

Abstract
Vimentin, a member of the intermediate filament protein family, is regulated both developmentally and tissue specifically. It is also a marker of the metastatic potential of many tumor cells. Pre viously, the human vimentin promoter has been shown to contain multiple elements for the binding of both positive- and negative-acting regulatory factors. Transient transfection analysis of various vimentin 5′-end promoter sequences and mutants thereof fused to a reporter gene further defined two regulatory elements, a positive element that binds Sp1 and a negative element that binds the protein ZBP-89. ZBP-89 has been shown to be either a repressor or an activator of gene expression, depending on the promoter. Here, we show that for vimentin, both ZBP-89 and ZBP-99 repress reporter gene expression in Schneider (S2) cells. Deletion constructs confirm that the glutamine-rich region of Sp1 is required to enhance vimentin transcription, whereas the N-terminus of ZBP-89 is required to interact with Sp1 and repress gene expression. The overexpression of hTAFII130 can alleviate ZBP-89 repression in S2 cells, suggesting how ZBP-89 might serve to block gene expression.
INTRODUCTION
The eukaryotic cytoskeleton is composed of three different networks, the microtubules, the microfilaments and the intermediate filaments (IFs). The intermediate filament protein (IFP) family includes a variety of proteins such as cytokeratins found in epithelial cells, glial fibrillary acidic protein (GFAP) in glial cells, desmin in muscle, vimentin in mesenchyme-derived cells, neurofilaments in neural cells and lamins in the nucleus. All IFPs share a common organization composed of three domains, a central α-helical core, flanked by globular N- and C-terminal domains. Yet subtle functional differences must exist requiring the variety of IFP family members. The vimentin network extends from the nuclear membrane to the plasma membrane (1). Thus, vimentin has been hypothesized to be involved in maintaining the overall integrity of the cytoplasm and cell membrane as well as a signal transducer transmitting extracellular signals from the plasma membrane to the nucleus (2–4). In order to understand how the various IFP genes are regulated, we have begun to investigate the requirements for vimentin expression.
Vimentin exhibits a complex pattern of gene regulation during embryonic development and cell proliferation and in neoplasia. Normally vimentin is expressed in cells of mesenchymal origin such as fibroblasts, myoblasts, endothelial or bone marrow cells (5). Initially, vimentin and desmin are co-expressed in early stages of muscle development, but then vimentin expression is turned off during terminal differentiation (6,7). A similar expression pattern is noted in the terminal differentiation of glial and neuronal cells (8,9). In addition, the vimentin gene is growth regulated and its expression can be induced by serum, phorbol ester, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) (10,11). More importantly, vimentin expression is associated with the occurrence of tumor metastasis such as melanoma and mammary tumors, and is a marker for the metastatic potential of many tumor cells (12–16). Thus, it is important to determine how the vimentin gene is selectively down- regulated during terminal differentiation of some cell types, remains expressed in others, or is aberrantly expressed in metastatic cells, 90% of which derive from epithelial cells, which initially express the cytokeratins (17).
To date, the regulation of vimentin gene expression involves multiple regulatory elements, which include several enhancers and at least one repressor element. Initially, these elements were identified within the chicken vimentin gene promoter. A proximal promoter region, which included several GC-boxes but lacked a TATA-box, provided a basal, constitutive level of gene activity. Further upstream were three homologous silencer elements (SE1–SE3) and an antisilencer element (18,19). Although this element was located 1 kb upstream of SE3, it could override the negative effect of the multiple SEs. Since it showed no enhancer activity by itself when fused to either the homologous vimentin or heterologous thymidine kinase promoter, it was termed an antisilencer (ASE). More recently, similar cis-elements have been found in the promoter region of the human vimentin gene. These include a TATA-box, eight putative GC-boxes, an NF-κB site (at –218 to –227), a PEA3-binding site (–153 to –161) and a repressor site (17,20–24). Further upstream are tandem AP-1-binding sites and the ASE (25,26). GC-box 1 (–64 to –55) has been shown to be indispensable for gene expression (23). PEA3 activates vimentin by binding to the PEA3 site (17). The NF-κB site is responsible for Tax-induced vimentin expression, and AP-1 family members participate in the serum response by binding to the two tandem AP-1 sites (21,25,27).
Previously, in vivo DNA footprinting experiments showed that a HeLa nuclear factor binds to the proximal GC-box 1, but not to the other putative GC-boxes (23). The sequence of GC-box 1 (TGGGaGGGGa) bears an 8 out of 10 identity to the Sp1 consensus site (T/GGGGCGGG/AG/AG/T), which is an important cis-acting element required for the appropriate expression of housekeeping as well as many tissue-specific and viral genes. The general transcription factor Sp1 binds to such GC/GT boxes and is, therefore, thought to be a ubiquitous protein essential for many different functions within the mammalian nucleus (28). However, Sp1 is not the only transcription factor that can bind to a GC/GT element, and vimentin’s GC-box 1 is not a perfect match to the Sp1 consensus sequence. Moreover, Sp1 is only one member of the Sp/XKLF family, which comprises at least 16 different members all containing a highly conserved DNA-binding domain consisting of three zinc fingers (29,30). Some Sp family members such as Sp1 are ubiquitously expressed, whereas others show tissue-specific expression, i.e., Sp4 is localized to the brain. Within the Sp family, Sp1, 2, 3 and 4 are closely related. Thus, it is important to determine which of the Sp family members is involved in regulating the human vimentin gene.
In addition, a known regulatory region was divided further into two subdomains: (i) a proximal silencer element (PS, at –288 to –303), which binds a 105 kDa protein by UV cross-linking and southwestern blots; and (ii) an adjacent enhancer element (–319 to –339) previously referred to as Δ19 (20,24). In vitro experiments suggested that ZBP-89 binds to the PS element. Sp1 itself does not bind to the PS element in vitro, but it can interact with ZBP-89 bound to PS DNA (24).
ZBP-89 (BFCOL1, BERF-1, ZNF 148) is a Krüppel-type, zinc finger transcription factor that binds to GC-rich elements and represses or activates known target genes. For example, ZBP-89 activates the expression of such genes as human stromelysin, the pre-T-cell receptor (TCR) α, mouse type I collagen, p21waf1 and the lymphocyte-specific protein tyrosine kinase (lck) (31–35). In other cases, ZBP-89 acts as a repressor for human gastrin, human ornithine decarboxylase (ODC), rat β-enolase, bovine adrenodoxin, chemokine epithelial neutrophil-activating peptide-78 (ENA-78) and the human β2 integrin CD11b genes (36–41). In all of these later cases, ZBP-89 appears to repress promoter activity by opposing the effect of Sp1 by binding to the same, overlapping or adjacent DNA element. Thus, it has been proposed that competitive binding to shared promoter elements may mediate transcriptional regulation by ZBP-89, as has been seen for other factors. However, in the human vimentin promoter, the PS element is a separate element ∼230 bp upstream from GC-box 1. Thus, competition for binding to the same or overlapping DNA element cannot account for the repression exhibited by ZBP-89 on the human vimentin promoter.
In addition to ZBP-89, ZBP-99 is another member of this zinc finger protein family (42). ZBP-99 is located on 1q32.1, whereas ZBP-89 is found on chromosome 3q21 (43). ZBP-99 is structurally and functionally homologous to ZBP-89. Its four zinc fingers share 91% amino acid sequence similarity and 79% sequence identity with those of ZBP-89. In addition, C-terminal segments of the two genes are highly conserved in amino acid sequences. Thus, ZBP-99 may also play an important role in regulating cell proliferation, differentiation and oncogenesis, as hypothesized for ZBP-89.
Previous experiments suggested that ZBP-89 binds to vimentin’s PS element in vitro (24). However, the functional consequences of such putative in vitro interactions were not confirmed by in vivo transient transfection experiments. Thus, additional experiments were necessary to confirm the functional interaction of Sp1 and ZBP-89 and to determine how these proteins might interact to control vimentin expression.
MATERIALS AND METHODS
Cell culture and reagents
The Drosophila Schneider cell line (S2) from Invitrogen Corporation was maintained in Drosophila medium with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% FBS, glutamine and 1% penicillin–streptomycin (Gibco-BRL).
Plasmids and constructs
The pUC18CAT expression vector, a pEMBL derivative containing the bacterial chloramphenicol acetyltransferase (CAT) gene, the ampicillin resistance gene and the pUC18 multicloning site, was used to assess the transcriptional activity of various vimentin promoter fragments. Different vimentin 5′-flanking sequences were fused to the CAT gene as previously described, and identified by their 5′-most nucleotide (24). Construct 83WT was cloned from 353WT by digestion with SacI and SacII. The hSE mutant construct was made using the Quick-change, site-directed mutagenesis kit (Stratagene) with the following two primers, 5′-CCCAGGCGGACCCATTCCTCACCGCGCGAC-3′ and 5′-GTCGCGCGGTGAGGAATGGGTCCGCCTGGG-3′. The content of the various vimentin promoter CAT constructs is shown in Figure 2A and the sequence of all clones was verified by DNA sequencing.
Figure 2.

