

Abstract
Prions are believed to be infectious self-propagating polymers of otherwise soluble host-encoded proteins1,2. This concept is now strongly supported by the recent findings that amyloid fibrils of recombinant prion proteins from yeast3-5, Podospora anserina6, and mammals7 can induce prion phenotypes in the corresponding hosts. However, the structural basis of prion infectivity remains largely elusive because acquisition of atomic resolution structural properties of amyloid fibrils represents a largely unsolved technical challenge. HET-s, the prion protein of P. anserina, contains a C-terminal prion domain comprising residues 218-289. Amyloid fibrils of HET-s(218-289) are necessary and sufficient for the induction and propagation of prion infectivity6. Here, we have used fluorescence studies, quenched hydrogen exchange NMR and solid state NMR to determine the sequence specific positions of secondary structure elements of amyloid fibrils of HET-s(218-289). This revealed four β-strands constituted by two pseudo repeat sequences, each forming a β-strand-turn-β-strand motif. We show that this conformation is the functional and infectious entity of the HET-s prion by using a structure-based mutagenesis approach. These results correlate for the first time distinct structural elements with prion infectivity.
The prion form of the protein HET-s is involved in a programmed cell death phenomenon termed heterokaryon incompatibility8,9. This reaction occurs in filamentous fungi when cells of incompatible genotype fuse and form a mixed cell. In P. anserina, two incompatible genotypes, called het-s and het-S, encode for the proteins HET-s and HET-S. They are both 289 amino acids long and differ in only 13 residues10. However, only HET-s can form a prion11: P. anserina cells expressing the HET-s protein exist either in a prion state called [Het-s], or in a non-prion state called [Het-s*]. Infection of a [Het-s*] cell by a [Het-s] cell occurs by simple contact. The cell death reaction is triggered if a cell in the [Het-s] prion state fuses with a cell expressing HET-S. In contrast, a prion-free [Het-s*] cell can readily fuse with a cell expressing HET-S to form a viable mixed cell. Hence, the formation of the [Het-s] prion can easily be assayed by measuring its function in heterokaryon incompatibility. Both HET-s and the prion domain fragment HET-s(218-289) form amyloid fibrils in vitro that are infectious to [Het-s*] cells of P. anserina6,12,13. Transition to the prion state in vivo can also be detected by following the aggregation of a HET-s-green fluorescent protein (GFP) fusion protein14,15.
To elucidate the fold of the amyloid fibrils of HET-s(218-289), we first identified the sequence specific positions of regular secondary structural elements using an improved technique of quenched hydrogen exchange measured by solution NMR16 (see Methods). This technique allows the identification of solvent-protected backbone amide protons involved in hydrogen bonds. The [15N,1H] correlation NMR spectrum (Fig. 1a) contained one assigned cross-peak for each backbone amide of HET-s(218-289) with the exception of 289, enabling a residue-specific determination of the hydrogen exchange rates. Upon exchange in D2O buffer for 6 weeks the intensity of about 45% of the resonances was significantly reduced or absent from the spectrum (Fig. 1b). This suggested that the corresponding amides have undergone exchange with solvent deuterons, which are not visible in this experiment.
Figure 1.

