific membrane microdomains, and together, both of them make important contributions to RSV morphogenesis and host-RSV signaling.
The cellular role or significance of stress fibers in general has remained uncertain. In fact, stress fibers could very well be artifacts of adherent growth on glass or plastic surfaces, as they are rarely found in cells in tissues. As we stated earlier, the absence of stress fibers in the A549 cell suggests that stress fibers per se are not essential for RSV growth either, and that one or more steps of the Virus (F) → RhoA → profilin → actin signaling pathway is perhaps missing or nonfunctional in A549. We have not found any obvious difference in the expression of RSV proteins, including F, between the stress-fiber responsive and unresponsive cells (for example, compare the "0" siRNA lanes of Panel C and D in Fig. 4). The cellular proteins in this pathway are also evolutionarily conserved, and indeed, HEp-2 and A549 cells express comparable amounts of RhoA, profilin, and actin (data not shown). The functionality of the RhoA of A549 cells is substantiated by a number of previous studies, such as the need for RhoA in bradykinin-mediated activation of NF-kappa B [35], and the abrogation of H2O2-mediated activation of JNK by RhoA antisense oligonucleotide [36]. Profilin purified from A549 cells is also indistinguishable from HEp-2 cell profilin, in terms its molecular weight, pI, affinity for poly-proline, and ability to stimulate actin-dependent transcription of RSV genome in vitro [11]. Thus, the inability of A549 cells to elicit actin stress fibers is likely due to an as yet unidentified factor or pathway that may be altered or missing in the A549 cells. Evidently, a detailed analysis of the relevant pathways in A549 and HEp-2 cells promises to shed light on this difference and lead to the identification of the missing function.
Regarding the syncytium, it is well established that the F protein plays an active role in its formation. Based on the premise described above, it is conceivable that the membrane microdomains may serve as nucleation points for the initial mixing of the membrane lipids with help from F, and this process may then travel through the membrane, eventually producing a completely fused cell mass (syncytium). In this paper, we have shown an importance of profilin in this process. If this function of profilin is mediated through actin, results from A549 cells suggest that the actin filaments (F-actin) involved in syncytium must be smaller that stress fibers, and not discernible by light microscopy. It is possible that short F-actin polymers, anchored at the microdomains, trigger syncytium. This is so proposed in Fig. 9.
Although syncytium is a common and one of the most visible morphological outcomes of infection by many viruses, including Paramyxoviruses [12], its relevance in virus replication has remained speculative. Extrinsic chemical blockers of the F protein inhibited the very first step in infection, i.e., attachment of the virus to the cell [37], suggesting an important role of F in this step. In another study, a different approach was taken to inhibit the intracellular function of F. The domain of RhoA that binds to F was mapped to RhoA residues 77–95 [14]. In cell culture, a peptide of this sequence inhibited syncytium and cell-to-cell spread of the virus, suggesting that the two might be related, and that syncytium formation required signaling through RhoA [14]. The same peptide, however, also inhibited human parainfluenza virus type 3 (HPIV-3), the reason for which was not obvious. Moreover, it remained unclear whether the signals that eventually led to syncytia had any effect of intracellular virus replication or maturation. To answer these questions, we have taken yet another approach in this paper by targeting profilin. However, we observed profilin-dependence of viral maturation even in sub-confluent monolayers where the cells were well separated, as well as in single-burst experiments (data not shown). Thus, we suggest that profilin has a direct role in virus maturation independent of its role in cell fusion. We would like to believe that syncytium is an outcome of viral infection, and as such, is unimportant for viral maturation in single cells. We do not rule out, however, that cell fusion may accelerate virus spread through closely packed cells such as in an infected tissue. Again, it is quite possible that the microdomain-anchored short actin filaments, proposed above, may allow the docking and assembly of subviral particles inside the infected cell. Perhaps the observed interaction of profilin with N and P facilitates the transfer of the viral ribonucleoprotein to these filaments. In the final phase of virus replication, these same short F-actins may also assist in virus locomotion and exit through the cell membrane. As mentioned, recent studies have indeed shown that budding of progeny RS virions occurs primarily through the membrane microdomains [34].
