


Abstract
Suppression subtractive hybridization performed on Down syndrome (DS) fetal brains revealed a differentially expressed gene, FABP7, mapped to 6q22–23. FABP7 overexpression in DS brains was verified by real-time PCR (1.63-fold). To elucidate the molecular basis of FABP7 overexpression and establish the relationship with chromosome 21 trisomy, the FABP7 promoter was cloned by genomic inverse PCR. Comparison to the mouse ortholog revealed conservation of reported regulatory elements, among them a Pbx/POU binding site, known to be the target of PBX heteromeric complexes. PBX partners include homeobox-containing proteins, such as PKNOX1 (PREP1), a transcription factor mapping at 21q22.3. We report here: (i) overexpression of PKNOX1 in DS fetal brains; (ii) in vitro specific binding of PKNOX1 to the Pbx/POU site of the FABP7 promoter; (iii) in vivo FABP7 promoter trans-activation in cultured neuroblastoma cells caused by PKNOX1 overexpression. To our knowledge this is the first report of a direct relation between dosage imbalance of a chromosome 21 gene and altered expression of a downstream gene mapping on another chromosome. Given the role of FABP7 in the establishment, development and maintenance of the CNS, we suggest that the overexpression of FABP7 could contribute to DS-associated neurological disorders.
INTRODUCTION
The Down syndrome (DS) is the most common autosomal aneuploidy in humans, affecting up to 1 in 700 neonates (1,2). Caused by total or partial trisomy of chromosome 21, it appears associated with more than 80 clinical traits, including typical facial features, anomalies of the intestinal tract, muscular hypotonia, increased risk of leukemia, congenital heart defects and mental retardation (3). Currently, despite remarkable efforts, the molecular basis of DS is not well understood. According to the gene-dosage effect hypothesis, the imbalance of a single gene or group of genes in the trisomic region is responsible for a particular DS feature. This could be either a direct or an indirect effect, through the altered expression of genes located on other chromosomes. A direct approach to the molecular basis of DS involves the identification and functional characterization of chromosome 21 genes. To this end, the recent completion of the 21 chromosome sequence (4) was a crucial contribution and has allowed the subsequent identification and systematic expression analysis of many chromosome 21-gene orthologs in mice (5,6). A promising alternative strategy aims to identify differentially expressed genes irrespective of their chromosomal location. In this respect, reported methodologies include subtractive hybridization (SH) (7–11), serial analysis of gene expression (12), differential display PCR (13,14) and micro-array based analyses (reviewed in 15). To date, several differentially expressed genes in DS individuals have been reported. Some of them are located on chromosome 21, such as Cu/Zn superoxide dismutase (SOD1), encoding a key enzyme in the metabolism of oxygen free radicals (16,17), and the gene for the amyloid precursor protein (APP), implicated in DS-associated early-onset Alzheimer’s disease (18). Others that have shown altered expression in DS have been located on other chromosomes, such as TRX (involved in cell oxidative status), XRCC1 (repair functions) and dynamin (neural tissue development) (19). Recently, another approach to identify differentially expressed genes based on a transcriptome analysis of a partial trisomy 16 mouse model of DS was performed (20).
Mental retardation is a universal phenotypic feature of DS. Thus, we focused on differential gene expression in fetal brains and performed suppression subtractive hybridization (SSH). Our results, verified by real-time PCR, clearly showed DS-associated overexpression of fatty acid-binding protein 7 (FABP7). This gene, also named brain fatty acid-binding protein (BFABP) or brain lipid binding protein (BLBP), had been previously characterized and located at 6q22–23 by FISH (21–24).
Fatty acid-binding proteins (FABPs) are small peptides (14–16 kDa), previously named according to the tissue of origin. At least nine FABP forms, each encoded by a different gene, have been characterized (brain or retina, liver, heart or muscle, intestine, epidermis, adipocyte, ileum, myelin and testis). Most family members (including FABP7) show dual cytoplasmic and nuclear localization, thus supporting their involvement in the uptake, storage and/or delivery of fatty acids and retinoids into the nucleus (25–27). In particular, FABP7 is expressed in radial glial cells and immature astrocytes of the developing central nervous system and later becomes restricted to the glia limitans, radial glial cells of the hippocampus and Bergmann glial cells in the adult mouse brain. This expression pattern is consistent with its predicted role in the organization and maintenance of the radial glial fiber system required for the correct migration of immature neurons (21,22). More recently, FABP7 expression in a subtype of radial glial precursor cells during neuro- and glio-genesis in the developing cerebral cortex has been correlated to the fate of their progeny and considered a marker for multipotentiality (28). Besides, FABP7 shows high affinity for docosahexaenoic acid (DHA), the most abundant polyunsaturated long chain fatty acid in the brain phospholipid pool (29). Deficiencies in DHA cause severe and progressive neurological disorders (30,31).