GC-box 1 within the human vimentin promoter is required for activation by either Sp1 or USp3. (A) Schematic structure of the human vimentin promoter. The numbers indicate the 5′-end of different vimentin promoter constructs fused to the reporter gene CAT. (B) The various wild-type (WT) vimentin promoter constructs (5 µg), 83WT, 189WT, 261WT and 353WT were either transfected alone (white box) or co-transfected with 0.2 µg of pPacSp1 (black boxes) or pPacUSp3 (gray boxes) into S2 cells. CAT activity is expressed as the mean ± SE. (C) The sequences of wild-type or mutant NF, PEA3 or Sp1 elements within the promoter region –353 fused to the CAT gene. (D) The vimentin (353WT) or mutant constructs (353PEA3, 353NF or 353Sp) were co-transfected with 0.2 µg of either pPacSp1 (black boxes) or pPacUSp3 (gray boxes) into S2 cells. CAT activity is determined as described in (B). (E) A DNA affinity precipitation assay using biotinylated oligonucleotides. The content of WCEs isolated from non-transfected (blank) or S2 cells transfected with 10 µg of pPacSp1 (input) is analyzed by western blot analysis with antibodies to Sp1. Double-stranded, wild-type GC-box 1 DNA (WT-BP) with one strand biotinylated at its 5′-end was used to ‘pull-out’ proteins from S2 WCEs transfected with pPacSp1 (input). A mutant GC-box 1 DNA sequence (MT-BP) as denoted in (C) was analyzed similarly. WCEs and each biotinylated labeled dsDNA sequence were incubated at 4°C for 3 h. DNA–protein complexes were removed by binding to SA-PMP beads and washed three times with 1× lysis buffer as described (34). The content of bound proteins was analyzed by western blots with anti-Sp1 antibody.
pPac-β-gal, pPacSp1 and the various deletion constructs of Sp1 were kindly provided by Dr Robert Tjian (University of California at Berkeley). pPacSp2 was kindly supplied by Dr John Noti (Guthrie Research Institute), and pPacSp3, pPacUSp3 and pPacSp4 were from Dr Guntram Suske (Institute fur Molekular Biologie und Tumorforschung). The schematic structural comparisons of Sp1–Sp4 are shown in Figure 1A. All of these plasmids are in pPac vectors under the control of the Drosophila actin 5C promoter. pBSK(+)-hTAFII130 was kindly provided by Dr Naoko Tanese (New York University). Two complementary oligonucleotides containing the myc tag (underlined) were cloned into the BamHI and EcoRI sites of this plasmid. The sequence of the two DNA strands is as follows: 5′-GATCCGAGCAGAAACTCATCTCTGAAGAGGATCTGCTAGCG-3′ and 5′-AATTCGCTAGCAGATCCTCTTCAGAGATGAGTTTCTGCTCG-3′, respectively. The positive clone was digested by BamHI and XhoI to release the hTAFII130 fragment, which was then inserted into the pPac vector at the BamHI and XhoI sites to generate pPac-hTAFII130.
Figure 1.

Sp1 and USp3 can activate expression of the human vimentin promoter. (A) Schematic structural comparison of Sp1–Sp4 family members with the number of amino acids (aa) for each protein indicated. (B and C) Effect of increasing amounts (0–10 µg) of Sp1–Sp4 expression plasmids on reporter gene activity. Drosophila S2 cells were co-transfected with the 261WT construct (5 µg) plus pPacSp1 (black triangles in B), pPacUSp3 (black squares in B), pPacSp2 (black diamonds in C), pPacSp3 (black triangles in C) or pPacSp4 (black squares in C) separately. CAT activity was normalized to β-gal (pPac-β-gal, 1 µg) as an internal control and determined as discussed in Materials and Methods. Values are expressed as the mean ± SE. (D) Synergism between Sp1 and USp3 in S2 cells. pPacSp1 (0.2 µg) and 261WT (5 µg) were co-transfected alone (black box) or with 0.2 or 5 µg of either pPacSp3 or pPacUSp3 (gray boxes) in S2 cells. CAT activity was determined as in (A). (E) Western blot analysis of WCEs (40 µg) prepared from S2 cells (blank) or transfected with pPacSp2 (10 µg) as described above and incubated with antibodies to Sp2 as described in Materials and Methods. The position of migration of the molecular weight markers is noted. (F) Western blot analysis of NEs prepared from HeLa cells as a positive control (lane 1), cellular extract prepared from S2 cells transfected with pPacUSp3 (10 µg) and pPacZBP-89 (10 µg) (lane 2), non-transfected (lane 3), pPacUSp3 (10 µg) alone (lane 4) and pPacSp3 (10 µg) (lane 5). The western blot was incubated with anti-Sp3 antibodies, and the migration position of molecular weight markers is noted.
The pcDNA3-ZBP-89 plasmid encoding the full-length human ZBP-89 (1–794) was kindly provided by Dr Juanita Merchant (University of Michigan). pPacZBP-89 was cloned from pcDNA3-ZBP-89 into the pPac vector digested with BamHI (then blunted) and XhoI. To generate hemagglutinin (HA)-tagged pPacZBP-89 (pPacZBP-89-HA), the following primers were used: forward primer 5′-TCGAGCTAGCATACCCATACGACGTCCCAGACTACGCTTCTAGATAATGA-3′ and reverse primer 5′-TCGATCATTATCTAGAAG CGTAGTCTGGGACGTCGTATGGGTATGCTAGC-3′. The HA-tagged primers were inserted into pPacZBP-89 digested with XhoI. For constructs containing the various deletion constructs of ZBP-89, different cloning strategies were used (Fig. 6A). To generate ΔAD (dA, a deletion of the acidic domain), pPacZBP-89-HA was digested with SmlI (blunt) and XhoI, then the ZBP-89 cDNA fragment was ligated to pPacZBP-89-HA digested with BstXI (blunt) and XhoI. To generate ΔBD2+C-ter (dB2C, a deletion from the basic domain to the C-terminus), pPacZBP-89-HA was digested by BspHI (blunt) and BstXI, and the resulting fragment was ligated to pPacZBP-89-HA digested by XhoI (blunt) and BstXI. To generate ΔSer-rich (dS, a deletion of the serine-rich region), pPacZBP-89-HA was digested by KpnI (blunt) and SacI (blunt) and ligated by T4 DNA ligase. To generate ΔC-Ter (dC, a deletion of the C-terminus after the serine-rich region), pPacZBP-89-HA was digested with NheI (blunt) and SacI (blunt) and ligated by T4 DNA ligase. To generate ΔSR+C-Ter (dSC, a deletion of the serine-rich and C-terminal regions), pPacZBP-89-HA was digested with KpnI (blunt) and XhoI (blunt) and ligated by T4 DNA ligase. To generate ΔAD+BD1 (dAB1, a deletion of the acidic domain and the first basic domain, BD1), the following primers were used to amplify the corresponding region by PCR: forward primer 5′-GGATGGATCCCTTGGTTTGAAAACCCCT-3′ and reverse primer 5′- TAGTCGTTTGGGCGAGAACT-3′. The PCR product was digested with BamHI and then ligated into the pPac vector digested with BamHI and XhoI. The content of ZBP-89 deletion constructs is shown in Figure 4A and was verified by DNA sequencing.
Figure 6.

The N-terminal region of ZBP-89 is required for repression. (A) A schematic of ZBP-89 and its various deletion mutants. The amino acid position of each deletion is noted. (B) The various ZBP-89 deletion mutants (gray boxes) in the pPac vector (1 µg) were co-transfected with pPacSp1 (0.2 µg) and 353WT (5 µg) into S2 cells. Transfection of pPacSp1 alone is indicated by the black box. CAT activity is expressed as the mean ± SE. (C) Western blot analysis of WCEs prepared from S2 cells transfected with pPacZBP-89 and its various deletion mutants (HA-tagged) incubated with anti-HA antibody as follows: lane 1, non-transfected control; lane 2, transfected with pPacZBP-89-HA; lane 3, transfected with dA; lane 4, transfected with dAB1; lane 5, transfected with dB2C; lane 6, transfected with dS; lane 7, transfected with dSC; lane 8, transfected with dC. The position of migration of molecular weight markers is noted.
Figure 4.

Analysis of the ZBP-89 DNA-binding site by EMSAs and functional assays. (A) Several mutations (M1–M10, bold letters) within the base sequence of the PS element (upper case letters) were constructed as underlined. Lower case letters are the extra bases added for cloning. (B) The effect of various PS mutants on PS DNA binding ability was analyzed by EMSAs. Recombinant purified ZBP-89 protein (1 µg) was incubated with 32P-labeled PS in reaction buffer. A 50-fold excess of either unlabeled PS DNA or mutant PS DNA was pre-incubated with purified, recombinant ZBP-89 protein (1 µg) for 15 min prior to the addition of 32P-labeled PS DNA. DNA–protein complexes were resolved on a 4% polyacrylamide gel as follows: lane 1, 32P-labeled PS alone; lane 2, 32P-labeled PS incubated with purified ZBP-89; lane 3, as for lane 2 with a 50-fold molar excess of cold PS element; lanes 4–13, as for lane 2, but containing a 50-fold molar excess of each dsDNA mutant sequence (M1–M10); lane 14, as for lane 2, but containing a 50-fold molar excess of the DM-PS mutant. (C) The effect of DM-PS and hSE mutants on reporter gene activity in HeLa cells. HeLa cells were transfected with 5 µg of p319, DM-PS or hSE constructs. p18-CAT was included as a negative control. CAT activity is determined as discussed in Figure 1. (D) The effect of the DM-PS- and hSE mutant-containing CAT constructs co-transfected with pPacSp1 (black box) or pPacSp1 plus pPacZBP-89 (gray box) in S2 cells as in (C). Co-transfection of the 353WT construct with pPac Sp1 alone (black box) or plus ZBP-89 (gray box) is included as a positive control for repression.
