


Abstract
Homologous gene targeting is the ultimate tool for reverse genetics, but its use is often limited by low efficiency. In a number of recent studies, site- specific DNA double-strand breaks (DSBs) have been used to induce efficient gene targeting. Engineering highly specific, dedicated DNA endonucleases is the key to a wider usage of this technology. In this study, we present two novel, chimeric meganucleases, derived from homing endonucleases. The first one is able to induce recombination in yeast and mammalian cells, whereas the second cleaves a novel (chosen) DNA target site. These results are a first step toward the generation of custom endonucleases for the purpose of targeted genome engineering.
INTRODUCTION
Genome engineering has become one of the major challenges of the post-genome era, and the development of gene targeting in the 1980s (1–3) has provided the means to perform precise genome surgery. However, classical methods can prove to be very inefficient, depending on the target locus and the organism. Meganuclease-induced gene targeting is a technology wherein homologous recombination is induced by a site-specific DNA cleavage. In mammalian cells, meganuclease-induced targeted double-strand breaks (DSBs) enhanced gene targeting yields by several orders of magnitude (4–8) [up to 10–2 of the total transfected cells can in fact be targeted (8)]. DSBs induced by meganucleases also allow efficient targeting in plants and insects (9–12).
Meganucleases are endonucleases, which recognize large (12–45 bp) DNA target sites (13). In the wild, meganucleases are essentially represented by homing endonucleases, a large family of DNA nucleases found in phages, bacteria, archaebacteria and various eukaryotes (14–16). Homing endonucleases are often encoded by introns or inteins behaving as mobile genetic elements. They recognize sites that usually correspond to intron-free or intein-free genes, where they produce a DSB. Eventually, DSB repair by homologous recombination with an intron- or intein- containing gene results in the insertion of the intron or intein where DSB occurred. Early studies on HO-induced mating type switching in budding yeast (17,18), and the I-SceI (19–22) and I-TevI (23–26) homing endonucleases have paved the way for meganuclease-induced gene targeting (4–8).
Meganuclease-induced gene targeting has, however, one major limitation: the target locus must contain a meganuclease cleavage site. Therefore, as a first step in all current applications, it is required to introduce that cut site. Wider uses of natural meganucleases as universal tools for gene targeting applications would depend on the possibility of finding enzymes whose target sequences are already contained at the target loci. This is most unlikely, since the natural repertoire of homing endonucleases is currently limited to about 300 proteins, most of them still hypothetical or uncharacterized. It thus becomes necessary to resort to novel, artificial meganucleases, for which specificity has been tuned according to target choice. One approach is to focus on structure-based, rational design.
Detailed three-dimensional structures have been obtained for four proteins of the LAGLIDADG family (27–36), the largest of the four homing endonucleases families. The hallmark of these proteins is a well conserved LAGLID ADG peptide motif, termed (do)decapeptide, found in one or two copies. Homing endonucleases with only one such motif, such as I-CreI (37) or I-CeuI (38), function as homodimers. In contrast, larger proteins bearing two (do)decapeptide motifs, such as I-SceI (19), PI-SceI (39) and I-DmoI (40), are single chain proteins. Despite an apparent lack of sequence conservation (overall pairwise sequence homology below 25%), structural comparisons indicate that LAGLIDADG proteins, should they cut as dimers or single chain proteins, adopt a similar active conformation. In all structures, the LAG LIDADG motifs are central and form two packed α-helices where a 2-fold (pseudo-)symmetry axis separates two monomers or apparent domains. On either side of the LAGLIDADG α-helices, a four-stranded β-sheet provides a DNA binding interface that drives the interaction of the protein with a half site of the target DNA sequence [clearly shown by structures of I-CreI with target DNA (28,29)].
In order to challenge the modularity of the interaction between DNA and domains of LAGLIDADG endonucleases, we designed a first series of novel meganucleases by swapping or fusing different homing endonucleases domains. I-CreI was chosen as the template, as this protein remains active in a broad range of temperature conditions (37) and its interaction with DNA is well characterized (28,29). A single chain version of I-CreI (scI-CreI) was modeled and engineered. scI-CreI cleaves its cognate DNA substrate in vitro and induces homologous recombination both in yeast and mammalian cells. In addition, we created a chimeric endonuclease (DmoCre) by fusing to I-CreI one of the two I-DmoI domains. DmoCre is functional, and cleaves the expected hybrid DNA target.
These results and other recent work (41) demonstrate that functional novel meganucleases can be designed by domain swapping or fusion of homing endonuclease domains, and suggest that many other nucleases with novel specificities could be designed in this way for the purpose of genome engineering.
MATERIALS AND METHODS
Modeling
All modeling and drawings of protein structures were realized using ICM (42). The structures from I-CreI and I-DmoI correspond to PDB entries 1g9y and 1b24, respectively. Residue numerations in the text always refer to these structures, except for residues in the second I-CreI protein domain of the homodimer (residue numbers were set as for the first domain).
Gene synthesis
PCR-based assembly of a series of overlapping oligonucleotides was used to construct a gene coding for I-DmoI, while the single chain synthetic I-CreI gene was designed and sent for synthesis to Entelechon GmbH. An I-CreI gene was then obtained by PCR amplification of a fragment corresponding to the second protein domain within the single chain I-CreI gene. The DmoCre hybrid gene was built by PCR-assembly using a pair of overlapping primers corresponding to the swap region. All genes were written with codons favoring protein expression in prokaryotic systems (I-DmoI and I-CreI sequences correspond to the protein sequences in structures 1b24 and 1g9y, respectively). Sequences can be obtained upon request.
Protein expression and purification
His-tagged proteins were over-expressed in Esherichia coli BL21 (DE3) cells using pET-24d (+) vectors (Novagen). Induction with IPTG (1 mM) was performed at 25°C. Cells were sonicated in a solution of 25 mM HEPES (pH 8) containing protease inhibitors (complete EDTA-free tablets, Roche) and 5% (v/v) glycerol. Cell lysates were centrifuged twice (15 000 g for 30 min). His-tagged proteins were then affinity-purified, using 5 ml Hi-Trap chelating columns (Amersham) loaded with cobalt. Several fractions were collected during elution with a linear gradient of imidazole (up to 0.25 M imidazole, followed by plateau at 0.5 M imidazole and 0.5 M NaCl). Protein-rich fractions (determined by SDS–PAGE) were concentrated with a 10 kDa cut- off centriprep Amicon system. The resulting sample was eventually purified by exclusion chromatography on a Superdex75 PG Hi-Load 26-60 column (Amersham). Fractions collected were submitted to SDS–PAGE and analysed for cleavage activity (for I-CreI and I-DmoI). Selected protein fractions were concentrated and dialyzed against a solution of 25 mM HEPES (pH 7.5) and 20% (v/v) glycerol.