To understand the molecular basis of FABP7 overexpression in DS we have undertaken a functional analysis of its promoter region. In the Fabp7 murine counterpart three distinct regulatory elements have been highlighted: a radial glial specific element (RGE, located at –800 to –300 bp); a sequence controlling Fabp7 expression in dorsal root ganglion and notochord (DRGE, located at –800 to –1200 bp); a silencer for transcriptional suppression in dorsal spinal cord (DSCS, located at –1200 to –1600 bp) (32). The region spanning –800 to –300 bp is necessary and sufficient for developmentally regulated expression throughout the fetal CNS (32), and it contains a hybrid Pbx/POU binding site (–370 to –362 bp), recognized by PBX-1, BRN-1 and BRN-2 (33). PBX transcription factors (PBX1, PBX2 and PBX3) are homeobox proteins that form stable heterodimeric complexes with other homeobox-containing proteins, such as HOX and PREP1 (34–41). Depending on the partner, the PBX-containing complexes display differential affinities for binding promoter motifs, even with antagonistic properties. Several genes, such as the urokinase plasminogen activator (35), somatostatin (37), glucagon (40) and alpha 2(V) collagen (COL5A2) (41) are regulated by PREP1/PBX heterodimeric complexes. Interestingly, the human PREP1 (Pbx regulating protein 1) ortholog, named PKNOX1 (Pbx/knotted homeobox1), is located on chromosome 21q22.3 (42,43).
Here we show that the Pbx/POU binding site in the human FABP7 promoter is conserved and we provide evidence of PKNOX1 involvement in FABP7 transcriptional regulation, thus indicating a direct link between trisomy 21 and FABP7 overexpression. DS brain abnormalities include functional disturbances in glial cells, delayed myelination and diminished neuronal density in the brain cortex (reviewed in 44). As FABP7 function is necessary for neuro- and glio-genesis, particularly for the establishment of the glial fibers and the proper migration of immature neurons to cortical layers, we suggest that overexpression of FABP7 may contribute to the DS-associated neurological disorders.
MATERIALS AND METHODS
Samples
Twenty-two male fetal half-brains obtained from pregnancy weeks 18–23 were used in this study. Twelve of these tissue samples were diagnosed as trisomic for chromosome 21 by amniocentesis. The remaining samples (10) did not show any chromosomal anomaly and were used as disomic controls. Six samples (three trisomic versus three disomic) were used for SSH. Differential expression was confirmed using 16 new samples (nine trisomic versus seven disomic) for real-time PCR quantification and densitometric analysis of northern blot hybridizations. This research followed the tenets of the Declaration of Helsinki. Informed consent was obtained from all the parents involved in this study.
mRNA extraction and cDNA synthesis
Total RNA was extracted from six male fetal half-brains (three DS versus three control) as described elsewhere (45). Two micrograms of mRNA were obtained from total RNA using biotinylated oligo-d(T) (Promega) and streptavidin-magnetic beads (Dynal), following the manufacturer’s instructions. Poly(A)+ RNA was retrotranscribed at 42°C for 1.5 h using MMLV-RT (Promega) according to the supplier’s instructions. The cDNA second-strand was obtained using the enzyme cocktail and protocol provided by the PCR-SELECT cDNA subtraction kit (Clontech).
Subtractive hybridization
SSH was performed on DS and control mRNAs (obtained as described above) using the PCR-SELECT cDNA subtraction kit following the manufacturer’s instructions. These minor modifications were introduced: RsaI digestion time was increased to ensure complete digestion and treatment with the Klenow enzyme was performed previously to the adaptor ligation, which was performed overnight at 13°C and then for 2 h at room temperature. Differentially expressed cDNAs were amplified by two rounds of suppression PCR amplification using the Advantage Klentaq polymerase (Clontech). Twenty primary PCR cycles and 30 secondary PCR cycles were performed according to the manufacturer’s instructions.