The pBSII-ZBP-99 gene encoding the full-length human ZBP-99 (residues 1–895) was kindly provided by Dr Juanita Merchant (University of Michigan). pPacZBP-99 was removed from pBSII-ZBP-99 by XbaI (blunt) and XhoI and inserted into the pPac vector digested with BamHI (blunt) and XhoI. The content was verified by DNA sequencing.
Transient transfection and CAT assay
At 48 h before transfection, S2 cells were plated in 6-well plates at a density of 1 × 106 cells/ml. Cells were transfected using the calcium phosphate method according to the manufacturer’s manual (Invitrogen Inc.). pPac-β-gal was co-transfected to normalize the CAT assay values to β-galactosidase activity. The amount of each plasmid was optimized by transfecting increasing amounts of plasmid DNA (see Fig. 1 for titration of Sp1–Sp4). Titration data for ZBP-89 or ZBP-99 expression plasmids are not shown. In these experiments, pUC18-CAT containing no promoter sequences served as the negative control. Plasmids from at least two different preparations were used, and transfections were repeated at least three times. The standard error bar is shown above each column.
HeLa cells were transfected using lipofectin (Gibco-BRL) as described (17). The cells were harvested 48 h after transfection, washed with phosphate-buffered saline (PBS) buffer, and lysates were prepared as for S2 cells. To standardize between different transfections, 1 µg of pCMV-β-gal was co-transfected to serve as an internal control, and 10 µl of the resulting cell extract was used for determining β-galactosidase activity.
The CAT activity of the transfected cells was assessed using a liquid scintillation counting assay that measures the conversion of [14C]chloramphenicol to n-butyryl [14C]chlor amphenicol according to the manufacturer’s protocol (Promega). The values presented are the average of more than three separate transfections.
Western blot analysis
Antibodies directed against Sp1 (H-225: sc-14027), Sp2 (K-20: sc-643), Sp3 (D-20: sc-644), HA (Y-11: sc-805) and c-Myc (A-14: sc-789) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Whole-cell extracts (WCEs) were prepared from S2 cells by the freeze–thaw method, and expression of selected target proteins was assayed by immunoblot analysis as described (24).
Nuclear extract preparation
Nuclear extracts (NEs) were prepared from HeLa cells as described (44). The protein concentration was measured using the Bradford method, and aliquots were stored in Dignam buffer D at –70°C.
Electrophoresis mobility shift assay (EMSA)
For PS DNA, two complementary oligonucleotides were synthesized, purified and annealed using 10× annealing buffer (1 M NaCl, 0.1 M Tris pH 7.8 and 10 mM EDTA pH 8.0). The resulting double-stranded (ds) DNA (1 µg) was 32P-labeled by T4 DNA kinase. EMSA was performed as described (24). For competition assays, a 50-fold excess of unlabeled dsDNA fragment was incubated with the NE at room temperature for 15 min prior to adding the 32P-labeled DNA.
DNA affinity precipitation assays using biotinylated oligonucleotides
S2 cells were harvested 48 h after transfection with pPacSp1 in 500 µl of 1× lysis buffer (20 mM HEPES pH 7.9, 10% glycerol, 50 mM KCl, 0.2 mM EDTA, 1.5 mM MgCl2, 20 µM ZnCl2, 0.1% Tween-20). The cell extract was prepared by the freeze–thaw method. The biotinylated DNA probes were made by PCR with the primers 5′-GCTAGGTCCC GATTGGCT-3′ and 5′-CGAGGGCGCTGTTTTTAT-3′. Fragments from 353WT and 353Sp digested with EcoRI served as templates for PCRs. The resulting PCR product is 92 bp in length and includes the TATA-box, GC-box 1 and GC-box 2. The DNA affinity precipitation assay was performed as described, except that Streptavidin MagneSphere Paramagnetic Particles (SA-PMPs) replaced streptavidin– agarose (34).
RESULTS
Sp1 and full-length Sp3, but not Sp2, activate vimentin promoter transcription in S2 cells
Previously, EMSAs showed that the proximal GC-box 1 specifically binds a protein in HeLa NEs (23). Moreover, mutation of GC-box 1 practically eliminated reporter CAT gene activity, which was not restored by the mere inclusion of upstream DNA containing additional GC-rich sequences. Because vimentin’s GC-box 1 is not a perfect match to the consensus binding site for Sp family members, it became important to determine which Sp protein(s) can bind to vimentin’s promoter region and activate transcription in vivo. S2 cells were used in these experiments, since they are known to lack all endogenous Sp1-like family proteins (37,45). From the phylogenic tree of Sp/XKLF, Sp1, Sp2, Sp3 and Sp4 are closely related in molecular weight as shown, mostly bind GC-rich sequences and share many similar features, which include a DNA-binding domain composed of four zinc fingers and multiple serine/threonine-rich or glutamine-rich transactivation domains (Fig. 1A). Because GC-box 1 is comprised of GC-rich sequences, other XKLF family members which prefer GT-rich sequences were ignored for this initial study. To determine which of the four Sp members regulates vimentin expression, a series of titrations with Sp1, Sp2, Sp3 and Sp4 were done (Fig. 1B and C). Sp-containing plasmids are co-transfected with 5 µg (a predetermined optimal amount) of the vimentin promoter CAT construct, 261WT (see Fig. 2A), into S2 cells. The results show that Sp1 and full-length Sp3 (USp3) are strong positive regulators of the vimentin promoter, showing saturation at 0.2 µg (Fig. 1B). Sp4 is intermediate, requiring a higher dose of plasmid (5 µg), while Sp2 and the shorter form of Sp3 (Sp3) fail to activate gene expression (Fig. 1C). Therefore, we chose to focus on Sp1 and USp3. In addition, we found a synergism between Sp1 and USp3, but not with the shorter form of Sp3 (Fig. 1D). Independently of concentration, USp3 can increase CAT activity 2.5- to 3-fold. Western blots confirm expression of Sp1 (Fig. 6C), Sp2 (Fig. 1E), and both the full-length and shorter forms of Sp3 in transfected S2 cells (Fig. 1F). Although the Sp3 protein is expressed at a lower level than USp3, there is still sufficient protein to produce a response in the transfection assays, if the shorter form of Sp3 was capable of activating transcription. This was verified by transfecting 10 µg of Sp3 plasmid, which yielded high levels of protein compared with the transfection of 0.2 µg of plasmid USp3 on western blots, yet Sp3 still failed to support reporter gene activity (data not shown).
Determination of the minimal vimentin promoter required for Sp1 activation
To determine the minimal vimentin promoter (Fig. 2A) required for Sp1 or USp3 activation, a series of vimentin promoter CAT constructs, including 83WT, 189WT, 261WT and 353WT, were co-transfected with pPacSp1 or pPacUSp3 (Fig. 2B). Reporter gene activity showed that 189WT, 261WT and 353WT constructs were all equally activated by either Sp1 or USp3, whereas the 83WT construct was not. It was felt that the failure to obtain CAT activity with 83WT might be due to the fact that this promoter fragment contains a single GC-box and TATA-element, which by themselves might not be sufficient to recruit the transcription apparatus successfully.
GC-box 1 is indispensable for Sp1 activation, while the NF-κB and PEA3 sites are not required
In addition to the four GC-boxes, the vimentin promoter also contains NF-κB and PEA3 sites, which have been shown to contribute to vimentin expression (Fig. 2A). To determine which of these elements are important for gene expression, each element was mutated to a known, non-functional sequence within an otherwise normal, active promoter of 353 bp, and designated as 353WT, 353PEA3, 353NF and 353Sp to denote which sequence was mutated (Fig. 2C). Each construct was co-transfected with pPacSp1 or pPacUSp3 (Fig. 2D). As expected, transfection with the pUC18CAT vector (no insert) yields no measurable gene activity. However, constructs containing either the NF-κB or PEA3 mutation were as active as 353WT with co-expression of either Sp1 or USp3. In fact, there was no significant difference between the wild-type promoter and these two mutant constructs. Previously, it had been shown that S2 cells contain homologs of NF-κB and PEA3 proteins, which implies that Sp1 or USp3 activation is independent of the NF-κB and PEA3 sites and their associated factors (Fig. 2D) (46,47). However, for the GC-box 1 mutant (353Sp), CAT activity dropped by >80%, which shows that Sp1 activation is dependent on GC-box 1, wild-type sequence.
The inability of mutant GC-box 1 DNA to bind Sp1 was tested further in vitro by a DNA affinity precipitation assay using biotinylated oligonucleotides (Fig. 2E). Here, wild-type GC-box 1 DNA can bind Sp1 from pPacSp1-transfected S2 extracts (WT-BP). However, the mutant GC-box 1 (MT-BP) was severely compromised for Sp1 binding, further documenting the requirement for a GC-box 1 DNA sequence.
ZBP-89 or ZBP-99 can repress Sp1 activation in vivo
Previously, the PS element was shown to bind a 105 kDa protein by UV cross-linking and southwestern blot analysis (24). The core sequence of this element (ggaCCcCCcCC) is similar to the consensus DNA-binding site for ZBP-89 (gccCCtCCxCC). Moreover, the DNA–protein complex found in HeLa NEs could be supershifted by anti-ZBP-89, and recombinant purified protein was capable of binding to the PS element in vitro. All these results suggest that ZBP-89 is the protein binding to the PS element. To confirm the repression of ZBP-89 by transient transfection analysis, pPacZBP-89 or pPacZBP-99 is co-transfected with pPacSp1 or pPacUSp3 in S2 cells (Fig. 3A). These results clearly show that either ZBP-89 or ZBP-99 can repress Sp1 or USp3 activation 5-fold in vivo, which confirms the conclusion that ZBP-89 is a repressor of vimentin gene expression. Whereas the ODC promoter was independently shown to be repressed by both ZBP-89 (37) and ZBP-99 (42), this is the first direct comparison of the gene-repressing activities of these ZBP family members.