Far-UV circular dichroism and temperature-induced equilibrium unfolding
Circular dichroism measurements were performed on a Jasco J-810 spectropolarimeter using a 0.1 cm path length quartz cuvette. Equilibrium unfolding was induced increasing temperature at a rate of 1°C/min (using a programmable Peltier thermoelectric). Samples were prepared by dialysis against 25 mM potassium phosphate buffer, pH 7.5, at protein concentrations of 17, 11, 37.5 and 41 µM for I-CreI, scI-CreI, I-DmoI and DmoCre, respectively.
In vitro cleavage assays
pGEM plasmids with single meganuclease DNA target cut sites (see Fig. 5A for the sequences) were first linearized with XmnI. Cleavage assays were performed at 37 or 65°C in 12.5 mM HEPES (pH 8), 2.5% (v/v) glycerol and 10 mM MgCl2. The concentrations were 10 nM for the substrate, 0.7 µM for I-DmoI, scI-CreI and DmoCre, 1.5 µM for I-CreI. Reactions were stopped after 1 h by addition of 0.1 vol of 0.1 M Tris–HCl (pH 7.5), 0.25 M EDTA, 5% (w/v) SDS and 0.5 mg/ml proteinase K and incubation at 37°C for 20 min. Reaction products were examined following separation by electrophoresis in 1% agarose gels.
Figure 5.





(Previous page and above) In vitro cleavage assays. Plasmid linearized with XmnI is 3.2 kb long. Subsequent cutting with meganucleases results in two bands of 2.1 and 1.1 kb. (A) I-CreI, I-DmoI and hybrid DNA target cut sites. The cleavage positions are indicated on each strand for the I-CreI target cut by I-CreI or scI-CreI (both proteins giving a similar pattern of cleavage), the I-DmoI target cut by I-DmoI, and the C1D2 target cut by DmoCre. (B) Cutting of an I-CreI DNA target site by I-CreI and scI-CreI at 37°C. (C) Cutting activity of the DmoCre hybrid at 37 and 65°C. (D) Kinetics of cleavage, at 37°C, for I-CreI and scI-CreI. Initial substrate DNA concentrations were 21.8 nM, and enzyme concentrations were varied as indicated; the time-dependent fluorescence increases were used to determine kobs (from equation 1). (E) Plots of kobs versus enzyme concentrations used to determine single turnover catalytic rates. Continuous lines represent the dependence on enzyme concentration modeled with equation 2 and best-fit parameters.
On-line cleavage experiments
The method of Eisenschmidt et al. (43) was adapted to monitor cleavage of double-stranded I-CreI target DNA. Quenched fluorescent DNA substrate was prepared by hybridization of the following three oligonucleotides (purchased from Eurogentec): (i) fluorescein-(linker)-5′-TCAAAACGTCTCGAGACAGTTTGGAAGCTTATGCTTAGTTGG-3′; (ii) 5′-CCAACTAAGCATAAGCTTCCAAACT GTCTCGAGACGTTTTGAGATGCGCCGCGTGCGGCT-3′ and (iii) 5′-AGCCGCACGCGGCGCATC-3′-(linker)-DarkQuencher (the I-CreI target site is underlined). Cleavage reactions were carried out at 37°C with excess enzyme over substrate (single turnover kinetics) (44), in the wells of a flat bottom 96-well microtiter plate. Fluorescence was acquired during cleavage reactions using a fluorescence-plate reader (Fluoroskan Ascent from Thermolabsystem) with excitation and emission wavelength, respectively, set at 485 and 538 nm. Reactions were initiated by addition of substrate DNA and magnesium to enzyme samples. Final samples contained 12.5 mM HEPES (pH 7.5), 10 mM MgCl2 and 21.8 nM substrate DNA (total sample volumes were 110 µl). For each enzyme concentration, at least three separate experiments were used to obtain kobs values (we assume that cleavage is a first-order process) by modeling fluorescence curves data with equation 1:
fluorescence = a + bt + c [1 – exp(–kobs t)]1
where t is the reaction time (min), a the initial sample fluorescence (a.u.), c the fluorescence increase upon total product formation and the term bt is used to correct for an apparent non-specific linear increase in fluorescence. Eventually, modeling the dependence of kobs on enzyme concentrations [E] with equation 2 allowed determining apparent kcat* and Km*:
kobs = (kcat* [E])/(Km* + [E])2
Mapping of the cleavage sites
After cutting with XmnI and I-CreI, I-DmoI, scI-CreI or DmoCre, the digestion products were run on agarose gels. In each case, the two XmnI-meganuclease bands were excised and purified from the gel, and sequenced with SP6 and T7 primers. The sequences obtained in this way corresponded to the sequence of the top strand, 5′ of the cleavage site, and the sequence of the bottom strand, 3′ of the cleavage site. In each case, the sequence was found to be swiftly interrupted at a position that we inferred to correspond to the cleavage site.
Yeast colorimetric assay
Our yeast transformation method has been adapted from previous protocols (45). For staining, we used a classic qualitative X-Gal Agarose Overlay Assay. Each plate was covered with 2.5 ml of 1% agarose in 0.1 M sodium phosphate buffer, pH 7.0, 0.2% SDS, 12% dimethyl formamide, 14 mM β-mercaptoethanol, 0.4% X-Gal, at 60°C. Plates were incubated at 37°C.
Mammalian cells assays
COS cells were transfected with Superfect transfection reagent according to the manufacturer’s (Qiagen) protocol. Seventy-two hours after transfection, cells were rinsed twice with 1× PBS and incubated in lysis buffer (Tris–HCl 10 mM pH 7.5, NaCl 150 mM, Triton X-100 0.1%, BSA 0.1 mg/ml, protease inhibitors). Lysate was centrifuged and the supernatant used for protein concentration determination and β-galactosidase liquid assay. Typically, 30 µl of extract was combined with 3 µl Mg 100× buffer (MgCl2 100 mM, β-mercaptoethanol 35%), 33 µl ONPG 8 mg/ml and 234 µl sodium phosphate 0.1 M pH 7.5. After incubation at 37°C, the reaction was stopped with 500 µl of 1 M Na2CO3 and OD was measured at 415 nm. The relative β-galactosidase activity is determined as a function of this OD, normalized by the reaction time and the total protein quantity.