Cloning and sequencing of putatively differentially expressed cDNAs
PCR products generated by SSH were subcloned into pBLUESCRIPT SK+ vector (Stratagene) by standard procedures. Sequencing was performed using an automated ABI 377 DNA sequencing system (Applied Biosystems). DNA sequence comparisons were performed at BLAST servers at NCBI (http://www.ncbi.nlm.nih.gov/BLAST/), BCM (http://searchlauncher.bcm.tmc.edu/seq-search/nucleic_acid-search.html) and links therein.
Quantification of differential gene expression by real-time (quantitative) PCR
Quantification of differential gene expression was performed on nine DS and seven control male human fetal brain samples for FABP7, and on eight samples (four disomic and four trisomic) for PKNOX1, SOD1 and USP25. Total RNA from 5 mg of tissue was obtained using the ABI PRISM 6700 Automated Nucleic Acid Workstation (Applied Biosystems). RT–PCRs were produced using the Taqman Reverse transcription reagents (Roche Molecular Systems). Quantitative PCRs were performed with the Universal Master Mix (Applied Biosystems) under the manufacturer’s conditions. The real-time amplification was analyzed by the ABI PRISM 7700 sequence detection system (Applied Biosystems). The primers and Taqman probe (using FAM as reporter and TAMRA as quencher) were designed following the Primer Express software. To normalize the FABP7, PKNOX1, SOD1 and USP25 quantitative determinations, G3PDH (primers and probe from Applied Biosystems) was used as an endogenous control.
Inverse PCR amplification
The protocol used for inverse PCR is adapted from Averof (46). Human blood genomic DNA (1 µg) was digested overnight with EcoRI. After heat inactivation of the restriction enzyme, the digested DNA was diluted to a final concentration of 1 µg/ml. The circularization of molecules was obtained in a 1 ml final volume by overnight ligation at 13°C and then 2 h at room temperature. Inverse PCR conditions were: 94°C for 20 s and 68°C for 5 min for 40 cycles using LE High Fidelity Taq polymerase (Roche). The amplified band was cloned into pBLUESCRIPT SK+ vector (Stratagene), sequenced and compared to DNA databases as described above.
Primers used to perform the inverse PCR amplification. FABP7.1 (reverse): 5′-CACAGAAAGCCTCCACCATCCT-3′ (complementary to the ATG codon). FABP7.2 (forward): 5′-GCTACCTGGAAGCTGACCAACA-3′ (sequence from the first exon, downstream the ATG codon).
Nuclear cell extracts
Nuclear SH-SY5Y extracts were obtained according to Schreiber et al. (47) with minor modifications (buffer C: 10% glycerol and 1.5 mM MgCl2). Protein concentration was measured by the Bradford method (reagents purchased from Bio-Rad).
Recombinant PKNOX1 purification
Human PKNOX1 cDNA was obtained by PCR on total human brain cDNA (obtained as described) using forward and reverse primers containing the ATG and STOP codons, respectively. The coding region of PKNOX1 was cloned into the pGEX- 4T-1 expression vector (Amersham Pharmacia) to synthesize a GST–PKNOX1 fusion protein. BL21 Escherichia coli cells were transformed with this construct and induced with IPTG (100 mM) for 3 h at 30°C. Cells were sonicated for 3 min and the GST–PKNOX1 protein was purified according to the manufacturer’s instructions, using glutathione–Sepharose 4B followed by thrombin cleavage to separate PKNOX1 from GST.
Electrophoretic mobility shift assay (EMSA)
Two double-stranded (ds) oligonucleotide probes were designed on the human FABP7 promoter sequence. The first (wt probe) encompassed the 9-bp Pbx/POU binding site reported in the homologous mouse promoter (31). The second, used to confirm the specificity of the binding, contained the same FABP7 promoter sequence except for the deletion of the 9-bp Pbx/POU binding site (del probe). The annealing reaction was performed in 100 mM NaCl for 5 min at 80°C and the solution was then allowed to reach room temperature. The ds probes were end-labeled using [γ-32P]dATP and T4 polynucleotide kinase (Promega). Protein binding reactions were performed in a total volume of 20 µl by combining 1 µg of poly(dI–dC) (Amersham Pharmacia Biotech), 6 µg of acetylated BSA (Promega) and 0.01% Nonidet-40 with 10 µg of SH-SY5Y nuclear extract and/or 10 ng of purified recombinant PKNOX1 protein (as indicated in the corresponding figures). The binding buffer contained 20 mM HEPES pH 7.9, 60 mM KCl, 1 mM EDTA, 1 mM DTT and 10% glycerol. After 10 min on ice, the labeled probe (100 000 c.p.m.) and, if required, the specific antibodies anti-PKNOX1 (sc-6245; Santa Cruz) or anti-GST (sc-138; Santa Cruz), were added to the reaction mixture, which was incubated for a further 30 min at room temperature. Competition assays were performed in molar excess of unlabeled ds oligonucleotides. In specific competitions the unlabeled wild-type or deleted ds oligonucleotides (wt probe and del probe, respectively) were added in 100-, 1000- and 10 000-fold molar excess. In non-specific competition assays, an unlabeled ds oligonucleotide containing the GRE binding site was used in 10 000-fold molar excess. Samples were loaded onto non-denaturing 5% acrylamide/bis-acrylamide (29:1) and 2.5% glycerol gels and resolved at 18 mA at 4°C. Gels were dried and autoradiographed.