Figure 3.

ZBP-89 or ZBP-99 can repress activation by either Sp1 or USp3, but requires a functional PS element. (A) The construct 353WT (5 µg) was transfected into S2 cells with pPacSp1 (0.2 µg), pPacUSp3 (0.2 µg), pPacZBP-89 (1 µg) or pPacZBP-99 (1 µg) in various combinations as indicated on the x-axis. CAT activity is expressed as the mean ± SE. (B) The 353 construct (5 µg) containing the DM-PS mutation (DM-PS) was co-transfected into S2 cells with the same expression plasmids as in (A).
Functional PS element is required for ZBP-89 or ZBP-99 repression
Previously, EMSAs showed that bacterially expressed ZBP-89 could shift the PS element, but not the non-functional, double mutant PS (DM-PS) element (24). In addition, ZBP-89 could be co-immunoprecipated with Sp1 in the presence of PS DNA, whereas Sp1 by itself failed to bind to the PS element, although the PS element (GgaCCcCCCCC) exhibits an 8 out of 11 match to a GC-box consensus sequence [(A/G)(T/C)(T/C)CCGCCCC(A/C)]. This implies that Sp1 is held in the DNA–protein complex via protein–protein interaction with ZBP-89. To confirm the consequences of this failed interaction in vivo, pPacZBP-89 or pPacZBP-99 is co-transfected with pPacSp1 or pPacUSp3 and the 353 construct containing the DM-PS mutant into S2 cells (Fig. 3B). The results show that neither ZBP-89 nor ZBP-99 can repress Sp1 or USp3 activation of the DM-PS construct. This confirms that a functional PS element is required for either ZBP-89 or ZBP-99 repression in vivo.
Identification of bases essential for binding of ZBP-89
Since the consensus sequence for ZBP-89 is gccCCtCCxCC and the sequence of the PS element is ggaCCcCCcCC within the region of the non-functional DM-PS mutant, a series of mutant oligonucleotides (M1–M10) were synthesized to determine ZBP-89’s binding specificity for vimentin’s PS element (Fig. 4A). Each mutant contains a substitution of 2–3 adenine or thymidine bases within the GC-rich region. Competition EMSAs were done with 32P-labeled PS DNA in the presence of a 50-fold excess of non-radioactive, wild-type or mutant PS element (as dsDNA) incubated with His-tagged, purified recombinant ZBP-89. Unlabeled wild-type PS element (lane 3) and mutants M1, M3, M4, M7, M8, M9 and M10 compete with the 32P-labeled PS for formation of the DNA–protein complex (Fig. 4B). This indicates that these mutant bases are not important for binding. However, mutants M2 (lane 5), M5 (lane 9) and M6 (lane 10) are unable to compete with the 32P-labeled PS element, suggesting that these bases are essential for binding. The DM-PS element (lane 14), which was known to abolish protein binding, is used as a control (24). As expected, it did not compete with the 32P-labeled PS as seen for M2, M5 and M6. Similar results were obtained with HeLa NEs (data not shown). We believe the lower shifted band in lanes 2, 8 and 9 comes from the premature release of ZBP-89 translation products in Escherichia coli due to ZBP-89’s content of rare codons, as this band was not detected with HeLa extracts. Moreover, the His-tagged, purified recombinant ZBP-89 showed a contaminating band on SDS–polyacrylamide gels, which cross-reacted with antibodies to ZBP-89 on a western blot. In summary, these results show that the CCC sequence in the middle of the PS element (ggacccCCCcc) is required for ZBP-89 protein binding, which differs somewhat from the stated ZBP-89 consensus element (gccCCtCCxCC).
The effect of PS mutations on transcription
To prove that these bases are indeed important for binding ZBP-89 in vivo, functional assays were done. The three bases (as found in M2 from Fig. 4A) were mutated within the PS element of an otherwise normal 319WT construct and called hSE. HeLa cells were transfected with wild-type p319, 319DM-PS or the 319hSE mutant (Fig. 4C). The hSE mutant failed to repress reporter gene activity. In fact, these three base changes were as deleterious to transcriptional repression as the nine base changes found in the original DM-PS mutant. Similar experiments were also done in S2 cells, where it was found that co-expression of ZBP-89 could not repress either the hSE mutant or the DM-PS construct (Fig. 4D), but could repress expression from the 353WT construct. These results prove that these bases are important for ZBP-89 binding and narrow down considerably the relevant binding site within the PS element for ZBP-89.
Deletion mutants of Sp1 show that either glutamine-rich region is indispensable for activation of the vimentin promoter
A series of Sp1 deletion constructs (Fig. 5A) were prepared in order to determine which domains are required for activation by Sp1 and ultimately repression by ZBP-89. All Sp1 deletions retain the three zinc finger DNA-binding domains located at the C-terminus and thus are capable of binding to GC-box 1. Each Sp1 construct or deletion thereof was co-transfected with 353WT into S2 cells (Fig. 5B, gray boxes). Reporter gene assays showed that Δint349, 516CΔint122, 440CΔint122 and Δint162 could all activate the vimentin promoter to comparable levels. Only 516CΔint266 failed to activate vimentin expression. Since 516CΔint266 does not contain much of a glutamine-rich region, we conclude that either glutamine-rich region is required for Sp1 activation. Western blots confirm that all Sp1 deletion constructs are equally expressed in S2 cells (Fig. 5C). Since Δint349 (lane 6) is deleted for the internal region, which contains the anti-Sp1 epitope, its expression cannot be verified. However, since it is exhibiting a comparable, if not better, effect on reporter gene activity as other Sp1 deletions (outside 516CΔint266), we assume that this mutant protein must be expressed to the same extent as other Sp1 deletions.
Figure 5.

At least one glutamine-rich region of Sp1 is required for activation of the vimentin promoter as well as repression by ZBP-89. (A) A schematic of Sp1 and its various deletion mutants. The number of amino acids (aa) included in each Sp1 deletion and the position of the Sp1 epitope are noted. (B) Sp1 or its various deletion mutants in the pPac vector (0.2 µg) were transfected with 5 µg of the 353WT construct (gray boxes) or plus 1 µg of pPacZBP-89 (black boxes) into S2 cells. CAT activity is expressed as the mean ± SE. (C) Western blot analysis of WCEs prepared from S2 cells transfected with Sp1 and its various deletion mutants and incubated with anti-Sp1 as follows: lane 1, non-transfected control; lane 2, transfected with 516c-Δint112; lane 3, transfected with 516c-Δint266; lane 4, transfected with 440c-Δint122; lane 5, transfected with Δint162; lane 6, transfected with Δint349; lane 7, transfected with pPacSp1.
Deletion mutants of Sp1 show that the glutamine-rich region is also indispensable for ZBP-89 repression
In order to determine what domain of Sp1 is required for ZBP-89 repression, this same series of Sp1 constructs (Fig. 5A) were co-transfected with 353WT and ZBP-89 (Fig. 5B, black boxes). The resulting CAT assays show that the activation of all constructs is repressed 2- to 3.5-fold by co-expression with ZBP-89. All these deletion constructs contain at least one glutamine-rich domain, which suggests that ZBP-89 represses Sp1 by interacting with the same glutamine-rich domain that is required for gene activation. Since at least one glutamine-rich domain is also required for Sp1 activation (no activity is obtained with 516CΔint266), it is impossible to verify this conclusion by transient transfection analysis. Thus, by elimination, we suggest that ZBP-89 must be interacting with the same glutamine-rich domain(s) required for Sp1 activation.
Deletion mutants of ZBP-89 show that the N-terminus is required for gene repression
ZBP-89 is also a zinc finger DNA-binding protein (Fig. 6A). Unlike Sp1, the four zinc fingers are internal and closer to the N-terminus. They are surrounded by two basic domains (BD1 and BD2) and an additional acidic domain (AD) located closest to the N-terminal end. To determine which domain(s) is required for ZBP-89’s repression of Sp1, various ZBP-89 deletion mutants were constructed and inserted into the pPac vector as shown (Fig. 6A). Each retained the zinc finger DNA-binding domain and thus was capable of binding DNA in vivo (Fig. 6A). Each was co-transfected with pPacSp1 and 353WT into S2 cells, and the resulting effect on reporter gene activity was measured (Fig. 6B). The retention of gene activity shows that deletion of the AD (dA) or AD plus BD1 (dAB1) regions results in the loss of repression, whereas deletion of BD2 or the C-terminal region does not affect gene repression. The equivalent expression of all deletion mutants was confirmed by western blot analysis with anti-HA antibody (Fig. 6C). These studies suggest that the acidic and basic domain closest to the N-terminus are required for ZBP-89 repression, while the C-terminal and BD2 regions are not.