Sequence specific determination of regular secondary structure and its topology in HET-s(218-289) fibrils. a,b, Fast HMQC-spectra of homogenously 15N-labeled HET-s(218-289) in d6-DMSO containing 0.1 % d1-TFA, corresponding to fully protonated (a), and partially hydrogen exchanged (b) amyloid fibrils. Sequence specific chemical shift assignments are labeled. Red lines encircle cross-peaks that show a virtually complete loss of intensity after t = 6 weeks. c,d, Homonuclear 13ex C-13C DREAM correlation spectra of 13C,15N-labeled, HET-s(218-289) fibrils. The carbonyl region of the DREAM spectra (c) was recorded at 40 kHz MAS. Positive contours (blue) were taken from a spectrum recorded with an up-down tangential DREAM sweep, negative contours (red) were taken from a similar spectrum but within down-up sweep. Contour levels start at ∼2.5 times rms noise level and increase by a factor 1.4. Representative traces through the 2D spectra are given in Figure S5. The aliphatic region of a DREAM spectrum (d) was recorded at 25 kHz MAS. Sequence specific chemical shift assignments are labeled. e, Plots of the observed quenched hydrogen exchange rates kex / h−1. The measurable upper limit was 5 h−1. Error bars indicate deviations from a monoexponential fit. Asterisks denote residues that could not be analyzed, blue arrows the location of β-strands. f, Plot of Δ(δ(13Cα))-Δ(δ(13Cβ)). δ(13Cα) and δ(13Cβ) are the difference between experimental 13Cα and 13Cβ chemical shifts and the corresponding ‘random coil’ chemical shifts. The line-width dependent accuracy for each value is indicated. g, Solvent accessibility of single cysteine mutants. The fluorescence intensity of Alexa Fluor 488 crosslinked to the cysteine side chains is given relative to a positive control (see methods) at the position at which the cysteine was introduced. Even-numbered positions are shown in green.
The hydrogen exchange was followed over a total period of 3 months. All residues displayed a mono-exponential decay (Fig. S1) indicating that the structure of the fibrils was well defined and homogeneous. The summarized hydrogen exchange data (Fig. 1e) show that due to exchange rates faster than 5·h-1, the 8 N-terminal residues, the 5 C-terminal residues and residues 247-261 are only weakly or not protected and may therefore be conformationally disordered. Four segments were observed that displayed slow exchange rates of 10−2·h−1 to 10−5·h−1 and were thus considered to be involved in hydrogen bonds. They comprise residues 226-234, 236-246, 262-270, and 272-282. Similarly slow exchange rates have been observed for Aβ (1-42) fibrils17 and reflect the extraordinary stability and compactness amyloid fibrils can achieve. Because circular dichroism experiments have established that the HET-s(218-289) fibrils contain mainly β-sheet secondary structure13, it is likely that the four protected segments represent four distinct β-strands. Hydrogen exchange data was also accumulated for amyloid fibrils of the full length HET-s. The protection pattern of the peptide segment 218-288 in HET-s was essentially identical to that of HET-s(218-289) (Fig. S2). This suggests that the prion domain acts as an independent folding unit.
To determine which residues are actually involved in β-sheet secondary structure, high-resolution solid-state NMR spectra of the HET-s(218-289) fibrils were recorded. The 13C-13C homonuclear DREAM18 correlation spectrum of uniformly 15N and 13C isotope labeled amyloid fibrils is shown in Fig. 1c and 1d. The resonance lines are strikingly narrow (down to 0.2 ppm) and are comparable to those of microcrystalline proteins, indicating a highly ordered atomic structure for part of the fibrils. Sequence specific chemical shift assignment could be obtained for residues 226-248 and 262-282 (with the exception of 274). The missing resonances of the remaining residues are probably a consequence of dynamical or structural heterogeneity19. Deviations of the combined 13Cα/13Cβ chemical shifts from random coil values were used to identify the type of secondary structure present in HET-s(218-289) fibrils. Negative deviations (Fig. 1f) indicated β-sheet secondary structure20 for residues 226-234, 237-244, 262-271 and 273-282, respectively. Exceptions were observed at residues 229 and 265 (see below) and, to a lesser degree, at 277. Chemical shift assignment by solid-state NMR was achieved for all residues that showed slow hydrogen exchange (Fig. 1e). The striking correlation between the exchange data (Fig. 1e) and the chemical shift data (Fig. 1f) allowed for the confident establishment of secondary structure in the HET-s(218-289) amyloid fibrils: Four β-strands comprising residues ∼226-234 (β1), ∼237-245 (β2), ∼262-270 (β3), and ∼273-282 (β4) are connected by two short loops between β1 and β2, and β3 and β4, respectively, and by an unstructured 15-residue long segment between β2 and β3.