Sequences of the ds oligonucleotides used (only forward strands are shown, Fig. 2): Pbx/POU binding site (wt probe): 5′-CATTAACGGAAATCAATCTGAATGCCCCAT-3′; mutated Pbx/POU binding site (del probe): 5′-CATTAACGGAAAATGCCCCAT-3′; GRE: 5′-TAATGAGAGAAGAT TCTGTTCTAATGACCA-3′.
Figure 2.

(A) Sequence alignment of murine and human FABP7 (BFABP) promoters. Nucleotide identities are boxed in black. Conserved regulatory motifs (the conserved HRE, the Pbx/POU binding site and the TATA box), are indicated by a black bar above. The dotted underline indicates the sequence of the probe used for EMSA. The transcription initiation site is marked by an arrow. (B) Diagram depicting the organization and relative position of the conserved regulatory elements in the human promoter.
DNA constructs for the luciferase assay
The pGL3-FABP7prom-luciferase was obtained by cloning the 1.2-kb PstI fragment (nucleotide 442 to nucleotide 1684, Fig. 2) from the human FABP7 promoter (including the Pbx/POU binding site) into the pGL3 Basic luciferase reporter vector (Promega), in order to drive the luciferase expression by the FABP7 promoter. The pGL3-mutantFABP7prom-luciferase, which lacked the Pbx/POU binding site (9 bp), was obtained by site-directed mutagenesis. To generate this deletion, a first PCR using a forward (5′-TTTAATTGGGAGGGGAGAG-3′) and a reverse mutated primer (5′-ATGGGGCATTTTCCGTTAATG-3′) amplified 846 bp of the promoter 5′ region. A second PCR using a forward mutated (5′-CATTAACGGAAAATGCCCCAT-3′) and a reverse primer (5′-CTCAGAAGACCCTTTACACCT-3′) amplified 418 bp of the promoter 3′ region. Both PCR products were purified, mixed in equimolar concentrations, denatured, allowed to anneal for 30 min at room temperature and filled-in with Taq polymerase plus nucleotides (5 min). After addition of the non-mutated forward and reverse primers, a 25-cycle PCR (94°C for 30 s, 60°C for 30 s and 72°C for 2 min) was performed to amplify the final 1.2-kb promoter fragment with the deletion.
The pSV-PKNOX1 construct was obtained by replacing the β-galactosidase gene of the pSV-β-Gal vector (Promega) by the coding region of PKNOX1. In this construct, PKNOX1 expression was controlled by the constitutive CMV promoter. The pSV-β-Gal vector was used to measure the efficiency of each transfection and the pSV-empty vector (without the β-galactosidase gene) was used to equilibrate the DNA concentrations and as a control for basal luciferase activity in transfections without the PKNOX1 construct.
Luciferase activity assay
Human SH-SY5Y neuroblastoma cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin and 1 µg/ml streptomycin (reagents from GIBCO-BRL) in a 5% CO2 humidified atmosphere at 37°C. Approximately 2 × 106 cells in a 60 mm plate were transiently transfected with 8.5 µg of DNA (see DNA constructs below) and 12.5 µl of FuGENE™ Transfection Reagent (Roche). Twenty-four hours after transfection, luciferase (Luciferase Assay System; Promega) and β-galactosidase (Chemi luminescent β-Gal reporter Gene Assay; Roche) activities were measured in a TD-20/20 Luminometer (Turner Designs).