Previously, the exchange of the zinc finger DNA-binding domain within the mouse homolog of ZBP-89 for the equivalent region from GAL4 resulted in the localization of a repression domain to the basic-rich region of ZBP-89 (38). Although the identity of the protein(s) that interacted with the GAL4-ZBP-89 fusion protein expressed in C2C12 muscle cells was not confirmed in this study, it was clear that this interaction was independent of the type of DNA-binding domain. Thus, we conclude that ZBP-89 and Sp1 do not interact via the zinc finger domains and these domains are not required for repression once protein is bound to DNA.
hTAFII130 can reverse the repression caused by ZBP-89
The above results suggest that ZBP-89 can repress Sp1 activation by binding to either glutamine-rich transactivation domain. Interestingly, these are the same domains that Sp1 uses to recruit the transcription complex by interacting specifically with hTAFII130 (48). Therefore, is it possible that overexpression of hTAFII130 could restore Sp1 activation? To address this question, increasing amounts of the pPac-hTAFII130 plasmid (0.5–2 µg) were co-transfected with ZBP-89 and Sp1 into S2 cells (Fig. 7A, black boxes). The resulting reporter gene activity shows that transcription is restored by co-expression of hTAFII130. In fact, even higher levels of gene activity were obtained with 2 µg of the pPac-hTAFII130 plasmid compared with the Sp1-only control (lane 4 compared with lane 1). However, co-transfection of ZBP-89 and hTAFII130 with an Sp1 mutant lacking a glutamine- rich domain (516c-Δint266) did not restore gene activity, confirming that hTAFII130-mediated rescue is dependent on Sp1 protein containing a functional glutamine-rich domain (Fig. 7A, gray boxes). The expression of hTAFII130 was confirmed by western blotting with anti-Myc antibody (Fig. 7B).
Figure 7.
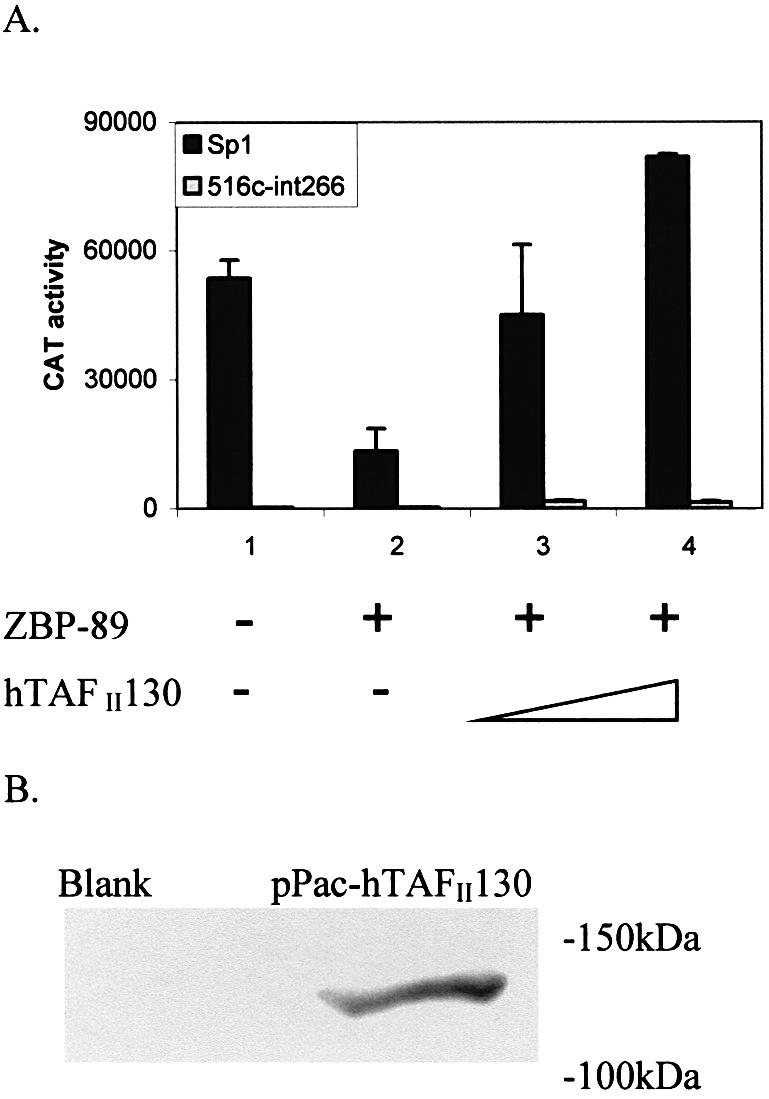
Human TAFII130 can reverse the repression of ZBP-89 on Sp1. (A) Increasing amounts (0.5 and 2 µg) of pPac-hTAFII130 were co- transfected with 353WT (5 µg), pPacSp1 (0.2 µg) and pPacZBP-89 (1 µg) into S2 cells (black box). A second set of transfections replaced pPacSp1 with the Sp1 mutant 516c-Δint266 lacking both glutamine-rich domains (gray box). CAT activity is expressed as the mean ± SE. (B) Western blot analysis of WCEs prepared from S2 cells non-transfected (blank) or transfected with pPac-hTAFII130 (myc-tagged) and incubated with anti-Myc antibody. The position of migration of molecular weight markers is noted.
DISCUSSION
The ubiquitously expressed Sp1 protein has been implicated in the activation of a large number of genes and in the regulation of the cell cycle by interacting with such factors as retinoblastoma protein (RB) or E2F (49–51). Interestingly, vimentin gene expression is regulated in a developmental, tissue-specific and cell cycle-dependent way (20,25). Sp1 is essential in early stages of embryonic development, which correlates with the time frame for vimentin synthesis (52). Hence, we have analyzed the regulation of vimentin gene expression by Sp1 and ZBP-89 by functional, transient transfection assays in vivo.
Sp1 and full-length Sp3 are strong positive activators for vimentin expression
A variety of previous studies indicated that a protein binds specifically to the proximal GC-box 1 within vimentin’s promoter, but not to the other putative GC-boxes (23). Here, it was found that both Sp1 and USp3 are strong positive activators for vimentin transcriptional regulation in S2 cells (Fig. 1B). Gene activity from any vimentin promoter CAT construct required supplementation with these specific Sp family members. Moreover, a mutation in GC-box 1 abolishes most, if not all, transcriptional activity. Thus, GC-box 1 binds either Sp1 or USp3, and this binding is required to activate gene expression. These in vivo transfection results are consistent with our previous in vitro results (23,24).
Interestingly, the shorter form of Sp3 failed to activate the vimentin promoter in S2 cells. Although Sp1 and Sp3 are very similar in structure, Sp3 is more complicated due to the existence of three Sp3 isoforms (Fig. 1A). USp3 (115 kDa) is initiated at a non-AUG codon (53), while the two smaller isoforms of Sp3 (70 kDa) arise from initiation at two different, but juxtaposed internal, initiation sites (54). Originally, Sp3 was found to repress Sp1-mediated activation by competing for the same binding site, thus preventing the subsequent binding and activation of Sp1 (55). However, now it is apparent that whether or not Sp3 represses Sp1 activation is promoter specific and indeed in some cases Sp3 can be an activator (30). USp3 carries both sets of serine/threonine- and glutamine-rich activation domains, whereas the smaller species contain only the second activation region (domain B) and thus can only act as weak activators. Since only USp3 and not the shorter forms can activate vimentin transcription (Fig. 1C), it implies that the N-terminal region of USp3 plays an important role in activating vimentin gene expression, which cannot be fulfilled by the shorter forms. In addition, synergism exists between Sp1 and USp3, but not with the Sp3 shorter forms (Fig. 1D). Sp1 can form multimeric complexes that are thought to exert transcriptional superactivation by binding to basal transcription factors, but this does not seem to be the case for Sp3 (51). Thus, Sp1 activation is a complex mechanism involving not only protein–DNA interactions, but also the interactions of multiple modular domains of Sp1 with co-activator proteins (56). A synergistic interaction between Sp1 and USp3 was also found for the murine Ctpct (CTP:phosphocholine cytidylytransferase-α) promoter (57). Here, synergism was explained by the presence of similar Sp1 complexes that were able to induce conformational changes by interacting with both basal transcription factors and Sp3. However, in this promoter, superactivation was not found at lower concentrations of transfected Sp3 plasmid (0.25– 0.5 µg), but only when concentrations exceeded 1 µg. On the other hand, USp3 causes superactivation of the vimentin promoter at either lower (0.2 µg) or higher concentrations (5 µg). Thus, for the vimentin promoter, a conformational change may also account for Sp3 superactivation, but this is apparent at all concentrations of Sp3 tested. However, the short form of Sp3 cannot synergize with Sp1. Thus, it seems that the N-terminal region of USp3 is required not only for activation in general, but for superactivation of Sp1 as well. Thus, we conclude that the N-terminal region of Sp3 is required for multiple effects upon vimentin gene regulation, which cannot be substituted for by the shorter Sp3 form(s). Deletion mapping experiments further support the importance of either Sp1 glutamine-rich region for activation of the reporter gene in S2 cells (Fig. 5A). Yet it is apparent that for Sp3, not all glutamine-rich regions are equal, since the shorter form of Sp3 is not active. Thus, we conclude that only the N-terminal, serine/threonine- and glutamine-rich domains support transcriptional activity of the vimentin promoter.
Our results also showed that Sp2 failed to activate vimentin reporter gene expression. It is known that Sp2 does not bind to the classical Sp1 GC-box, but rather to a GT-rich element (GGTGTGGGG) as found in the TCR Vα promoter (53). This might explain why Sp2 does not activate the vimentin gene with its GC-rich sequence (Fig. 1C).