To determine the topology of β-strands in HET-s(218-289) fibrils, we made use of the architecture of β-sheets, in which the side chains of the even-numbered residues face in one direction, and those of the odd-numbered residues in the opposite direction. Therefore, single cysteine residues were introduced at the odd- and even-numbered sides of each β-strand. Solvent accessibility was then probed by chemical crosslinking21 with a fluorescent dye using Alexa Fluor 488 C5 maleimide. The extent of solvent accessibility of 11 Cys residues is shown in Fig. 1g. The even numbered residues of each strand displayed high crosslinking rates, whereas all odd numbered sides remained unlabeled. This indicated that all β-sheets have one solvent-accessible side.
The acquired structural data (Fig. 1) allowed us to propose a fold for the HET-s(218-289) fibrils (Fig. 2): The core of the fibrils consists of two β-strand-turn-β-strand motifs comprising β1 and β2 and β3 and β4, respectively, which are interconnected by a long loop formed by residues 246-261. The two segments interact through hydrogen bonding between β1 and β3, and β2 and β4, respectively, forming two layers of parallel β-sheets. The β-sheets continue along the fibril axis through intermolecular hydrogen bonding between neighbouring molecules (Fig. 2). The proposed fold is consistent with the observed pattern of solvent accessibility, the distribution of polar and hydrophobic residues on each strand, and the short loops between β1 and β2, and β3 and β4, respectively. It also takes into account the remarkable sequence identity of 28% between the segments β1-β2 and β3-β4, which suggests a repetitive arrangement (Fig. S3). The only charged residues facing towards the inside of the fibril are K229 in β1 and E265 in β3, which are located in adjacent positions in the proposed fold, and therefore able to compensate their opposite charges. The reduced protection against hydrogen exchange of these residues as well as their non-beta solid-state chemical shifts (Fig. 1e,f) indicate a structural disturbance of the β-sheets at the positions of the proposed salt bridge.
Figure 2.
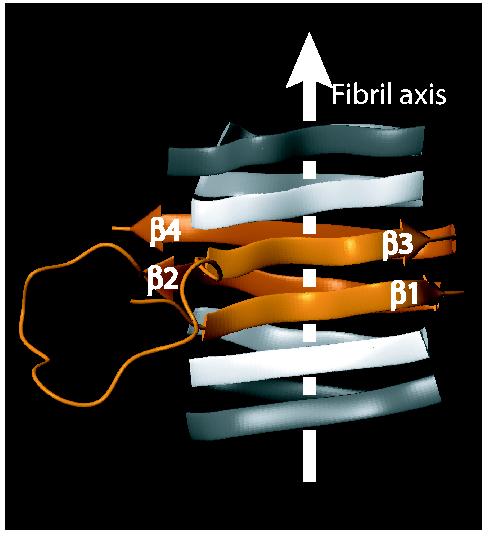
The proposed fold of the infectious conformation of HET-s(218-289) amyloid fibrils. Extend and position in primary sequence of the β-sheets is obtained from NMR data, the relative spatial position of the β-sheets is modeled taking into account the hydrophobicity of the residues, the high sequence identity between beta sheets 1 and 3, and solvent accessibility data. There are, however no direct intersheet distance constraints. A model is shown as an orange ribbon diagram, flanked by neighboring molecules indicated in white. β-sheets are indicated by arrows, non regular secondary structures by spine curves through the Cα-atoms of the corresponding residues and the fibril axis by a white arrow. The β-sheets are labeled.
In order to correlate the proposed fold of the HET-s(218-289) fibrils with [Het-s]-function and infectivity, a mutational approach was used (Wood et al. 1995). Because the fold is based on β-sheet secondary structure, we introduced proline residues, which are known to act as local β-strand breakers. A series of 13 mutants was generated with a single proline-substitution approximately every 5 residues in the 218-289 region of full length HET-s (Fig. 3a). A number of short deletion mutants affecting the central loop and the β-strand segments were also generated (Fig. 3b). P. anserina transformants expressing the mutant proteins were prion-infected by contact with a wild-type [Het-s] colony. The presence of [Het-s] prions in these transformants was then assayed by monitoring their ability to (i) produce a cell death reaction when fused to a HET-S-expressing colony and to (ii) infect a prion-free [Het-s*] colony. [Het-s]-infectivity was assayed immediately after contact of the transformants with the wild-type [Het-s] donor and again after a 3 day sub-culture (Fig. 3a and Supplementary Table 1). In order to monitor the formation of HET-s aggregates in vivo, the same mutations were introduced in a HET-s-GFP fusion protein. The mutant fusion proteins were over-expressed both in P. anserina Δhet-s knock-out strains and in a [Het-s] strain in order to determine whether co-expression with endogenous wild-type prions would lead to aggregation of the mutant fusion proteins (Fig. 3c).