RESULTS
FABP7 overexpression in DS patients
Male fetal half-brain samples from three DS and three control individuals were used for SSH forward and reverse experiments. The forward assay allowed the identification of presumptively overexpressed mRNAs in DS brains, whereas the reverse strategy amplified down-regulated transcripts. Two rounds of the forward SSH to enrich the sample in the differentially expressed transcripts were followed by two rounds of PCR amplification. After this procedure, several bands were amplified. One of them, of 550 bp, corresponded to a FABP7 cDNA fragment.
FABP7 overexpression in DS fetal brains was confirmed by quantitative real-time PCR. Sixteen samples (nine DS and seven controls) were quantified and G3PDH was used as an internal control (Fig. 1). According to this analysis, DS fetal brains overexpressed FABP7 by 1.63-fold. Similar results were obtained by densitometric analysis of northern blots (data not shown). We also quantified the expression of other genes, located on chromosome 21 [SOD1, USP25 and PKNOX1 (see below)], as internal controls (Fig. 1).
Figure 1.

Average expression of FABP7 and other chromosome 21 genes (PKNOX1, SOD1 and USP25) in control and DS fetal brains, as detected by real-time PCR. All values are given in arbitrary units and normalized using G3PDH as an endogenous control. The statistical error is also shown. The asterisks indicate that the expression differences between trisomic and disomic brains are statistically significant (Mann–Whitney test, P < 0.05).
Cloning of the human FABP7 promoter
To examine the FABP7 overexpression associated with DS, we analyzed the promoter region of the human gene. In the murine promoter, the region stretching from –300 to –800 bp is necessary and sufficient for developmentally regulated expression throughout the fetal CNS (32). Two domains have been described within this region: a hormone-responsive element (HRE) and a Pbx/POU binding site (–370 to –362 bp).
Since the human genomic sequence of this promoter was then not available, an inverse PCR was devised to isolate and clone this genomic fragment. Genomic DNA was digested with EcoRI, diluted and ligated to produce circular molecules. For the PCR, the reverse primer was complementary to the initial ATG codon, while the forward primer contained part of the first exon downstream the ATG. The PCR consistently amplified a 2.7-kb band, which was subsequently cloned and sequenced. The amplified fragment encompassed 1.8 kb of a sequence highly homologous to the Fabp7 mouse promoter (the nucleotide sequence for the human FABP7 promoter has been deposited in the GenBank database under GenBank accession no. AY070217) and 0.9 kb containing the first and second FABP7 exons plus some intron sequences. The alignment between the human and mouse regions revealed that all the murine regulatory elements were conserved in the human promoter, including the presumptive Pbx/POU binding site at a similar location (Fig. 2).
PKNOX1, a PBX partner, is overexpressed in DS
One of the reported PBX partners is encoded by the PKNOX1 gene (PREP1 in mice), located on chromosome 21q22.3. We postulated that if PKNOX1 binds and trans-activates the human FABP7 promoter through a PBX/PKNOX1 complex, FABP7 overexpression in DS brains will be directly correlated to the gene-dosage imbalance caused by the 21 trisomy. To test this hypothesis, we assessed PKNOX1 overexpression in DS fetal brains, and performed the specific binding assay of PKNOX1 to the FABP7 promoter.
Real-time PCR in eight male fetal brain samples (four disomic and four trisomic) showed 1.88-fold overexpression of PKNOX1 in DS fetal brain samples with respect to controls (Fig. 1).
PKNOX1 binds weakly to the Pbx/POU motif of the FABP7 promoter
We assessed PKNOX1 binding to the Pbx/POU site in the human FABP7 promoter by in vitro EMSAs. To this end, recombinant PKNOX1 protein was purified from E.coli cells expressing a GST–PKNOX1 fusion construct. In addition, we designed two probes derived from the FABP7 promoter sequence (Fig. 2): (i) a 30-bp ds oligonucleotide encompassing the Pbx/POU (ATCAATCtg) site (wt probe); (ii) the same sequence without the Pbx/POU target site (del probe, 21 bp).
A retarded band appeared only when high amounts (1.25 µg) of recombinant PKNOX1 were added to the binding mixture with the wt probe (Fig. 3, lane 3). However, no bands were detected with the deleted Pbx/POU probe in the same conditions (Fig. 3, lane 6). These in vitro assays revealed that PKNOX1 binds to the reported site, but only under protein excess conditions. This is consistent with previous reports showing that, on their own, murine PREP1 (the PKNOX1 ortholog) and its PBX partners bind the DNA target weakly. In contrast, the formation of a PBX/PREP1 complex drastically increases the DNA-binding affinity of both subunits and broadens the DNA target selectivity (34,35).