RESULTS
Design of the single chain I-CreI meganuclease
I-CreI from Chlamydomonas reinhardtii (Fig. 1A) is a small LAGLIDADG homing endonuclease that has a single (do)decapeptide motif and dimerizes into a structure similar to that of larger, single chain LAGLIDADG proteins. To engineer a single chain version of I-CreI (scI-CreI), two I-CreI copies were fused. This required placing a linker region between the two domains, and a significant part of the I-CreI protein had to be removed at the end of the domain preceding the linker (Fig. 2).
Figure 1.

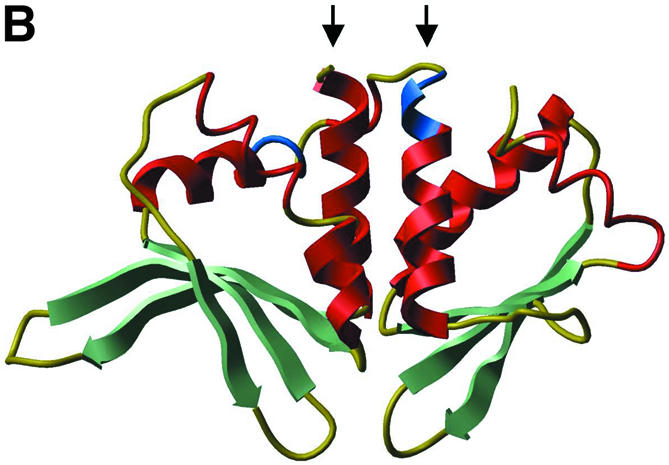
(A) Ribbon representation of I-CreI with target DNA (PDB code 1g9y). Residues 93–95 (first domain) and 7–9 (second domain) are shown in blue. (B) Ribbon representation of I-DmoI (PDB code 1b24). Residues 93–95 (first domain) and 104–106 (second domain) are shown in blue. In both (A) and (B), as well as following figures, β-strands are in green and α-helices in red. Beginnings of LAGLIDADG α-helices (residues Asn6 to Asp20 of I-CreI and Ser8 to Gly20 or Glu105 to Glu117 in I-DmoI) are marked with arrows.
Figure 2.


(A) Ribbon representation of I-CreI (PDB code 1g9y). (B) A model of single chain I-CreI. Parts drawn in blue in (A) (the C-terminal α-helices, from residue 94 in the first I-CreI domain and up to residue 7 in the second one) are excluded from the single chain protein, where a linker taken from I-DmoI, shown in blue in (B), replaces them.
The three-dimensional structure of I-DmoI from Desulfurococcus mobilis (Fig. 1B) is comparable with that of I-CreI, with the exception that I-DmoI comprises a linker region that leads from one apparent domain to the other. The boundary of that linker finely matches related main chain atoms of the I-CreI dimer. In the first domain, residues 93–95 from the third α-helices of I-CreI and I-DmoI (prior to the linker) are structurally equivalent and at the beginning of the second LAGLIDADG α-helix (second domain), I-DmoI residues 104–106 correspond to I-CreI residues 7–9 (compare Fig. 1A and B). In addition, Leu95 and Glu105 from I-DmoI have conserved identities in I-CreI, and I-DmoI residue Arg104 aligns with another basic residue in I-CreI (Lys7). Thus, the scI-CreI was designed by inserting the I-DmoI region from residue 94–104 (sequence MLERIRLFNMR) between a first I-CreI domain (terminated at Pro93) and a second I-CreI domain (starting at Glu8), as shown in Figure 2.
Detailed structural analysis of how the new linker connects the scI-CreI protein domains (in a modeled structure) revealed no potential incompatibility (not shown). For example, the side chains of non-polar amino acids taken from I-DmoI, Met94, Ile98 and Phe101 point inside fitting cavities of I-CreI. A single mutation was made (P93A), however, to promote regularity of the backbone in the α-helix prior to the linker region.
Design of the DmoCre hybrid meganuclease
Superimposition of the main chain atoms from the LAGLIDADG sequence motifs from I-DmoI and I-CreI results in low r.m.s. deviation (0.66 Å, Fig. 3A and B). This is a strong indication of the similarity between the two LAGLIDADG packing interfaces. Such similarity could be sufficient to allow swapping DNA binding domains between I-DmoI and I-CreI, thereby creating proteins that cleave hybrid DNA targets. We thus replaced the C-terminal I-DmoI sequence (2nd domain) with that of I-CreI, starting at the second LAGLIDADG motif.
Figure 3.
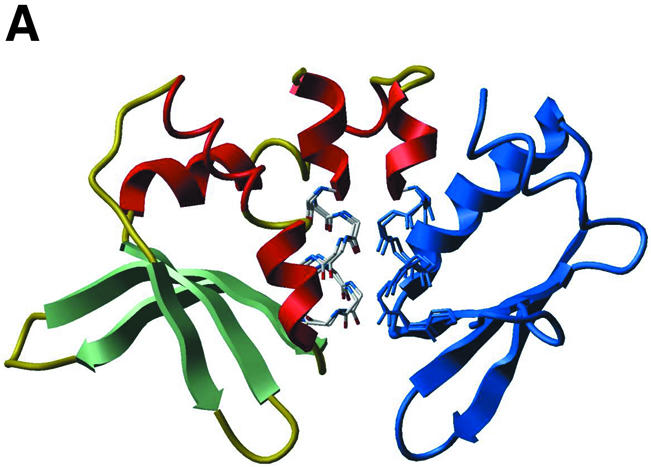


(A) Ribbon representation of I-DmoI (PDB code 1b24). The part of the second protein domain (starting with the second LAGLIDADG motif) that is replaced with I-CreI in the DmoCre protein is shown in blue. (B) Ribbon representation of I-CreI (PDB code 1g9y). The first I-CreI domain and part of the second domain, shown in blue, are replaced with I-DmoI in the DmoCre protein. In both (A) and (B), the main chain atoms corresponding to the superimposed LAGLIDADG motifs from either protein are shown in stick representation. (C) Ribbon representation of a model of the DmoCre hybrid protein. The ribbon is interrupted at the swap point separating I-DmoI and I-CreI sequences, with Phe109 and Leu13, respectively, the last residue from I-DmoI and the first from I-CreI, colored in cyan.