Figure 3.

In vivo prion formation of HET-s proline and deletion mutants. a, proline mutants were introduced into a Δhet-s P. anserina strain and the transformants prion infected by confrontation with a [Het-s] colony. Given are the percentage of transformants producing a cell death reaction when confronted with a het-S tester colony (black) and the percentage of transformants displaying [Het-s] infectivity immediately after infection (grey) and after a 3 day sub-culture (white). Controls: vector alone (−) and wild-type HET-s (wt), mutants are labeled by the position of the proline. Zero indicates that all transformants were negative. b, Deletion mutants of HET-s were expressed in a Δhet-s strain and the transformants prion infected as above. Color code as in a. c, In vivo aggregation of HET-s-GFP fusion proteins with proline-substitutions. Wild-type and mutant HET-s-GFP fusion proteins were expressed either in a Δhet-s knock-out strain or a wild-type prion infected [Het-s] strain. Transformants of the Δhet-s background were prion infected by contact with a wild-type [Het-s] strain prior to microscopic observation (scale bar: 4 μm). Mutants located in β-strand elements are underlined. A complete data set with the numbers of individual transformants tested is given in Table S1.
In striking correlation with the structural data, proline-substitutions located in the β-strand regions strongly affected [Het-s] function, infectivity (Fig. 3a) and HET-s-GFP foci formation (Fig. 3c). In contrast, proline-substitutions in the N- or C-terminal flexible tails or the central loop did not affect [Het-s]-activity, infectivity, or HET-s-GFP foci formation. Similarly, the deletions Δ225-230, Δ244-248, Δ258-262 and Δ276-279, which affected the β-strand elements, abolished [Het-s]-infectivity. Short deletions of no more than three amino acids in the central loop did not affect [Het-s], but a larger deletion, Δ248-256, led to loss of [Het-s] function and infectivity. This indicates that a minimal length in the central loop might be required.
For some mutations in β-strand positions, [Het-s] prion infectivity could be detected transiently, but was lost upon subculturing (for instance HET-s 240P, Fig. 3a). Concomitantly, GFP foci formation of HET-s 240P was detected only when the mutant protein was co-expressed with wild-type [Het-s]-prions (Fig. 3b). This suggests that these mutant proteins retained a certain ability to form [Het-s] prions, but that they were mitotically very unstable. In addition, a number of proline-substitutions in β-strand regions and in the short connecting loop did not lead to a total loss of [Het-s]-function or infectivity, indicating that they interfered only partially with amyloid formation. Indeed, recombinant HET-s 226P and 266P retained a residual ability to form amyloid fibrils in vitro and to infect prion-free [Het-s*] colonies when introduced biolistically (Fig. S3 and Table S2). Interestingly, HET-s 246P retained [Het-s] activity. Residue 246 is still involved in hydrogen bonding, but has an α-helical rather than a β-sheet chemical shift. This suggests that it is specifically the β-sheet secondary structure that is required for HET-s function and infectivity.
Taken together, the local disruption of any of the four β-strands reduced or even abolished the formation and function of the [Het-s] prion, while the central loop only required a certain minimal length to sustain prion formation.