Figure 3.
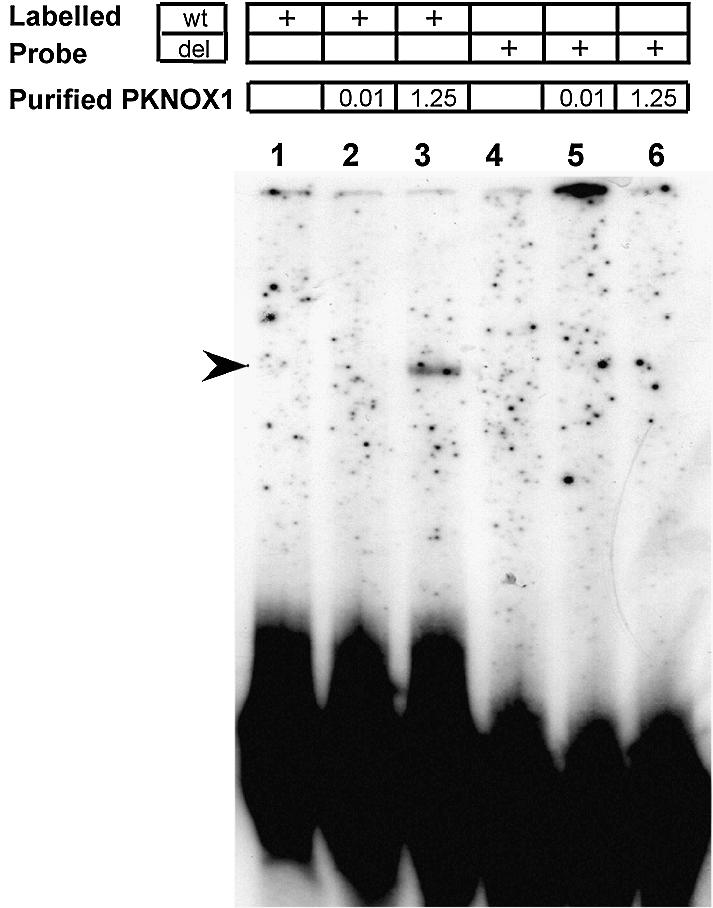
EMSA analysis using recombinant PKNOX1 protein and two different labeled ds oligonucleotides (as indicated above): wt probe, which encompasses the Pbx/POU binding site of the FABP7 promoter, and del probe, which contains the same sequence but for the deletion of the Pbx/POU binding site. Lanes 1 and 6, probes with no protein added; lanes 2 and 4, probes plus 10 ng of purified recombinant PKNOX1; lanes 3 and 5, probes plus 1.25 µg of purified recombinant PKNOX1.
PKNOX1 complexes from SH-SY5Y nuclear extracts bind to the Pbx/POU motif of the FABP7 promoter
In order to provide the partners required for the formation of PKNOX1 complexes, nuclear protein extracts from human neuroblastoma SH-SY5Y cells were added to a new set of EMSAs. On their own, SH-SY5Y nuclear extracts produced retarded bands, indicating that they contained several proteins that could bind the probe. The two most prominent bands are indicated by arrows (Fig. 4A, lane 3). The banding pattern was not affected when PKNOX1 was added to the binding mixture (Fig. 4A, lane 4 compared to lane 3), which could be explained by the presence of endogenous PKNOX1 in SH-SY5Y cells (as previously assessed by western analysis; data not shown). The specificity of the binding was confirmed by competition with cold Pbx/POU wt probe (at 100-, 1,000- and 10 000-fold molar excess) (Fig. 4A, lanes 5–7) and the absence of competition with the non-specific GRE oligonucleotide (10 000-fold molar excess) (Fig. 4A, lane 8). The oligonucleotide lacking the Pbx/POU site (del probe) did not compete out any of the DNA–protein complexes, which highlighted the relevance of this 9-bp motif (Fig. 4B).
Figure 4.