Sp1 and Sp3 are ubiquitously expressed transcription factors (51), whereas Sp4 is highly expressed in the developing central nervous system, and is abundant in epithelial tissues, developing teeth and testes (58). In the case of vimentin, Sp4 moderately activates reporter gene expression (Fig. 1C), but only at a much higher level of transfected plasmid (0.2 µg for Sp1 or USp3 versus 5 µg for Sp4), which might be due to any of the following reasons. First, we did not document the level of Sp4 expressed protein, so this family member may not be as highly expressed as Sp1 or USp3, thus accounting for the requirement for transfecting a higher amount of plasmid before detecting an increase in gene activity. Secondly, the phylogenic tree of Sp/XKLF shows that the structures of Sp1, Sp3 and Sp4 are more closely related to each other than Sp2. In addition, Sp4 also prefers binding to a GC-box, which is similar to both Sp1 and Sp3, but not Sp2 (29). Lastly, vimentin is not expressed in terminally differentiated neuronal, glial or epithelial cells, but is found in some progenitor cells, neuroblastomas, gliomas or most, if not all, metastatic cancer cells, which arise from epithelial origins. Thus, in these special cases, activation by Sp4 may play more of a role in supporting vimentin gene expression.
ZBP-89 represses vimentin transcription via interaction with Sp1
Our previous in vitro studies suggested that vimentin’s PS element-binding protein could be ZBP-89 (24). Here, we found that not only is vimentin’s PS element (ggaCCC CCCCC) similar to the consensus DNA-binding site for ZBP-89 (ggcCCtCCxCC) as found in other genes, but the x must be a C and the three Cs encompassing this portion of the binding site are essential for vimentin’s PS element to bind ZBP-89 and subsequently repress transcription (Fig. 4). Previously, we found the addition of purified Sp1 to a PS–ZBP-89 complex produced a new band with slower mobility on EMSA gels than PS–ZBP-89 alone (24). Thus, Sp1 could be found in a PS–protein complex, but only after the prior addition of ZBP-89. Sp1 could not bind to PS DNA by itself. Moreover, antibodies to ZBP-89 or Sp1 were capable of supershifting the larger PS DNA–protein complex, while unrelated antibodies gave no supershift.
Here, we verified that both ZBP-89 and its homolog ZBP-99 are capable of inhibiting Sp1 activation of the vimentin promoter in S2 cells by transient transfection analyses in vivo (Fig. 3A). ZBP-99 is thought to be structurally and functionally homologous to ZBP-89 (42). Its four zinc fingers share 91% amino acid sequence similarity and 79% sequence identity with those of ZBP-89. Besides that, ZBP-99 contains a basic-rich domain at the N-terminus, but also contains an extra 100 amino acids containing glycine-, proline- and histidine-rich regions not found in ZBP-89. Although the basic domain is 55% similar between the two proteins, ZBP-99 does not contain much of an acidic domain compared with ZBP-89. Yet both proteins serve to repress Sp1 activation equally well, leading to the conclusion that it must be the basic-rich region which is the most important for repression.
ZBP-89 has been shown to regulate the expression of several genes where it can function as either an activator or a repressor. Thus, its mechanism of action must be promoter specific. In all cases where ZBP-89 serves to repress gene expression, it appears to compete with another protein, such as Sp1, Sp3, Sp4, WT1 or Krox, for binding to an overlapping site in DNA (33,37,43). Quite differently from all other promoters repressed by ZBP-89, the PS element in the vimentin promoter is not overlapping with an Sp1-binding site. In fact, there is a considerable distance (235 bp) between the PS element and GC-box 1. Thus, it is improbable that Sp1 and ZBP-89 compete for DNA binding. Rather EMSAs suggest that both proteins can be found in complex with PS DNA, meaning that GC-box 1 and the PS element might need to reside on the same face of adjacent nucleosomes. In this context, an optimal spatial location for their respective binding sites could exist, permitting the protein–protein interaction detected by EMSAs.
To analyze further the mechanism of interaction between Sp1 and ZBP-89, we created a series of Sp1 and ZBP-89 deletion constructs. Functional assays with these constructs transfected into S2 cells suggested that the N-terminal region of ZBP-89 and a glutamine-rich region of Sp1 were required for repression (Figs 5 and 6). From these results, it is tempting to speculate that ZBP-89’s binding to Sp1 may abolish activation of the vimentin gene by prohibiting Sp1’s ability to recruit the transcription complex. This is different from what has been suggested for other promoters repressed by ZBP-89. For example, in the case of the ENA-78 promoter, binding to site A was mutually exclusive between ZBP-89 and Sp1 (40). When ZBP-89 was bound, minimal transcription occurred. Upon replacement by Sp1, basal expression was restored. Surprisingly, in the bovine adrenodoxin (Adx) gene, ZBP-89 could repress activation of the Adx reporter gene construct, which contained a mutant Adx-binding element (39). Thus, in this case, ZBP-89 could repress Sp1’s activation without being bound to DNA. However, the vimentin promoter is quite different from these examples. In vimentin, the binding of ZBP-89 to the PS element is indispensable for the repression of ZBP-89, since mere expression of ZBP-89 could not repress the vimentin promoter construct containing a mutant PS element, as either DM-PS or hSE (Figs 3B, and 4C and D). Our previous results with purified components in EMSAs or PS DNA affinity columns with crude NE show that Sp1 can be held in a PS–ZBP-89 complex via protein–protein interaction, but not for the DM-PS mutant (24).
Thus, the regulation of vimentin expression by ZBP-89 and Sp1 appears to be unique compared with previous reports. The interesting question is how could the binding of ZBP-89 to the PS element repress Sp1 activation. By both in vitro and now in vivo transfection experiments, we have shown that ZBP-89 bound to DNA can interact with Sp1 (24). We propose that this interaction may block Sp1’s ability to make the necessary contacts with the transcriptional machinery, thus inhibiting transcription. Because Sp1 has been shown to interact with YY1, TBP, NF-κB, STAT-1, hTAFII130 and dTAFII110, it has been suggested that Sp1 is a mediator between sequence-specific and general transcription factors (29,30,51). More over, it has been shown that hTAFII130 interacts with various cellular activators such as Sp1 or CREB, suggesting that it may serve to bridge these factors to the transcription machinery (48,59). Although the data were not shown, it was reported that BFCOL1 (the mouse homolog of ZBP-89) can interact with dTAF110, the Drosophila homolog of hTAFII130 (60,61). Recently, it was proposed that mutant huntingtin inhibits Sp1-mediated transcription by interfering with Sp1/hTAFII130 function (59). It was postulated that this disruption could lead to changes in gene expression, thereby contributing to the Huntington’s disease phenotype. Similarly, we found that the overexpression of hTAFII130 could indeed reverse the repression caused by ZBP-89 with the vimentin promoter (Fig. 7A). Thus, it is possible for ZBP-89 to repress Sp1 by interfering with the required interaction between Sp1 and hTAFII130. Since both ZBP-89 and hTAFII130 interact with the glutamine-rich region of Sp1, a competition might exist between ZBP-89 and hTAFII130 for Sp1 binding. Overexpression of hTAFII130 relieves this competition and permits a permissive interaction with Sp1, which overrides ZBP-89 repression. However, hTAFII130-mediated rescue is dependent on the glutamine-rich domains of Sp1, since a mutant Sp1 protein deleted for these domains cannot be rescued by overexpression of hTAFII130. In addition, hTAFII130 further enhances Sp1 activation 1.5-fold even in the presence of ZBP-89. This result implies that hTAFII130 can further elevate Sp1 activation in addition to relieving ZBP-89 repression.
Negative regulation of Sp1
Sp1 has been shown to be negatively regulated by a number of different mechanisms. For example, some proteins such as Sp1-I or p74 have been shown to bind directly to Sp1 and thereby repress Sp1 activity. Binding of Sp1-I appears to block the subsequent binding of Sp1 to DNA, but this blockage can be overcome by the addition of recombinant RB protein (49). Hence, Sp1-I is both an Sp1- and RB-binding protein, which together regulate Sp1 transactivation. p74 binds to the N-terminal serine/threonine-rich subdomains of Sp1, thereby resulting in the inhibition of Sp1-mediated transcription (62). Similar to p74, we found that ZBP-89 requires a glutamine-rich subdomain in order to repress Sp1 activation. On the other hand, myc represses the p21 promoter by binding to the zinc finger domain of either Sp1 or Sp3 (63). Unlike ZBP-89, none of these Sp1 repressor proteins displays any affinity for DNA. Thus, they regulate Sp1 activity by direct binding to either the activation or DNA-binding domain and subsequently block further Sp1 associations. In addition, Sp1 activity can be inhibited by histone deacetylase 1 (HDAC1) (64). Treatment with the HDAC inhibitors butyrate or trichostatin A (TSA) was shown to elevate the p21waf1/cip1 promoter dependent on ZBP-89’s ability to recruit p300 (34). In this case, activation was specific to p300 and not CBP, and the acidic domain of ZBP-89 was required.
Other proteins such as Gut-enriched Krüppel-like factor (GKLF), Basic Transcription element binding protein 3 (BTEB3), TGFβ-Inducible Early Gene 2 (TIEG2) or islet-specific transcription factor (βTF-1) have been shown to repress Sp1 activity by competing with Sp1 for binding to the same, overlapping or adjacent sites in DNA (65–67). In some cases, this mechanism can become very complicated, as recently reported for the regulation of ODC gene expression by Sp1, ZBP-89 and GKLF (65). Here, GKLF represses ODC gene expression by binding to a site that overlaps with the ZBP-89 site. Chromatin immunoprecipitation assays suggest that GKLF blocks Sp1 binding to DNA, but exactly how GKLF and ZBP-89 associate to regulate ODC gene expression is unknown.