Our data correlate strongly the amyloid conformation with infectivity. Hence, the proposed fold of the HET-s(218-289) fibrils shown in Fig. 2 represents an infectious protein conformation. Because the [Het-s] prion has a natural function in heterokaryon incompatibility, it appears plausible that this infectious conformation was evolutionary optimised for amyloid fibril formation. This view is supported by the extraordinarily well-ordered structure as manifested in the narrow line-width observed in the solid state NMR spectra, and the monoexponential hydrogen exchange rates. In contrast, other amyloid fibrils associated with misfolding diseases appear to be structurally more heterogeneous22-25. Furthermore, the dimer-like double β-strand-turn-β-strand motif formed by the pseudo-repeats β1-β2 and β3-β4 may have evolved to optimise the polymerisation: In the case of a single β-strand-turn-β-strand motif, the initial step of fibril growth requires the diffusion-dependant formation of an oligomeric nucleus26. The covalent nature of the pseudo-repeats is therefore likely to promote this nucleation event by reducing its diffusion dependency.
It is generally believed that most prions will share β-sheet rich amyloid fibrils as a common structural feature. Here, we have now shown that a β-sheet structure is indeed the infectious prion conformation of HET-s. This link between structure and infectivity constitutes a significant step towards the elucidation of the mechanism of prion formation and propagation.
Methods
Mutagenesis
All site-directed mutants were generated with the Quick-change kit (Stratagene) using the pCB1004-het-s, pGPD-het-s-GFP14 and the pET-21a-het-s vectors as template12.
Sample preparation of stable-isotope labeled HET-s(218-289) fibrils
Stable-isotope labeled HET-s(218-289) was expressed as described for other HET-s constructs13. The bacterial pellet was solubilized in 50 mM TRIS pH 8.0 and 150 mM sodium chlorid (TCl) containing 6 M guanidinium hydrochlorid. The supernatant was centrifuged for 30 min at 18000 g. The protein was purified from the supernatant by His6-affinity chromatography and concentrated to approx. 0.5-1 mM. Fast buffer exchange to TCl-buffer yielded monomeric HET-s(218-289), which started to aggregate immediately into amyloid fibrils.
H/D exchange
15N-labeled HET-s(218-289) was used. To reduce the size of large clumps of fibrils, a suspension of the fibrils in liquid nitrogen was bruised with a mortar. To start the H/D-exchange, the fibrils were sedimented at 3600 × g for 10 minutes, washed with 50 mM TRIS pH* 8.0 containing 150 mM sodium chlorid, 5 mM dithiothreitol and D2O as the solvent, and resuspended in the same buffer. At suitable intervals, aliquots were sedimented at 24 000 × g for 2 minutes, washed with D2O and frozen in liquid nitrogen to quench H/D exchange. For the NMR-analysis the fibrils were dissociated in perdeuterated dimethylsulfoxide (d6-DMSO) containing 0.1% deuterated trifluoroacetic acid (d1-TFA). The amount of residual D2O was about 3 %. Immediately after dissolving the fibrils, a series of 80 2D [15N,1H] -correlation spectra were measured for 8 hours. To distinguish residues that display fast exchange in the fibrils, and residues that have high intrinsic exchange rates in DMSO, which would both result in absent peaks in the [15N,1H] correlation spectrum17, a second series of 80 2D spectra were measured upon addition of 3 % H20. Using this control measurement, residues 253 and 283 were excluded form the analysis. With the exception of the amide group of N289, a complete sequence specific assignment of the backbone HN/N cross peaks was obtained using the triple resonance experiments HNCA27 and HNCA(CO)NH28 applied to 13C,15N-labeled HET-s(218-289). The data were analyzed using the programs PROSA29 and CARA (http://www.nmr.ch), and a specially written Visual basic program in combination with Microsoft Excel17.