PKNOX1 is present in SH-SY5Y nuclear extracts and binds specifically to the Pbx/POU binding site of the FABP7 promoter. (A) Specific competitions (100-, 1000- and 10 000-fold molar excess) with cold wt probe and non-specific competition with the GRE oligonucleotide (10 000-fold molar excess). (B) Specific competitions with cold mutated del probe (100-, 1000- and 10 000-fold molar excess). (C) Disruption of the PKNOX1-binding complexes by co-incubation with an anti-PKNOX1 antibody. The asterisk on lane 5 indicates that the antibody was added from the beginning of the reaction instead of addition after the 10 min incubation on ice (see Materials and Methods). (D) PKNOX1 binds specifically to the Pbx/POU target site and this binding is abolished by the anti-PKNOX1 antibody. The presence of nuclear extract, purified recombinant PKNOX1 (10 ng), type of oligonucleotide probes, competitors and antibodies used are indicated above each lane. Specific retarded bands due to protein binding at the Pbx/POU target site are indicated by arrows. Note their disappearance after addition of the anti-PKNOX1 antibody.
Further EMSAs were performed to establish the contribution of PKNOX1 to the protein binding complexes. This was shown by preincubation of the binding mixture with an anti-PKNOX1 (anti-PREP1) antibody. This addition abolished the formation of the two specific complexes, while an unrelated antibody (anti-GST antibody) had no effect (Fig. 4C). Again, the pattern was not affected by the addition of the del probe at 10 000-fold molar excess (Fig. 4C, lane 7). Moreover, to confirm PKNOX1 binding to the 9-bp core of the Pbx/POU site, the wt and del probes were labeled and used in new EMSAs. The patterns obtained were compared in the presence or absence of the anti-PKNOX1 antibody (Fig. 4D). As expected, if PKNOX1 specifically recognized this site, no retarded bands appeared with the del probe and addition of the anti-PKNOX1 antibody had no effect (Fig. 4D, lanes 3 and 5).
Overall, these in vitro data support: (i) PKNOX1 binding to the Pbx/POU target of the FABP7 promoter; (ii) that this binding is greatly enhanced by PKNOX1 assembling into a functional complex with partners of the nuclear extracts.
PKNOX1 trans-activates FABP7 in vivo
Next, we analyzed the effect of PKNOX1 on the regulation of FABP7 transcription. SH-SY5Y neuroblastoma cells were transiently co-transfected with a vector constitutively expressing recombinant PKNOX1 and also, a reporter construct bearing the luciferase gene under the control of the FABP7 promoter. In addition, a β-galactosidase reporter construct was added to the liposome–DNA mixture to normalize the transfection efficiency. The ratio of the luciferase/β-gal activities allowed both comparison between samples and quantification of transcription trans-activation in arbitrary units. According to our data PKNOX1 overexpression induced luciferase expression by 3.7-fold (statistical significance, P < 0.001, Mann–Whitney test) and therefore, PKNOX1 did trans-activate the FABP7 promoter (Fig. 5). In addition, the deletion of the 9-bp Pbx/POU motif by site-directed mutagenesis decreased the trans-activation response of this promoter to PKNOX1 (Fig. 5). It should be noted that the deletion of the Pbx/POU target increased the basal transcription of the promoter by 1.5-fold and, thus, the effect of the addition of PKNOX1 on the deleted promoter may appear higher than it really was [2.2-fold increase if compared to the wild-type promoter but only 1.4-fold if compared to the basal expression of the mutant promoter without added PKNOX1 (statistical significance P < 0.05, Mann–Whitney test)].
Figure 5.

PKNOX1-dependent trans-activation of the FABP7 promoter on cultured transfected SH-SY5Y cells. Luciferase/β-gal activity ratios are given in arbitrary units with respect to the wild-type promoter without added PKNOX1. The average values and the standard deviation are obtained after nine replicas for the wild-type promoter with or without co-expression of recombinant PKNOX1 and four replicas for the mutant deleted promoter with or without PKNOX1. Statistical significance was calculated by the Mann–Whitney test (*P < 0.05; **P < 0.005; ***P < 0.001; the first level above the histogram: statistical significance when comparing the values to the wild-type promoter without added PKNOX1; second level above the histogram: statistical significance when comparing to the mutant deleted promoter without added PKNOX1; third level above the histogram: statistical significance of the trans-activation differences between the wild-type and mutant promoters after addition of PKNOX1).
DISCUSSION
The search for differentially expressed genes is a promising strategy to explore the molecular basis of the DS pathogenesis. We focused on DS fetal brains because of the high prevalence of mental retardation, albeit with great differences in the level of severity, in affected individuals. The SSH approach revealed overexpression of FABP7 (located at 6q22–23) in DS fetal brains, which was further verified and quantified by real-time PCR (1.63-fold) in a total of 22 samples. These data support that, far from gross alterations in the transcription levels of a few specific genes, most differences between trisomic and disomic individuals stem from moderate changes in the expression of many genes, as reported in a partial trisomy 16 mouse model of DS (20).