Finally, repressors of Sp1 could bind their own DNA element and directly block Sp1 activation, as shown here. Not many examples of this type of regulation have been identified. GKLF has been reported to suppress the activity of the cytochrome P-4501A1 promoter presumably by physically interacting with Sp1, yet a stable ternary complex between GKLF, Sp1 and BTE (the DNA-binding site) was not observed (68). Previously, it was suggested that ZBP-89 competed with Sp1 for binding to an overlapping GC-rich site on DNA. From our studies, it is obvious that this may not be the case for all promoters regulated by ZBP-89. Rather, we suggest that ZBP-89 binds to its own PS element, and represses transcription not by competing with Sp1 for binding to DNA, but more probably by protein– protein interactions, which serve to block Sp1 from making crucial contacts with the transcriptional apparatus. In support of this hypothesis, we found that repression could be overcome by the overexpression of hTAFII130, one of Sp1’s known partners required for transcription (48). Previously, with purified components, we could detect sequential binding of ZBP-89 followed by Sp1 to PS DNA (24). However, we have not been able to co-immunoprecipitate ZBP-89 and Sp1 from HeLa NEs even when these proteins were overexpressed. From these results, we conclude that a functional interaction between ZBP-89 and Sp1 is confined to DNA, which is very different from the examples of Sp1 regulation cited above. Therefore, we propose that ZBP-89 binds to its own DNA element, interacts with Sp1 when bound to DNA, and subsequently blocks it from activating gene expression. The interaction of two proteins with opposing activities, i.e. activator versus repressor, is a common theme in modulating cellular regulation. As more ZBP-89-regulated genes are identified, it will be interesting to determine which genes are regulated like vimentin and what further underlies the mechanism of ZBP-89 activation versus repression.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr Yongzhong Wu and Ms Yonghe Li for their expert technical assistance. We also thank Drs Robert Tjian, Guntram Suske, Reinhold Krug, Juanita Merchant, John Noti, Paul D. Gardner and Naoko Tanese for providing various expression plasmids. HeLa cells were purchased from the National Cell Culture Center. This work was supported by NHLBI, National Institutes of Health (NIH) grant HL-45422 to Z.E.Z.
REFERENCES
- 1.Franke W.W., Schmid,E., Osborn,M. and Weber,K. (1978) Different intermediate-sized filaments distinguished by immunofluorescence microscopy. Proc. Natl Acad. Sci. USA, 75, 5034–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y., Kobori,J.A. and Hood,L. (1993) The htβ gene encodes a novel CACCC box-binding protein that regulates T-cell receptor gene expression. Mol. Cell. Biol., 13, 5691–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skalli O. and Goldman,R.D. (1991) Recent insights into the assembly, dynamics and function of intermediate filament networks. Cell Motil. Cytoskel., 19, 67–79. [DOI] [PubMed] [Google Scholar]
- 4.Goldman R.D., Khuon,S., Chou,Y.H., Opal,P. and Steinert,P.M. (1996) The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell Biol., 134, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinert P.M. and Roop,D.R. (1988) Molecular and cellular biology of intermediate filaments. Annu. Rev. Biochem., 57, 593–625. [DOI] [PubMed] [Google Scholar]
- 6.Lazarides E. (1982) Intermediate filaments: a chemically heterogeneous, developmentally regulated class of proteins. Annu. Rev. Biochem., 51, 210–250. [DOI] [PubMed] [Google Scholar]
- 7.Duprey P. and Paulin,D. (1995) What can be learned from intermediate filament gene regulation in the mouse embryo. Int. J. Dev. Biol., 39, 443–457. [PubMed] [Google Scholar]
- 8.Pazanin L., Poljakovic,Z. and Palic,J. (1992) Vimentin and glial fibrillary acidic protein expression in relation to neoplastic cell differentiation in glial tumors. Neurol. Croat., 41, 191–203. [PubMed] [Google Scholar]
- 9.Kamada H., Itoh,T., Hyogo,T., Satoh,S., Ogasawara,T., Fujiwara,H., Ara,S., Hotta,T., Suematsu,K., Nakamura,J. et al. (1988) Immunohistochemical study of developing rat embryo—localization of vimentin, GFAP, neurofilament protein within rat embryo central nervous system. No To Shinkei, 40, 211–218. [PubMed] [Google Scholar]
- 10.Gibson C.W., Rittling,S.R., Hirschhorn,R.R., Kaczmarek,L., Calabretta,B., Stiles,C.D. and Baserga,R. (1986) Cell cycle dependent genes inducible by different mitogens in cells from different species. Mol. Cell. Biochem., 71, 61–69. [DOI] [PubMed] [Google Scholar]
- 11.Carey I. and Zehner,Z.E. (1995) Regulation of chicken vimentin gene expression by serum, phorbol ester and growth factors: identification of a novel fibroblast growth factor-inducible element. Cell Growth Differ., 6, 899–908. [PubMed] [Google Scholar]
- 12.Chu Y.W., Seftor,E.A., Romer,L.H. and Hendrix,M.J. (1996) Experimental coexpression of vimentin and keratin intermediate filaments in human melanoma cell augments motility. Am. J. Pathol., 148, 63–69. [PMC free article] [PubMed] [Google Scholar]
- 13.Zajchowski D.A., Bartholdi,M.F., Gong,Y., Webster,L., Liu,H.L., Munishkin,A., Beauheim,C., Harvey,S., Ethier,S.P. and Johnson,P.H. (2001) Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res., 61, 5168–5178. [PubMed] [Google Scholar]
- 14.Young A.N., Amin,M.B., Moreno,C.S., Lim,S.D., Cohen,C., Petros,J.A., Marshall,F.F. and Neish,A.S. (2001) Expression profiling of renal epithelial neoplasms: a method for tumor classification and discovery of diagnostic molecular markers. Am. J. Pathol., 158, 1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sommers C.L., Skerker,J.M., Chrysogelos,S.A., Bosseler,M. and Gelmann,E.P. (1994) Regulation of vimentin gene transcription in human breast cancer cell lines. Cell Growth Differ., 5, 839–846. [PubMed] [Google Scholar]
- 16.Gilles C. and Thompson,E.W. (1996) The epithelial to mesenchymal transition and metastatic progression in carcinoma. Breast J., 2, 83–96. [Google Scholar]
- 17.Chen J.H., Vercamer,C., Li,Z., Paulin,D., Vandenbunder,B. and Stehelin,D. (1996) PEA3 transactivates vimentin promoter in mammary epithelial and tumor cells. Oncogene, 13, 1667–1675. [PubMed] [Google Scholar]
- 18.Garzon R.J. and Zehner,Z.E. (1994) Multiple silencer elements are involved in regulating the chicken vimentin gene. Mol. Cell. Biol., 14, 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stover D.M. and Zehner,Z.E. (1992) Identification of a cis-acting DNA antisilencer element which modulates vimentin gene expression. Mol. Cell. Biol., 12, 2230–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paulin D., Lilienbaum,A., Duprey,P., Li Z. and Vicart,P. (1990) Regulatory elements of the human vimentin gene: activation during proliferation. Reprod. Nutr. Dev., 30, 423–429. [DOI] [PubMed] [Google Scholar]
- 21.Salvetti A., Lilienbaum,A., Li,Z., Paulin,D. and Gazzolo,L. (1993) Identification of a negative element in the human vimentin promoter: modulation by the human T-cell leukemia virus type I Tax protein. Mol. Cell. Biol., 13, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moura-Neto V., Kryszke,M.H., Li,Z., Vicart,P., Lilienbaum,A. and Paulin,D. (1996) A 28-bp negative element with multiple factor-binding activity controls expression of the vimentin-encoding gene. Gene, 168, 261–266. [DOI] [PubMed] [Google Scholar]
- 23.Izmailova E.S., Wieczorek,E., Perkins,E.B. and Zehner,Z.E. (1999) A GC-box is required for expression of the human vimentin gene. Gene, 235, 69–75. [DOI] [PubMed] [Google Scholar]
- 24.Wieczorek E., Lin,Z., Perkins,E.B., Law,D.J., Merchant,J.L. and Zehner,Z.E. (2000) The zinc finger repressor, ZBP-89, binds to the silencer element of the human vimentin gene and complexes with the transcriptional activator, Sp1. J. Biol. Chem., 275, 12879–12888. [DOI] [PubMed] [Google Scholar]
- 25.Rittling S.R., Coutinho,L., Amram,T. and Kolbe,M. (1989) AP-1/jun binding sites mediate serum inducibility of the human vimentin promoter. Nucleic Acids Res., 17, 1619–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izmailova E.S. and Zehner,Z.E. (1999) An antisilencer element is involved in the transcriptional regulation of the human vimentin gene. Gene, 230, 111–120. [DOI] [PubMed] [Google Scholar]
- 27.Munoz E. and Israel,A. (1995) Activation of NF-κB by the Tax protein of HTLV-1. Immunobiology, 193, 128–136. [DOI] [PubMed] [Google Scholar]
- 28.Kadonaga J.T., Carner,K.R., Masiarz,F.R. and Tjian,R. (1987) Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell, 51, 1079–1090. [DOI] [PubMed] [Google Scholar]
- 29.Philipsen S. and Suske,G. (1999) A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res., 27, 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suske G. (1999) The Sp-family of transcription factors. Gene, 238, 291–300. [DOI] [PubMed] [Google Scholar]