To sum up, profilin can act as a major liaison between viral proteins and actin, and articulate multiple outcomes for both the cell and the virus, involving signal transduction as well as cytoskeletal changes (Fig. 9). Studies in yeast and Drosophila indicated that profilin is generally nonessential for cellular viability, although profilin mutants have phenotypes. In yeast, profilin null mutations result in cells that are viable at permissive temperature, but grow more slowly, are larger than wild type, and are often multinucleate [38]. The chickadee (chic) gene encodes Drosophila profilin and chic mutations produce sterile female flies [39,40]. In both species, profilin mutations resulted in defects in actin polymerization [38-40]. In addition to regulating actin assembly, a large body of in vitro and in vivo evidence suggests a role for profilin in signal transduction, mainly involving PIP2, a major second messenger that strongly binds to profilin [41] and localizes to the membrane microdomains described earlier. Thus, in addition to the actin pathway, the phospholipase C → PIP2 pathway may also be involved in profilin's role in host-RSV interaction. In either case, the relative nonessentiality of profilin in cellular viability makes it an attractive drug target for the intervention of viral pathogenesis over a short window of time. It should be noted that besides profilin, there are many other cellular proteins known to modulate actin filamentation and cytoskeletal dynamics, and profilin itself binds to a large number proteins, many with poly-proline sequences [42,43]. Clearly, identification of the various members of the cytoskeletal signaling pathway will constitute an important step in understanding host-RSV interaction, and may lead to additional targets for pathological intervention. In this paper, we provide proof-of-concept that RNA interference can be a major tool in dissecting the pathways of host-virus interaction.
Conclusions
RNA interference using sequence-specific siRNA can be successfully used to knock down profilin, a major actin-modulating protein of the eukaryotic cell. Detailed studies of infection of such cells by RSV reveals that profilin is needed for cell fusion, which is a universal response RSV infection in all cell lines tested. Profilin is also essential for the final stages of viral maturation, as profilin-depleted cells fail to release infectious progeny virus. Profilin binds to viral transcriptional proteins, N and P, but makes only a small contribution in viral transcription; perhaps these interactions allow docking of the viral RNP complex to the actin filaments. In contrast, RSV-induced actin stress fibers form only in some cells but not in others, suggesting a nonessential role of stress fibers and an involvement of cell-specific signaling molecules in this pathway. Thus, profilin is involved in virus-mediated syncytium formation and stress fiber induction, and in viral morphogenesis. It remains to be seen whether these are due to profilin's ability to reorganize the cytoskeleton, or due to a direct participation of profilin in these processes. The determination of the various interactive domains of profilin and its partners in these pathways may allow one to design anti-RSV drugs that will disrupt specific host-virus interactions without extensive cytotoxicity.
Materials and Methods
Clones, proteins, and antibodies
His-tagged P protein was expressed by IPTG-mediated induction of a pET-15b-RSV P (Long) clone in E. coli BL21(DE3). The protein was soluble, and was purified by standard Ni-chelation chromatography [45]. Purified recombinant profilin and antibodies against profilin, RSV P protein, and F protein have been described previously [11,25]. Polyclonal antibodies to RSV proteins G, N, and M were purchased from Chemicon International (Temecula, CA) and the corresponding rhodamine-conjugated secondary antibody from Sigma. The pcDNA3 clone of RSV P protein has been described earlier [21]; the N gene was similarly cloned in this vector. In transient transfection experiments, optimal amounts of these DNA were transfected into monolayers-grown cells using the Fugene 6 reagent (Roche Diagnostics, Indianapolis, IN). All RSV infections were carried out at an m.o.i. of 3 using RSV Long grown on HEp-2 cells and purified as described previously [22].