The resulting hybrid protein, which we called DmoCre, corresponds to I-DmoI up to Phe109 followed by I-CreI from residue Leu13 (Fig. 3C). Compared with the connection created between scI-CreI domains, the DmoCre linker region retains more of I-DmoI. This preserves to some degree the packing interface between the first and second LAGLIDADG α-helices. For the remainder of the interface, the LAGLIDADG motif from I-CreI (sequence LLGLIIGDG) is now packed along the first of the two motifs from I-DmoI (sequence LAGFVDGDG). As a consequence of sequence homology, substituting the I-DmoI motif for that of I-CreI should not disrupt considerably the structural fit between the central α-helices. However, detailed examination of the residues at and around the packing interface indicates that a few residues may not allow proper interaction between the two protein domains (not shown). Ile107 from I-DmoI may clash (steric repulsion) with Phe94 from I-CreI. Residue Ile107 was thus mutated to leucine, the corresponding amino acid for that position in I-CreI. Also, Leu47, His51 and Leu55 from I-DmoI are too close to Lys96 and Leu97 from I-CreI. The three residues were then mutated to alanine, alanine and aspartic acid, respectively, in order to avoid strains between side chains (additionally, a salt-bridge may be formed between the novel aspartic acid and Lys96).
Biochemical and biophysical properties of the single chain I-CreI protein
A synthetic gene corresponding to the new enzyme was engineered and the scI-CreI protein over-expressed in E.coli.
The engineered protein was found to be well-folded (data not shown) and stable at temperatures up to 50°C, which is comparable with I-CreI. Both proteins unfold above 50°C (Fig. 4), unfolding of scI-CreI being apparently complete at 65°C. In contrast, the denaturation profile for I-CreI indicates that a population of folded proteins remains until temperature has reached 80°C. However, under the conditions used to induce unfolding, precipitates appear. Thus, folding properties of both I-CreI and scI-CreI at high temperature could not be obtained with confidence, i.e. melting points (Tm) could not be compared.
Figure 4.

Temperature-induced equilibrium unfolding of I-CreI, I-DmoI, scI-CreI and DmoCre. Unfolding was obtained by increasing temperature (1°C/min) and monitored by acquisition of the far-UV circular dichroism signal at 222 nm.
The ability of purified scI-CreI to cleave DNA substrates in vitro was tested, using linearized plasmids bearing a copy of the I-CreI homing site. Similarly to parent I-CreI (37), the novel enzyme cleaves an I-CreI target site at 37°C (Fig. 5B). To determine the cleavage site, we used a method based on the sequencing of the cleaved fragments (see Materials and Methods). When used to determine the cleavage sites of I-CreI and I-DmoI, this method confimed the results obtained previously (40,46). Cleavage with scI-CreI appeared to result in two staggered single-strand nicks, non-distinguishable from what is obtained with I-CreI (data not shown). Our method would not allow the detection of minor cleavage sites 5′ or 3′ of each nick, and we cannot exclude that scI-CreI might be less precise than I-CreI. Nevertheless, scI-CreI appears to behave mostly as its wild type counterpart.
Catalytic properties of scI-CreI and I-CreI were examined and compared. Double-stranded DNA cleavage reactions were monitored on-line, following the conversion of quenched fluorescent substrates into fluorescent products (43). Apparent Km* and kcat* were determined analyzing how the observed reaction rates (kobs) depend on enzyme concentrations (Fig. 5D and E) under single turnover conditions (with excess enzyme) (44). We found that the apparent catalytic rate (kcat*) of scI-CreI (∼0.03 min–1) is about three times lower than that of I-CreI (∼0.09 min–1). Our I-CreI kcat* (determined at 37°C) is also three times higher than the value quoted by Chevalier et al. (41). For both proteins, Km* were found to be in the order of, or above, the highest enzyme concentrations (182 nM) used in our experiments.
The DmoCre hybrid, as parent I-DmoI, is functional at high temperature
DmoCre was engineered and tested similarly to scI-CreI.
DmoCre, which is well-folded at 20°C (data not shown), is the least stable of the four proteins tested in this work. Above that temperature, the hybrid protein unfolds progressively with a thermal denaturation profile (Fig. 4) suggestive of reduced folding co-operativity (compared with either I-DmoI or I-CreI). Again, under the conditions used to induce unfolding, the apparition of precipitated materials unfortunately precludes further characterization of folding properties.
We found that I-CreI and I-DmoI homing sites are resistant to (in vitro) cleavage by DmoCre, and that the engineered enzyme has, as expected, a novel DNA cutting specificity (Fig. 5C). Combinations of either of the two I-DmoI homing half-sites (D1 and D2) with either of the two I-CreI homing half-sites (C1 and C2) result in four possible hybrid DNA substrates (Fig. 5A). Only vectors bearing either the C1D2 or the C2D2 combination could be efficiently cleaved at 65°C (Fig. 5C). Chevalier et al. obtained similar results earlier (41). As they pointed out, cleavage of substrates bearing the D2 half-site, by E-DreI or DmoCre, indicates that the N-terminal and C-terminal I-DmoI domains bind the second (D2) and first (D1) half-sites, respectively.
We sequenced the fragments resulting from cleavage of the C1D2 site. Interestingly, cutting appeared to result again from two staggered nicks, leaving two 4 bp 3′ overhangs. As pointed out for scI-CreI, the method we use does not allow detection of minor cleavage sites. Nevertheless, we can infer from the results that in DmoCre, the I-CreI and I-DmoI moieties have kept most of their cleavage specificities, the nick in the I-CreI and I-DmoI parts of the site being positioned exactly as in the wild type sites cut by the wild type proteins (see Fig. 5A).
As for wild type I-DmoI, cleavage activity for both C1D2 and C2D2 targets was barely detectable at 37°C (Fig. 5C). In addition, our on-line fluorescence detection method and procedure would not allow monitoring on-line cleavage reactions at high temperatures. Therefore, we have not been able so far to characterize further the biochemical properties of DmoCre.
Single chain I-CreI cleaves its DNA substrate in living cells
In order to test the functionality of scI-CreI in vivo, we designed an assay to monitor meganuclease-induced homologous recombination in yeast and mammalian cells. In yeast, Xenopus oocytes and mammalian cells, DNA cleavage between two direct repeats is known to induce a very high level of homologous recombination between the repeats (47–54). The recombination pathway, often referred to as single-strand annealing (SSA), removes one repeat unit and all intervening sequences (Fig. 6A) (55). We thus constructed a SSA reporter vector, with two truncated, non-functional copies of the bacterial LacZ gene and an I-CreI cut site within the intervening sequence, in a yeast replicative plasmid. Cleavage of the cut site should result in a unique, functional LacZ copy that can be easily detected by X-gal staining.