- 31.Ye S., Whatling,C., Watkins,H. and Henney,A. (1999) Human stromelysin gene promoter activity is modulated by transcription factor ZBP-89. FEBS Lett., 450, 268–272. [DOI] [PubMed] [Google Scholar]
- 32.Reizis E. and Leder,P. (1999) Expression of the mouse pre-T cell receptor α gene is controlled by an upstream region containing a transcriptional enhancer. J. Exp. Med., 189, 1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasegawa T., Takeuchi,A., Miyishi,O., Isobe,K.I. and deCrombrugghe,B. (1997) Cloning and characterization of a transcription factor that binds to the proximal promoters of the two mouse type I collagen genes. J. Biol. Chem., 272, 4915–4923. [DOI] [PubMed] [Google Scholar]
- 34.Bai L. and Merchant,J.L. (2000) Transcription factor ZBP-89 cooperates with histone acetyltransferase p300 during butyrate activation of p21waf1 transcription in human cells. J. Biol. Chem., 275, 30725–30733. [DOI] [PubMed] [Google Scholar]
- 35.Yamada A., Takaki,S., Hayashi,F., Georgopoulos,K., Perlmutter,R.M. and Takatsu,K. (2001) Identification and characterization of a transcriptional regulator for the lck proximal promoter. J. Biol. Chem., 276, 18082–18089. [DOI] [PubMed] [Google Scholar]
- 36.Merchant J.L., Iyer,G.R., Taylor,B.R., Kitchen,J.R., Mortensen,E.R., Wang,Z., Flintoft,R.J., Michel,J.B. and Bassel-Duby,R. (1996) ZBP-89, a kruppel-like zinc finger protein, inhibits epidermal growth factor induction of the gastrin promoter. Mol. Cell. Biochem., 16, 6644–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Law G.L., Itoh,H., Law,D.J., Mize,G.J., Merchant,J.L. and Morris,D.R. (1998) Transcription factor ZBP-89 regulates the activity of the ornithine decarboxylase promoter. J. Biol. Chem., 273, 19955–19964. [DOI] [PubMed] [Google Scholar]
- 38.Passantino R., Antona,V., Barbieri,G., Rubino,P., Melchionna,R., Cossu,G., Feo,S. and Giallongo,A. (1998) Negative regulation of β-enolase gene transcription in embryonic muscle is dependent upon a zinc-finger factor that binds to the G-rich box within the muscle-specific enhancer. J. Biol. Chem., 273, 484–494. [DOI] [PubMed] [Google Scholar]
- 39.Cheng P.Y., Kagawa,N., Takahashi,Y. and Waterman,M.R. (2000) Three zinc finger nuclear proteins, Sp1, Sp3 and a ZBP-89 homologue, bind to the cyclic adenosine monophosphate-responsive sequence of the bovine adrenodoxin gene and regulate transcription. Biochemistry, 39, 4347–4357. [DOI] [PubMed] [Google Scholar]
- 40.Keates A.C., Keates,S., Kwon,J.H., Arseneau,K.O., Law,D.J., Bai,L., Merchant,J.L., Wang,T.C. and Kelly,C.P. (2001) ZBP-89, Sp1 and NF-κB regulate epithelial neutrophil-activating peptide-78 gene expression in caco-2 human colonic epithelial cells. J. Biol. Chem., 276, 43713–43722. [DOI] [PubMed] [Google Scholar]
- 41.Park H., Shelley,C.S. and Arnaout,M.A. (2003) The zinc finger transcription factor ZBP-89 is a repressor of the human β2 integrin CD11b gene. Blood, 101, 894–902. [DOI] [PubMed] [Google Scholar]
- 42.Law D.J., Du,M., Law,L. and Merchant,J.L. (1999) ZBP-99 defines a conserved family of transcription factors and regulates ornithine decarboxylase gene expression. Biochem. Biophys. Res. Commun., 262, 113–120. [DOI] [PubMed] [Google Scholar]
- 43.Law D.J., Tarle,S.A. and Merchant,J.L. (1998) The human ZBP-89 homolog, located at chromosome 3q21, represses gastrin gene expression. Mamm. Genome, 9, 165–167. [DOI] [PubMed] [Google Scholar]
- 44.Dignam J.D., Lebowitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Courey A.J. and Tjian,R. (1988) Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell, 55, 887–989. [DOI] [PubMed] [Google Scholar]
- 46.Monte D.L., Coutee,J.L., Baert,I.A., Stehelin,D. and deLaunoit,Y. (1995) Molecular characterization of the ets-regulated human transcription factor ER81. Oncogene, 11, 771–779. [PubMed] [Google Scholar]
- 47.Gillespie S.K. and Wasserman,S.A. (1994) Dorsal, a Drosophila Rel-like protein, is phosphorylated upon activation of the transmembrane protein toll. Mol. Cell. Biol., 14, 3559–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saluja D., Vassallo,M.F. and Tanese,N. (1998) Distinct sub-domains of human TAFII130 are required for interactions with glutamine-rich transcriptional activators. Mol. Cell. Biol., 18, 5734–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L., Nishinaka,T., Kwan,K., Kitabayashi,I., Yokoyama,K., Fu,Y.H., Grunwald,S. and Chiu,R. (1994) The retinoblastoma gene product RB stimulates Sp1-mediated transcription by liberating Sp1 from a negative regulator. Mol. Cell. Biol., 14, 4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karlseder J., Rotheneder,H. and Wintersberger,E. (1996) Interaction of Sp1 with the growth- and cell cycle-regulated transcription factor E2F. Mol. Cell. Biol., 16, 1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lania L., Majello,B. and De Luca,P. (1997) Transcriptional regulation by the Sp family proteins. Int. J. Biochem. Cell Biol., 29, 1313–1323. [DOI] [PubMed] [Google Scholar]
- 52.Marin M., Karis,A., Visser,P., Grosveld,F. and Philipsen,S. (1997) Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell, 89, 619–628. [DOI] [PubMed] [Google Scholar]
- 53.Kingsley C. and Winoto,A. (1992) Cloning of GT box-binding proteins: a novel Sp1 multigene family regulating T-cell receptor gene expression. Mol. Cell. Biol., 12, 4251–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kennett S.B., Udvadia,A.J. and Horowitz,J.M. (1997) Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res., 25, 3110–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hagen G., Muller,S., Beato,M. and Suske,G. (1994) Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J., 13, 3843–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gill G., Pascal,A., Tseng,Z.H. and Tjian,R. (1994) A glutamine-rich hydrophobic patch in transcription factor Sp1 contacts the dTAFII110 component of the Drosophila TFIID complex and mediates transcriptional activation. Proc. Natl Acad. Sci. USA, 91, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bakovic M., Waite,K.A. and Vance,D.E. (2000) Functional significance of Sp1, Sp2 and Sp3 transcription factors in regulation of the murine CTP:phosphocholine cytidylyltransferase α promoter. J. Lipid Res., 41, 583–594. [PubMed] [Google Scholar]
- 58.Supp D.M., Witte,D.P., Branford,W.W., Smith,E.P. and Potter,S.S. (1996) Sp4, a member of the Sp1-family of zinc finger transcription factors, is required for normal murine growth, viability and male fertility. Dev. Biol., 176, 284–299. [DOI] [PubMed] [Google Scholar]
- 59.Dunah A.W., Jeong,H., Griffin,A., Kim,Y.M., Standaert,D.G., Hersch,S.M., Mouradian,M.M., Young,A.B., Tanese,N. and Krainc,D. (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science, 296, 2238–2243. [DOI] [PubMed] [Google Scholar]
- 60.Hasegawa T., Takeuchi,A., Miyaishi,O., Xiao,H., Mao,J. and Isobe,K. (2000) PTRF (polymerase I and transcript-release factor) is tissue-specific and interacts with the BFCOL1 (binding factor of a type-I collagen promoter) zinc-finger transcription factor which binds to the two mouse type-I collagen gene promoters. Biochem. J., 347, 55–59. [PMC free article] [PubMed] [Google Scholar]
- 61.Hoey T., Weinziere,R.O., Gill,G., Chen,J.L., Dynlacht,B.D. and Tjian,R. (1993) Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell, 72, 247–260. [DOI] [PubMed] [Google Scholar]
- 62.Murata Y., Kim,H.G., Rogers,K.T., Udvadia,A.J. and Horowitz,J.M. (1994) Negative regulation of Sp1 trans-activation is correlated with the binding of cellular proteins to the amino terminus of the Sp1 trans-activation domain. J. Biol. Chem., 269, 20674–20681. [PubMed] [Google Scholar]
- 63.Gartel A.L., Ye,X., Goufman,E., Shianov,P., Hay,N., Najmabadi,F. and Tyner,A.L. (2001) Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl Acad. Sci. USA, 98, 4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doetzlhofer A., Rotheneder,G., Lagger,G., Koranda,M., Kurtev,V., Brosch,G., Wintersberger,E. and Seiser,C. (1999) Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol., 19, 5504–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Z.Y., Shie,J.L. and Tseng,C.C. (2002) Gut-enriched Kruppel-like factor represses ornithine decarboxylase gene expression and functions as checkpoint regulator in colonic cancer cells. J. Biol. Chem., 277, 46831–46839. [DOI] [PubMed] [Google Scholar]
- 66.Chung D.C., Brand,S.J. and Tillotson,L.G. (1995) Mutually exclusive interactions between factors binding to adjacent Sp1 and AT-rich elements regulate gastrin gene transcription in insulinoma cells. J. Biol. Chem., 270, 8829–8836. [DOI] [PubMed] [Google Scholar]
- 67.Shie J.L., Chen,Z.Y., Fu,M., Pestell,R.G. and Tseng,C.C. (2000) Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res., 28, 2969–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W., Shields,J.M., Sogawa,K., Fuji-Kuriyama,Y. and Yang,V.W. (1998) The gut-enriched Kruppel-like factor suppresses the activity of the CYP1A1 promoter in an Sp1-dependent fashion. J. Biol. Chem., 273, 17919–17925. [DOI] [PMC free article] [PubMed] [Google Scholar]