RNA interference and RT-PCR analysis
The anti-profilin double-stranded RNA (siRNA) of the following sequence were made by Dharmacon http://www.dharmacon.com:
5' AGAAGGTGTCCACGGTGGTdTdT 3'(sense)
5' ACCACCGTGGACACCTTCTdTdT 3' (antisense)
The (N)19 "core" of the sense strand [24] corresponded to nucleotide 348–366 of human profilin I ORF (Accession No. NM_005022). Transfection of monolayer cells with the siRNA and subsequent infection of the cells with RSV were carried out essentially as described [25]. Interestingly, after designing the siRNA and successfully using it to knock down profilin in rat cells, we realized that the rat profilin sequence (Accession No. X96967) differs from the human sequence by one nucleotide; the underlined G of human is A in rat. The serendipitous success of the human siRNA against rat profilin suggested that small mismatch(es) may be tolerated in RNA interference, the details of which will be published elsewhere. Substantial tolerance of one or two nucleotide mismatch in siRNA-mediated silencing of human coagulation trigger tissue factor mRNA has been reported recently [46]. When siRNA transfected cells were infected with RSV, the virus was added 10 hours after transfection.
RSV negative-strand genomic RNA and N and F mRNA from infected cells were quantified by real-time RT-PCR using specific primers described previously [25] and a Bio-Rad iCycler iQ Real-time PCR Detection System (Bio-Rad Laboratories, Hercules, CA).
Protein-protein interaction and cytological studies
For in vitro binding studies, about 5 μg of (His)6P protein was bound to 25 μl (packed volume) of Ni+2-charged NTA column using the binding buffer provided by the manufacturer (Novagen, Madison, WI). The matrix was washed four times with 100 μl of buffer A (50 mM Tris-Cl, pH 7.5, 50 mM NaCl, 10% glycerol), and appropriate purified protein or cell extract in 50 μl buffer A was added to it. Following incubation at room temperature for 10 min, the bound proteins were eluted with buffer A plus 1 M imidazole, and then analyzed by SDS-PAGE and Western blot, as and where mentioned.
Immunoprecipitation of uninfected or RSV-infected (at 18 h p.i.) cell extracts and immunofluorescence staining of fixed cells were carried out as described previously [25]. Nuclei were stained with DAPI. Immunoblot (Western) detection by ECL was done with the SuperSignal Western blotting kit (Pierce Biotechnology, Rockford, IL) [10].
Acknowledgments
Acknowledgements
This research was supported in part by a grant from National Eye Institute (EY13826) and a NRSA Fellowship (AI49682) from the National Institute of Allergy and Infectious Diseases (NIH). We thank Dr. Anthony Gard for use his immunofluorescence microscope, and Titus Barik for help with computer technology. The kind gifts of the following reagents are gratefully acknowledged: anti-F antibody (Dr. James E. Crowe, Jr., Vanderbilt University, Tennessee), recombinant human platelet profilin (Dr. Steven C. Almo, Albert Einstein College of Medicine, Bronx, New York), and anti-profilin antibody (Drs. William Zeile and Frederick Southwick, University of Florida, Florida).
Contributor Information
Vira Bitko, Email: virabitko@hotmail.com.
Anja Oldenburg, Email: anja.oldenburg@gmx.net.
Nicolle E Garmon, Email: nkoch1992@yahoo.com.
Sailen Barik, Email: sbarik@jaguar1.usouthal.edu.
References
- Braga VM. Cell-cell adhesion and signalling. Curr Opin Cell Biol. 2002;14:546–556. doi: 10.1016/S0955-0674(02)00373-3. [DOI] [PubMed] [Google Scholar]
- Welch MD, Mullins RD. Cellular control of actin nucleation. Annu Rev Cell Dev Biol. 2002;18:247–288. doi: 10.1146/annurev.cellbio.18.040202.112133. [DOI] [PubMed] [Google Scholar]
- Ploubidou A, Way M. Viral transport and the cytoskeleton. Curr Opin Cell Biol. 2001;13:97–105. doi: 10.1016/S0955-0674(00)00180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MB. Actin-based motility of intracellular microbial pathogens. Microbiol Mol Biol Rev. 2001;65:595–626. doi: 10.1128/MMBR.65.4.595-626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins PL, Murphy BR. Respiratory syncytial virus: reverse genetics and vaccine strategies. Virology. 2002;296:204–11. doi: 10.1006/viro.2002.1437. [DOI] [PubMed] [Google Scholar]