Figure 6.

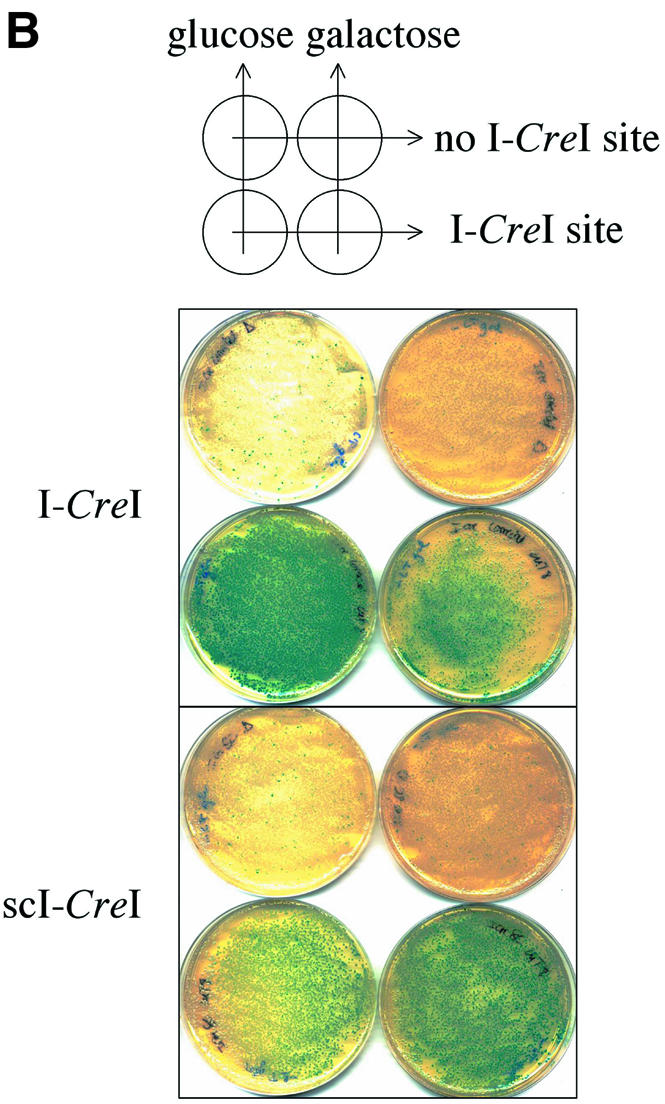



(Previous page) (A) Direct repeat recombination assay to detect meganuclease-induced recombination in yeast or mammals. Recombination occurs mostly by SSA. (B) Direct repeat recombination assay in yeast. The reporter and meganuclease expression vectors are marked by LEU2 and TRP1, respectively. The meganuclease expression is driven by an inducible Gal10 promoter. The LacZ tandem repeats share 800 bp of homology, and are separated by 1.3 kb of DNA. They are surrounded by ADH promoter and terminator sequences. The cells are co-transformed with the meganuclease expression vector and either the reporter plasmid with the cognate cut site (lower row), or a cut-site free version of the reporter (upper row). For I-CreI, we used a CTGGGTTCAAAACGTCGTGAGACAGTTTGGTCCA cut site. After transformation, equivalent amounts of cells are plated on double selective medium, with glucose or galactose as a carbon source, and X-Gal coloration was performed on colonies grown for 3 days. (C) Direct repeat recombination assay in mammalian COS cells. The meganuclease expression is driven by the GAS5 promoter and the LacZ tandem repeats are surrounded by a CMV promoter and a BGH terminator. The LacZ activity is monitored by a standard ONPG dosage 72 h after transfection. A representative experiment is shown. In this experiment, each transfection was made in duplicate. (D) Principle of inverted repeat recombination assay. Recombination between inverted repeats can occur by gene conversion, with a homologous donor template, to restore a functional LacZ gene. In the absence of donor template, the plasmid can be repaired by non-homologous end joining, resulting in a non-functional LacZ gene. The backbone, promoter and terminator were the same as in the SSA reporter plasmid. (E) Gene conversion assay in COS cell. The experimental scheme is the same as with the SSA reporter. Here, the two LacZ repeats share 2.5 kb of homology. In this experiment, each transfection was made in duplicate. a.u., arbitrary units; CS, cut site.
The reporter vector was used to transform yeast cells. A small fraction of cells appeared to express functional LacZ (not shown), probably due to recombination events during transformation (F. Pâques and J.E. Haber, personnal communication). Co-transformation with plasmids expressing either I-CreI or scI-CreI, in contrast, resulted in blue staining for all plated cells (Fig. 6B, lower row). These results could be confirmed by a quantitative assay (not shown) similar to the one used in mammalian cells (see below). Even in non-induced conditions (glucose), the residual level of protein was enough to induce SSA, suggesting that scI-CreI, as much as I-CreI, is highly efficient in yeast cells. Furthermore, SSA induction was truly dependent on cleavage of the target cut site by I-CreI proteins, as vectors devoid of that site display no increase in β-galactosidase activity compared with background levels (Fig. 6B, upper row). scI-CreI, though, does not affect cell growth, whereas upon over-expression (on galactose plates) of wild-type I-CreI only a small number of slow growing colonies (all of them blue) is observed. Dose-dependent toxicity of I-CreI had been observed previously in bacteria (56) and flies (12) where it seemed to result from non-specific cleavage in rDNA. However, lack of toxicity of scI-CreI could be due to a different level of expression or stability.
Our SSA assay was modified for tests in mammalian cells. The promoter and termination sequences of the reporter and meganuclease expression plasmid were changed, and plasmid recombination was evaluated in a transient transfection assay. Similar levels of induced recombination (2–3-fold increase) were observed with either scI-CreI or I-CreI. A typical experiment is shown in Figure 6C. As in the yeast experiment, recombination depends on an I-CreI cut site between the repeats, for no increase of the β-galactosidase was observed in the absence of this site.