Solid state NMR
The HET-s(218-289) fibrils were washed in H2O and centrifuged into MAS rotors sealed with two-component epoxy adhesive to prevent dehydration. Solid-state NMR spectra were recorded on a Bruker AV600 spectrometer at a static field of 14.09 T. A 2.5 mm Chemagnetics probe was used for the experiments at 25 kHz MAS and a 1.8 mm probe constructed by A. Samoson30 for the experiments at 40 kHz MAS. The sample temperature was kept at 5°-15° C. For the 13C-13C correlation spectra recorded using the DREAM scheme18, the contact time of the initial adiabatic cross-polarization was 1 ms and the rf-field strengths were 70 kHz and 90 kHz for 13C and 1H at 25 kHz MAS and 90 kHz and 130 kHz for 13C and 1H at 40 kHz MAS respectively. Continuous wave decoupling was applied during the DREAM mixing step, XiX decoupling during t1 and t2 both with a rf-field amplitude of 150 kHz. The measurement time was 33 h and 55 h for the experiments at 25 kHz and 40 kHz MAS respectively. These spectra we used for the identification of spin systems and the 13C side-chain assignment. For the backbone assignment we recorded the following heteronuclear 2D correlation spectra: NCA, NCO, N(CA)CO, N(CA)CB and N(CO)CA. The assignment was done using the CARA program (http://www.nmr.ch) and confirmed via a CA-CA correlation spectrum.
Side chain solvent accessibility studies
Proteins were purified as described above. Fibrils were reduced with 20 mM DTT prior to the final buffer exchange, and renatured under nitrogen. To label solvent accessible Cys-residues by chemical crosslinking, 85 μM fibrils were incubated with a 2.5-fold molar excess of Alexa Fluor 488 C5 maleimide (Molecular Probes) for 20 min at room temperature. The fibrils were washed extensively with TCl-buffer and solubilized with 8 M spectroscopically pure guanidine hydrochlorid. For fluorimetry, the samples were diluted at least 70-fold into TCl-buffer. The Alexa Fluor 488 fluorescence (excitation 493 nm, emission 516 nm) was measured relative to the fluorescence of a single Trp residue in HET-s(218-289) (excitation 295 nm, emission 353 nm). To determine the maximum labeling efficiency, the mutant HET-s(218-289) D288C was used, which is located in the presumably solvent accessible C-terminal region showing fast H/D exchange. Wild-type HET-s(218-389) fibrils were labeled to 1,9 ± 0.1 %.
In vivo analysis of HET-s mutants
het-s mutants alleles were introduced in the Δhet-s and wild-type het-s recipient strains10. [Het-s]-incompatibility function, [Het-s]-infectivity and HET-s-GFP foci formation were analyzed as described previously15. After their initial contact with the [Het-s] priondonor, the transformants were either sub-cultured for 3 days or directly confronted with the [Het-s*] recipient in order to be able to detect transient expression of the [Het-s] state. Under the tested conditions, spontaneous emergence of [Het-s] in [Het-s*] colonies is in the range of 2%.
Supplementary Material
Acknowledgements
R.R. is a Pew scholar. This research was supported in part by grants from the National Institute of Health, the U.S.Army, the ETH Zurich and the Swiss National Science Foundation.
Footnotes
Supplementary Information accompanies the paper on Nature's website (http://www.nature.com).