- Barik S. Transcription of human respiratory syncytial virus genome RNA in vitro: requirement of cellular factor(s) J Virol. 1992;66:6813–6818. doi: 10.1128/jvi.66.11.6813-6818.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik S, McLean T, Dupuy LC. Phosphorylation of Ser232 directly regulates the transcriptional activity of the P protein of human respiratory syncytial virus: phosphorylation of Ser237 may play an accessory role. Virology. 1995;213:405–412. doi: 10.1006/viro.1995.0013. [DOI] [PubMed] [Google Scholar]
- Yu Q, Hardy RW, Wertz GW. Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J Virol. 1995;69:2412–2419. doi: 10.1128/jvi.69.4.2412-2419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearns R, Collins PL. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J Virol. 1999;73:5852–5864. doi: 10.1128/jvi.73.7.5852-5864.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke E, Dupuy L, Wall C, Barik S. Role of cellular actin in the gene expression and morphogenesis of human respiratory syncytial virus. Virology. 1998;252:137–148. doi: 10.1006/viro.1998.9471. [DOI] [PubMed] [Google Scholar]
- Burke E, Mahoney NM, Almo SC, Barik S. Profilin is required for optimal actin-dependent transcription of respiratory syncytial virus genome RNA. J Virol. 2000;74:669–675. doi: 10.1128/JVI.74.2.669-675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutch RE, Jardetzky TS, Lamb RA. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci Rep. 2000;20:597–612. doi: 10.1023/A:1010467106305. [DOI] [PubMed] [Google Scholar]
- Techaarpornkul S, Barretto N, Peeples ME. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol. 2001;75:6825–6834. doi: 10.1128/JVI.75.15.6825-6834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastey MK, Gower TL, Spearman PW, Crowe JE, Jr, Graham BS. A RhoA-derived peptide inhibits syncytium formation induced by respiratory syncytial virus and parainfluenza virus type 3. Nat Med. 2000;6:35–40. doi: 10.1038/71503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Gower TL, Peeples ME, Collins PL, Graham BS. RhoA is activated during respiratory syncytial virus infection. Virology. 2001;283:188–196. doi: 10.1006/viro.2001.0891. [DOI] [PubMed] [Google Scholar]
- Pastey MK, Crowe JE, Jr, Graham BS. RhoA interacts with the fusion glycoprotein of respiratory syncytial virus and facilitates virus-induced syncytium formation. J Virol. 1999;73:7262–7270. doi: 10.1128/jvi.73.9.7262-7270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Zhang Y, Luxon BA, Garofalo RP, Casola A, Sinha M, Brasier AR. Identification of NF-kappaB-dependent gene networks in respiratory syncytial virus-infected cells. J Virol. 2002;76:6800–6814. doi: 10.1128/JVI.76.13.6800-6814.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler MA, Wernke-Dollries K. Incomplete regulation of NF-kappaB by IkappaBalpha during respiratory syncytial virus infection in A549 cells. J Virol. 1999;73:4502–4507. doi: 10.1128/jvi.73.5.4502-4507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde JG, He B, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-kappa B and NF-IL-6. J Infect Dis. 1996;174:262–267. doi: 10.1093/infdis/174.2.262. [DOI] [PubMed] [Google Scholar]
- Bitko V, Barik S. Persistent activation of RelA by respiratory syncytial virus involves protein kinase C, underphosphorylated IkappaBbeta, and sequestration of protein phosphatase 2A by the viral phosphoprotein. J Virol. 1998;72:5610–5618. doi: 10.1128/jvi.72.7.5610-5618.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitko V, Velazquez A, Yang L, Yang YC, Barik S. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-kappa B and is inhibited by sodium salicylate and aspirin. Virology. 1997;232:369–378. doi: 10.1006/viro.1997.8582. [DOI] [PubMed] [Google Scholar]
- Banerjee AK, Barik S, De BP. Gene expression of nonsegmented negative strand RNA viruses. Pharmacol Ther. 1991;51:47–70. doi: 10.1016/0163-7258(91)90041-J. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Bitko V, Barik S. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol. 2001;1:34. doi: 10.1186/1471-2180-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114:4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI200216390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A. Peering inside lipid rafts and caveolae. Trends Biochem Sci. 2002;27:435–437. doi: 10.1016/S0968-0004(02)02178-3. [DOI] [PubMed] [Google Scholar]