Another recombination assay, based on recombination between inverted repeats, was also used to monitor meganuclease-induced recombination in COS cells (Fig. 6D). While direct repeats can recombine by SSA, homologous recombination between indirect repeats requires a gene conversion event (see Fig. 6D) (55). Similar stimulation of gene conversion (3–4-fold) was observed with either scI-CreI or I-CreI. A typical experiment is shown in Figure 6E. As expected for a true homologous recombination event (see Fig. 6D), no enhancement was observed in the absence of a homologous donor template.
We have shown that DmoCre, our other artificial meganuclease, is functional at high temperature. As a probable consequence, DmoCre could not induce any detectable recombination in our yeast assay carried out at 30°C. Attempts to perform the assay at higher (non lethal) temperatures gave no better result.
DISCUSSION
In this study, we present hybrid endonucleases derived from homing endonucleases through domain swapping. Domain swapping is a powerful strategy to create hybrid enzymes (57). The simplest approach is to assemble distinct protein moieties, identified in their natural context (the protein they belong to) as functionally and structurally independent domains. Following this strategy, nucleases with new specificities and large cut sites have recently been engineered. Protein fusions combining a DNA binding domain (58–61) or a DNA binding protein (62–65) to the non-specific catalytic domain of FokI, a Class IIS restriction enzyme, were successfully tested in in vivo assays for cutting (62–65), mutagenesis (61) and even recombination (60). Chimeric meganucleases derived from the HO yeast endonuclease represent another interesting case (66). HO is an atypical LAGLIDADG protein, with two LAGLIDADG motifs, and a C-terminal zinc finger domain. Nahon and Raveh could change the specificity of HO by swapping the zinc finger domain (66).
However, typical LAGLIDADG homing endonucleases correspond to a more complex situation, and the functionality of artificial hybrid enzymes is not a trivial issue. First, in the natural proteins, the catalytic, active site is surrounded by two elements of their DNA binding motif. Linkage between catalysis and specific binding is thereby complete. Thus, domain swapping between related LAGLIDADG proteins most likely needs to maintain a high degree of linkage, the packing interface between the LAGLIDADG α-helices separating both domains being highly conserved at the sequence and structural levels. Our own data indicate that the scI-CreI protein is well-folded and relatively stable at high temperature. Besides, scI-CreI has enzymatic properties barely below those of its natural counterpart, I-CreI. In contrast, DmoCre appears to be less stable (although it is active at 65°C) perhaps due to unsatisfactory packing of the combined protein domains. Hence, further improvements of the design should be considered. Actually, a hybrid homing endonuclease homologous to our DmoCre protein was recently engineered (E-DreI) (41) and characterized at the structural level. E-DreI shares in vitro properties with our novel endonuclease, in terms of temperature requirement, and cleavage specificity. In addition, it adopts a three-dimensional structure comparable with those of its parent proteins (as intended by design). These structural results validate the engineering strategy, and will be useful for further engineering projects based on I-DmoI, as well as I-CreI.
Secondly, in LAGLIDADG homing endonucleases, the unique catalytic, active site comprises amino acid residues from both DNA binding domains. Their specific nature and spatial distribution is probably required for DNA cleavage, but it is difficult to assign functional roles to residues in the active site. The only thoroughly conserved residues are the last acidic amino acids (aspartate and/or glutamate amino acids) from both LAGLIDADG motifs. Mutations of those residues abolish catalysis, but not DNA binding (67,68). Besides, a hydration shell, consisting of several water molecules structurally organized by the amino acid side chains of acidic and basic residues, together with divalent cations, probably has an essential role in conducting the cleavage of DNA phosphodiester bonds (29). Consequently, it was not known whether domain swapping of those enzymes, which have distinct catalytic site residues, would restore functional, active proteins. In addition, the degree of homology between different LAGLIDADG interfaces, may not be sufficient to allow proper packing between any pair of LAGLIDADG α-helices.
Our results and those obtained with E-DreI (41) show that LAGLIDAGD proteins are truly made of separable DNA binding domains that can be recombined, and that these proteins could be the precursors of artificial, designed endonucleases. LAGLIDADG proteins belong to the largest (and best characterized) family of homing endonucleases. Hence, domain swapping between LAGLIDADG enzymes would provide a great number of novel proteins. Obtaining specific nucleases for each desired locus, however, will require additional strategies. We have shown that the two DNA binding domains behave rather independently. This will be essential, for instance if directed molecular evolution strategies are to be used to select from mutagenized libraries of either natural or chimeric homing endonucleases, for each domain could be modified independently.
There are many possible applications for novel endonucleases recognizing such large cleavage sites, but the first one is certainly their use in genome engineering, and more precisely, in meganuclease-induced gene targeting. In this case the artificial novel proteins have to be functional in living cells. scI-CreI was shown to induce homologous recombination in yeast and mammalian cells (our assays enable the testing of proteins that are active in the 30–37°C range). We could induce recombination between direct repeats, which most often occurs by SSA, but also between inverted repeats, which usually occurs by gene conversion (55). Gene conversion, which underlies all homologous targeting strategies, is enzymatically more demanding (55), and this experiment represents a further step toward genome surgery with engineered meganucleases. The detectable levels of scI-CreI-induced recombination in both kinds of assay confirm our in vitro results, but they also illustrate for the first time that man-made proteins derived from homing endonucleases will be useful for DNA targeting applications.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank V. Gallois, A. Perrin, D. Sourdive, A. Choulika and all members of the Cellectis R&D department for fruitful discussions, as well as B. Dujon and F. Jacob for critical reading of the manuscript. We are grateful to J. Bolard for giving us the opportunity to perform circular dichroism experiments at the LPBC-CSSB, UMR CNRS 7033, Pierre and Marie Curie University in Paris.