References
- 1.Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid? Nature. 1967;214:764–6. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Sparrer HE, Santoso A, Szoka FC, Jr., Weissman JS. Evidence for the prion hypothesis: induction of the yeast [PSI+] factor by in vitro- converted Sup35 protein. Science. 2000;289:595–9. doi: 10.1126/science.289.5479.595. [DOI] [PubMed] [Google Scholar]
- 4.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–23. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–8. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 6.Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A. 2002;99:7402–7. doi: 10.1073/pnas.072199199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Legname G, et al. Synthetic mammalian prions. Science. 2004;305:673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 8.Glass NL, Kaneko I. Fatal attraction: nonself recognition and heterokaryon incompatibility in filamentous fungi. Eukaryot Cell. 2003;2:1–8. doi: 10.1128/EC.2.1.1-8.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saupe SJ. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol Mol Biol Rev. 2000;64:489–502. doi: 10.1128/mmbr.64.3.489-502.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turcq B, Deleu C, Denayrolles M, Begueret J. Two allelic genes responsible for vegetative incompatibility in the fungus Podospora anserina are not essential for cell viability. Mol Gen Genet. 1991;228:265–9. doi: 10.1007/BF00282475. [DOI] [PubMed] [Google Scholar]
- 11.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A. 1997;94:9773–8. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dos Reis S, et al. The HET-s prion protein of the filamentous fungus Podospora anserina aggregates in vitro into amyloid-like fibrils. J Biol Chem. 2002;277:5703–6. doi: 10.1074/jbc.M110183200. [DOI] [PubMed] [Google Scholar]
- 13.Balguerie A, et al. Domain organization and structure-function relationship of the HET-s prion protein of Podospora anserina. Embo J. 2003;22:2071–81. doi: 10.1093/emboj/cdg213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coustou-Linares V, Maddelein ML, Begueret J, Saupe SJ. In vivo aggregation of the HET-s prion protein of the fungus Podospora anserina. Mol Microbiol. 2001;42:1325–35. doi: 10.1046/j.1365-2958.2001.02707.x. [DOI] [PubMed] [Google Scholar]
- 15.Balguerie A, et al. The sequences appended to the amyloid core region of the HET-s prion protein determine higher-order aggregate organization in vivo. J Cell Sci. 2004;117:2599–610. doi: 10.1242/jcs.01116. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino M, et al. Mapping the core of the beta(2)-microglobulin amyloid fibril by H/D exchange. Nat Struct Biol. 2002;9:332–6. doi: 10.1038/nsb792. [DOI] [PubMed] [Google Scholar]
- 17.Lührs T, et al. The 3D structure of Alzheimer's Aβ(1-42) fibrils. submitted to Nature. 2005 [Google Scholar]
- 18.Verel R, Ernst M, Meier BH. Adiabatic dipolar recoupling in solid-state NMR: The DREAM scheme. Journal of Magnetic Resonance. 2001;150:81–99. doi: 10.1006/jmre.2001.2310. [DOI] [PubMed] [Google Scholar]
- 19.Siemer AB, Ritter C, Ernst M, Riek R, Meier BH. High-resolution solid-state NMR of the prion protein HET-s in its amyloid conformation. Angew Chem Int Ed Engl. 2005 doi: 10.1002/anie.200462952. in press. [DOI] [PubMed] [Google Scholar]
- 20.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–80. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 21.Javitch JA, Shi L, Liapakis G. Use of the substituted cysteine accessibility method to study the structure and function of G protein-coupled receptors. Methods Enzymol. 2002;343:137–56. doi: 10.1016/s0076-6879(02)43131-x. [DOI] [PubMed] [Google Scholar]
- 22.Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Current Opinion in Structural Biology. 2004;14:96–103. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 24.Laws DD, et al. Solid-state NMR studies of the secondary structure of a mutant prion protein fragment of 55 residues that induces neurodegeneration. Proc Natl Acad Sci U S A. 2001;98:11686–90. doi: 10.1073/pnas.201404298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaguchi K, et al. Core and heterogeneity of beta2-microglobulin amyloid fibrils as revealed by H/D exchange. J Mol Biol. 2004;338:559–71. doi: 10.1016/j.jmb.2004.02.067. [DOI] [PubMed] [Google Scholar]
- 26.Harper JD, Lansbury PT., Jr. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 27.Grzesiek S, et al. 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-gamma. Biochemistry. 1992;31:8180–90. doi: 10.1021/bi00150a009. [DOI] [PubMed] [Google Scholar]
- 28.Bracken C, Palmer AG, 3rd, Cavanagh J. (H)N(COCA)NH and HN(COCA)NH experiments for 1H-15N backbone assignments in 13C/15N-labeled proteins. J Biomol NMR. 1997;9:94–100. doi: 10.1023/a:1018679819693. [DOI] [PubMed] [Google Scholar]
- 29.Guntert P, Dotsch V, Wider G, Wuthrich K. Processing of Multidimensional NMR Data with the New Software Prosa. Journal of Biomolecular Nmr. 1992;2:619–629. [Google Scholar]
- 30.Samoson A, Tuherm T, Past J. Rotation sweep NMR. Chemical Physics Letters. 2002;365:292–299. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.