- van der Goot FG, Harder T. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin Immunol. 2001;13:89–97. doi: 10.1006/smim.2000.0300. [DOI] [PubMed] [Google Scholar]
- Campbell SM, Crowe SM, Mak J. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J Clin Virol. 2001;22:217–227. doi: 10.1016/S1386-6532(01)00193-7. [DOI] [PubMed] [Google Scholar]
- Manie SN, Debreyne S, Vincent S, Gerlier D. Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J Virol. 2000;74:305–311. doi: 10.1128/jvi.74.1.305-311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S, Ali A, Hui EK, Adhikary L, Nayak DP. Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses. Virus Res. 2001;77:61–69. doi: 10.1016/S0168-1702(01)00266-0. [DOI] [PubMed] [Google Scholar]
- Brown G, Rixon HW, Sugrue RJ. Respiratory syncytial virus assembly occurs in GM1-rich regions of the host-cell membrane and alters the cellular distribution of tyrosine phosphorylated caveolin-1. J Gen Virol. 2002;83:1841–1850. doi: 10.1099/0022-1317-83-8-1841. [DOI] [PubMed] [Google Scholar]
- McCurdy LH, Graham BS. Role of plasma membrane lipid microdomains in respiratory syncytial virus filament formation. J Virol. 2003;77:1747–1756. doi: 10.1128/JVI.77.3.1747-1756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZK, Christiansen SC, Ptasznik A, Zuraw BL. Requirement of phosphatidylinositol 3-kinase activity for bradykinin stimulation of NF-kappaB activation in cultured human epithelial cells. J Biol Chem. 1999;274:9918–9922. doi: 10.1074/jbc.274.15.9918. [DOI] [PubMed] [Google Scholar]
- Roberts ML, Cowsert LM. Interleukin-1β and reactive oxygen species mediate activation of c-Jun NH2-terminal kinases, in human epithelial cells, by two independent pathways. Biochem Biophys Res Commun. 1998;251:166–172. doi: 10.1006/bbrc.1998.9434. [DOI] [PubMed] [Google Scholar]
- Razinkov V, Huntley C, Ellestad G, Krishnamurthy G. RSV entry inhibitors block F-protein mediated fusion with model membranes. Antiviral Res. 2002;55:189–200. doi: 10.1016/S0166-3542(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Haarer BK, Lillie SH, Adams AE, Magdolen V, Bandlow W, Brown SS. Purification of profilin from Saccharomyces cerevisiae and analysis of profilin-deficient cells. J Cell Biol. 1990;110:105–114. doi: 10.1083/jcb.110.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley L, Verheyen E, Ayers K. chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell. 1992;69:173–184. doi: 10.1016/0092-8674(92)90128-y. [DOI] [PubMed] [Google Scholar]
- Verheyen EM, Cooley L. Profilin mutations disrupt multiple actin-dependent processes during Drosophila development. Development. 1994;120:717–728. doi: 10.1242/dev.120.4.717. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont PJ, Machesky LM, Baldassare JJ, Pollard TD. The actin-binding protein profilin binds to PIP2 and inhibits its hydrolysis by phospholipase C. Science. 1990;247:1575–1578. doi: 10.1126/science.2157283. [DOI] [PubMed] [Google Scholar]
- Wear MA, Schafer DA, Cooper JA. Actin dynamics: assembly and disassembly of actin networks. Curr Biol. 2000;10:R891–5. doi: 10.1016/S0960-9822(00)00845-9. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Rosenberg HF. Cytokeratin 17 is expressed in cells infected with respiratory syncytial virus via NF-kappaB activation and is associated with the formation of cytopathic syncytia. J Infect Dis. 2000;182:1022–1028. doi: 10.1086/315841. [DOI] [PubMed] [Google Scholar]
- Dobson S, Kar B, Kumar R, Adams B, Barik S. A novel tetratricopeptide repeat (TPR) containing PP5 serine/threonine protein phosphatase in the malaria parasite, Plasmodium falciparum. BMC Microbiol. 2001;1:31. doi: 10.1186/1471-2180-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holen T, Amarzguioui M, Wiiger MT, Babaie E, Prydz H. Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res. 2002;30:1757–1766. doi: 10.1093/nar/30.8.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]