REFERENCES
- 1.Capecchi M.R. (1989) The new mouse genetics: altering the genome by gene targeting. Trends Genet., 5, 70–76. [DOI] [PubMed] [Google Scholar]
- 2.Capecchi M.R. (1989) Altering the genome by homologous recombination. Science, 244, 1288–1292. [DOI] [PubMed] [Google Scholar]
- 3.Smithies O. (2001) Forty years with homologous recombination. Nature Med., 7, 1083–1086. [DOI] [PubMed] [Google Scholar]
- 4.Rouet P., Smih,F. and Jasin,M. (1994) Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol., 14, 8096–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choulika A., Perrin,A., Dujon,B. and Nicolas,J.F. (1995) Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 1968–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott B., Richardson,C., Winderbaum,J., Nickoloff,J.A. and Jasin,M. (1998) Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cell. Biol., 18, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen-Tannoudji M., Robine,S., Choulika,A., Pinto,D., El Marjou,F., Babinet,C., Louvard,D. and Jaisser,F. (1998) I-SceI-induced gene replacement at a natural locus in embryonic stem cells. Mol. Cell. Biol., 18, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donoho G., Jasin,M. and Berg,P. (1998) Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol. Cell. Biol., 18, 4070–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puchta H., Dujon,B. and Hohn,B. (1993) Homologous recombination in plant cells is enhanced by in vivo induction of double strand breaks into DNA by a site-specific endonuclease. Nucleic Acids Res., 21, 5034–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rong Y.S. and Golic,K.G. (2000) Gene targeting by homologous recombination in Drosophila. Science, 288, 2013–2018. [DOI] [PubMed] [Google Scholar]
- 11.Rong Y.S. and Golic,K.G. (2001) A targeted gene knockout in Drosophila. Genetics, 157, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rong Y.S., Titen,S.W., Xie,H.B., Golic,M.M., Bastiani,M., Bandyopadhyay,P., Olivera,B.M., Brodsky,M., Rubin,G.M. and Golic,K.G. (2002) Targeted mutagenesis by homologous recombination in D.melanogaster. Genes Dev., 16, 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thierry A. and Dujon,B. (1992) Nested chromosomal fragmentation in yeast using the meganuclease I-Sce I: a new method for physical mapping of eukaryotic genomes. Nucleic Acids Res., 20, 5625–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belfort M. and Roberts,R.J. (1997) Homing endonucleases: keeping the house in order. Nucleic Acids Res., 25, 3379–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurica M.S. and Stoddard,B.L. (1999) Homing endonucleases: structure, function and evolution. Cell. Mol. Life Sci., 55, 1304–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chevalier B.S. and Stoddard,B.L. (2001) Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res., 29, 3757–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kostriken R., Strathern,J.N., Klar,A.J., Hicks,J.B. and Heffron,F. (1983) A site-specific endonuclease essential for mating-type switching in Saccharomyces cerevisiae. Cell, 35, 167–174. [DOI] [PubMed] [Google Scholar]
- 18.Haber J.E. (1998) Mating-type gene switching in Saccharomyces cerevisiae. Annu. Rev. Genet., 32, 561–599. [DOI] [PubMed] [Google Scholar]
- 19.Jacquier A. and Dujon,B. (1985) An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell, 41, 383–394. [DOI] [PubMed] [Google Scholar]
- 20.Colleaux L., D’Auriol,L., Galibert,F. and Dujon,B. (1988) Recognition and cleavage site of the intron-encoded omega transposase. Proc. Natl Acad. Sci. USA, 85, 6022–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plessis A., Perrin,A., Haber,J.E. and Dujon,B. (1992) Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics, 130, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perrin A., Buckle,M. and Dujon,B. (1993) Asymmetrical recognition and activity of the I-SceI endonuclease on its site and on intron-exon junctions. EMBO J., 12, 2939–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bell-Pedersen D., Quirk,S., Clyman,J. and Belfort,M. (1990) Intron mobility in phage T4 is dependent upon a distinctive class of endonucleases and independent of DNA sequences encoding the intron core: mechanistic and evolutionary implications. Nucleic Acids Res., 18, 3763–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell-Pedersen D., Quirk,S.M., Aubrey,M. and Belfort,M. (1989) A site-specific endonuclease and co-conversion of flanking exons associated with the mobile td intron of phage T4. Gene, 82, 119–126. [DOI] [PubMed] [Google Scholar]
- 25.Bell-Pedersen D., Quirk,S.M., Bryk,M. and Belfort,M. (1991) I-TevI, the endonuclease encoded by the mobile td intron, recognizes binding and cleavage domains on its DNA target. Proc. Natl Acad. Sci. USA, 88, 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller J.E., Smith,D. and Belfort,M. (1996) Exon coconversion biases accompanying intron homing: battle of the nucleases. Genes Dev., 10, 2158–2166. [DOI] [PubMed] [Google Scholar]
- 27.Heath P.J., Stephens,K.M., Monnat,R.J.,Jr and Stoddard,B.L. (1997) The structure of I-Crel, a group I intron-encoded homing endonuclease. Nature Struct. Biol., 4, 468–476. [DOI] [PubMed] [Google Scholar]
- 28.Jurica M.S., Monnat,R.J.,Jr and Stoddard,B.L. (1998) DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol. Cell., 2, 469–476. [DOI] [PubMed] [Google Scholar]
- 29.Chevalier B.S., Monnat,R.J.,Jr and Stoddard,B.L. (2001) The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nature Struct. Biol., 8, 312–316. [DOI] [PubMed] [Google Scholar]
- 30.Silva G.H., Dalgaard,J.Z., Belfort,M. and Van Roey,P. (1999) Crystal structure of the thermostable archaeal intron-encoded endonuclease I-DmoI. J. Mol. Biol., 286, 1123–1136. [DOI] [PubMed] [Google Scholar]
- 31.Hu D., Crist,M., Duan,X., Quiocho,F.A. and Gimble,F.S. (2000) Probing the structure of the PI-SceI-DNA complex by affinity cleavage and affinity photocross-linking. J. Biol. Chem., 275, 2705–2712. [DOI] [PubMed] [Google Scholar]
- 32.Poland B.W., Xu,M.Q. and Quiocho,F.A. (2000) Structural insights into the protein splicing mechanism of PI-SceI. J. Biol. Chem., 275, 16408–16413. [DOI] [PubMed] [Google Scholar]
- 33.Duan X., Gimble,F.S. and Quiocho,F.A. (1997) Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell, 89, 555–564. [DOI] [PubMed] [Google Scholar]
- 34.Werner E., Wende,W., Pingoud,A. and Heinemann,U. (2002) High resolution crystal structure of domain I of the Saccharomyces cerevisiae homing endonuclease PI-SceI. Nucleic Acids Res., 30, 3962–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moure C.M., Gimble,F.S. and Quiocho,F.A. (2002) Crystal structure of the intein homing endonuclease PI-SceI, bound to its recognition sequence. Nature Struct. Biol., 9, 764–770. [DOI] [PubMed] [Google Scholar]
- 36.Ichiyanagi K., Ishino,Y., Ariyoshi,M., Komori,K. and Morikawa,K. (2000) Crystal structure of an archaeal intein-encoded homing endonuclease PI-PfuI. J. Mol. Biol., 300, 889–901. [DOI] [PubMed] [Google Scholar]
- 37.Wang J., Kim,H.H., Yuan,X. and Herrin,D.L. (1997) Purification, biochemical characterization and protein–DNA interactions of the I-CreI endonuclease produced in Escherichia coli. Nucleic Acids Res., 25, 3767–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marshall P., Davis,T.B. and Lemieux,C. (1994) The I-CeuI endonuclease: purification and potential role in the evolution of Chlamydomonas group I introns. Eur. J. Biochem., 220, 855–859. [DOI] [PubMed] [Google Scholar]
- 39.Gimble F.S. and Wang,J. (1996) Substrate recognition and induced DNA distortion by the PI-SceI endonuclease, an enzyme generated by protein splicing. J. Mol. Biol., 263, 163–180. [DOI] [PubMed] [Google Scholar]
- 40.Dalgaard J.Z., Garrett,R.A. and Belfort,M. (1993) A site-specific endonuclease encoded by a typical archaeal intron. Proc. Natl Acad. Sci. USA, 90, 5414–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chevalier B.S., Kortemme,T., Chadsey,M.S., Baker,D., Monnat,R.J. and Stoddard,B.L. (2002) Design, activity and structure of a highly specific artificial endonuclease. Mol. Cell, 10, 895–905. [DOI] [PubMed] [Google Scholar]
- 42.Abagyan R.A., Totrov,M.M. and Kuznetsov,D.N. (1994) ICM—a new protein modeling and design. Applications to docking and structure prediction from the distorted native conformation. J. Comp. Chem., 15, 488–506. [Google Scholar]
- 43.Eisenschmidt K., Lanio,T., Jeltsch,A. and Pingoud,A. (2002) A fluorimetric assay for on-line detection of DNA cleavage by restriction endonucleases. J. Biotechnol., 96, 185–191. [DOI] [PubMed] [Google Scholar]
- 44.Halford S.E., Johnson,N.P., Grinsted,J. (1980). The EcoRI restriction endonuclease with bacteriophage λ DNA. Biochem. J., 191, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gietz R.D. and Woods,R.A. (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol., 350, 87–96. [DOI] [PubMed] [Google Scholar]
- 46.Thompson A.J., Yuan,X., Kudlicki,W. and Herrin,D.L. (1992) Cleavage and recognition pattern of aa double-strand-specific endonuclease (I-CreI) encoded by the chloroplast 23S rRNA intron of Chlamydomonas reinhadtii. Gene, 119, 247–251. [DOI] [PubMed] [Google Scholar]
- 47.Lin F.L., Sperle,K. and Sternberg,N. (1984) Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol. Cell. Biol., 4, 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin F.L., Sperle,K. and Sternberg,N. (1985) Recombination in mouse L cells between DNA introduced into cells and homologous chromosomal sequences. Proc. Natl Acad. Sci. USA, 82, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin F.L., Sperle,K. and Sternberg,N. (1990) Repair of double-stranded DNA breaks by homologous DNA fragments during transfer of DNA into mouse L cells. Mol. Cell. Biol., 10, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rudin N. and Haber,J.E. (1988) Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol. Cell. Biol., 8, 3918–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fishman-Lobell J., Rudin,N. and Haber,J.E. (1992) Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell. Biol., 12, 1292–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugawara N. and Haber,J.E. (1992) Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol. Cell. Biol., 12, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carroll D., Wright,S.H., Wolff,R.K., Grzesiuk,E. and Maryon,E.B. (1986) Efficient homologous recombination of linear DNA substrates after injection into Xenopus laevis oocytes. Mol. Cell. Biol., 6, 2053–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang F., Han,M., Romanienko,P.J. and Jasin,M. (1998) Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paques F. and Haber,J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seligman L.M., Stephens,K.M., Savage,J.H. and Monnat,R.J.,Jr (1997) Genetic analysis of the Chlamydomonas reinhardtii I-CreI mobile intron homing system in Escherichia coli. Genetics, 147, 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nixon A.E., Ostermeier,M. and Benkovic,S.J. (1998) Hybrid enzymes: manipulating enzyme design. Trends Biotechnol., 16, 258–264. [DOI] [PubMed] [Google Scholar]
- 58.Kim Y.G., Smith,J., Durgesha,M. and Chandrasegaran,S. (1998) Chimeric restriction enzyme: Gal4 fusion to FokI cleavage domain. Biol. Chem., 379, 489–495. [DOI] [PubMed] [Google Scholar]
- 59.Smith J., Bibikova,M., Whitby,F.G., Reddy,A.R., Chandrasegaran,S. and Carroll,D. (2000) Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res., 28, 3361–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bibikova M., Carroll,D., Segal,D.J., Trautman,J.K., Smith,J., Kim,Y.G. and Chandrasegaran,S. (2001) Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol., 21, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bibikova M., Golic,M., Golic,K.G. and Carroll,D. (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics, 161, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim M.K., Lee,J.S. and Chung,J.H. (1999) In vivo transcription factor recruitment during thyroid hormone receptor-mediated activation. Proc. Natl Acad. Sci. USA, 96, 10092–10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee C.H., Murphy,M.R., Lee,J.S. and Chung,J.H. (1999) Targeting a SWI/SNF-related chromatin remodeling complex to the beta-globin promoter in erythroid cells. Proc. Natl Acad. Sci. USA, 96, 12311–12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee J.S., Lee,C.H. and Chung,J.H. (1999) The beta-globin promoter is important for recruitment of erythroid Kruppel-like factor to the locus control region in erythroid cells. Proc. Natl Acad. Sci. USA, 96, 10051–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee J.S., Ngo,H., Kim,D. and Chung,J.H. (2000) Erythroid Kruppel-like factor is recruited to the CACCC box in the beta-globin promoter but not to the CACCC box in the gamma-globin promoter: the role of the neighboring promoter elements. Proc. Natl Acad. Sci. USA, 97, 2468–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nahon E. and Raveh,D. (1998) Targeting a truncated Ho-endonuclease of yeast to novel DNA sites with foreign zinc fingers. Nucleic Acids Res., 26, 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lykke-Andersen J., Garrett,R.A. and Kjems,J. (1997) Mapping metal ions at the catalytic centres of two intron-encoded endonucleases. EMBO J., 16, 3272–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gimble F.S. and Stephens,B.W. (1995) Substitutions in conserved dodecapeptide motifs that uncouple the DNA binding and DNA cleavage activities of PI-SceI endonuclease. J. Biol. Chem., 270, 5849–5856. [DOI] [PubMed] [Google Scholar]