Several data support the relevance of FABP7 overexpression in mental retardation. First, FABP7 is involved in the development, establishment and maintenance of the nervous system, mainly setting the grounds for proper neuronal migration (21,22). Secondly, the expression of FABP7 during neuro- and glio-genesis correlates with the fate of the progeny of radial glial precursor cells, since it is one of the markers that define multipotential precursors (28). Thirdly, DHA is the putative ligand of FABP7 and, as already established, DHA deficiencies cause severe neurological disorders (30,31). Hence, we aimed at the functional characterization of the FABP7 promoter to elucidate the molecular basis of its DS-associated overexpression. In particular, among the regulatory elements described on the murine promoter, we considered the Pbx/POU target site as the best candidate, because PBX factors form heterodimers with PREP1, whose human homolog gene, PKNOX1, is located on chromosome 21.
EMSAs with the recombinant PKNOX1 protein alone produced a weak band shift, and only under protein excess conditions, in accordance with previous reports showing that mouse PREP1 binds weakly to DNA and that high-affinity binding could only be attained when heterodimers with PBX and/or other partners were assembled (34,35). The most revealing data were achieved when using neuroblastoma nuclear protein extracts, which express both endogenous PKNOX1 and the partners required for the binding complexes. Two main bands were detected, which were specifically competed by cold wt oligonucleotide but not by the mutated del probe, lacking the Pbx/POU binding site. Moreover, the addition of an anti-PKNOX1 antibody specifically disrupted the binding of these complexes, thus clearly implicating PKNOX1 involvement. Other reports have identified PREP1 as a partner in several DNA binding complexes when the same anti-PREP1/PKNOX1 antibody (sc-6245) was used to disrupt them (38,40). Overall, these results are consistent with PKNOX1 specifically binding to the reported Pbx/POU site by assembling into at least two heteromeric complexes.
The in vitro data were confirmed in vivo by cell transfection assays, where PKNOX1 overexpression increased transcription from the FABP7 promoter by 3.7-fold. Again, specific binding of PKNOX1 to the Pbx/POU defined site was later confirmed, as the 9-bp target site deletion clearly decreased PKNOX1-dependent trans-activation. The finding that addition of PKNOX1 slightly trans-activated the mutant promoter suggests additional PKNOX1 target(s) in this promoter, without demeaning the relevance of the deleted site.
In summary, the in vitro EMSAs as well as the luciferase assays in live cells, strongly support a direct relationship between PKNOX1, at 21q22.3, and the FABP7 expression, at 6q22–23. PKNOX1 is known to interact with the PBX factors and the complexes formed regulate the expression of different genes. We here show the ability of PKNOX1 to bind to the Pbx/POU site at the FABP7 promoter and, thus, regulate FABP7 transcription. If PKNOX1 is a limiting factor in certain tissues, three allelic doses may significantly affect the level of transcription of target genes. To our knowledge, this is the first report directly relating the dosage imbalance of a particular chromosome 21 gene to altered expression of a downstream gene. As DS brains suffer from widespread abnormalities in neuronal cytoarchitecture and function (including disturbances in glial cell function, delayed myelination and diminished neuronal density in cortical layers), and FABP7 plays a crucial role in the establishment of the radial glial fiber system required for the migration of immature neurons to the brain cortex, we postulate that FABP7 overexpression may contribute to, or even enhance the DS-associated neurological disorders.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Olga González-Angulo for technical assistance, N. Garcia-Giralt for helpful comments on EMSA assays, N. Torán (Hospital de la Vall de Hebrón) for providing the fetal brain samples. We are grateful to Robin Rycroft and the Servei d’Assessorament Linguístic for revising the English. We also thank the Serveis Científico-Tècnics de la Universitat de Barcelona for the use of the 377 ABI PRISM, the ABI PRISM 6700 Automated Nucleic Acid Workstation and the ABI PRISM 7700 sequence detection system. M.F.S.-F. was in receipt of a FPI fellowship from the (Ministerio de Educación y Ciencia) and A. Bosch-Comas is grant-aided by the MECD (Ministerio de Educación, Cultura y Deporte). This study was funded by PM99-0168 (Ministerio de Ciencia y Tecnología) to R.G.-D.
DDBJ/EMBL/GenBank accession no. AY070217
REFERENCES
- 1.Hassold T. and Jacobs,P. (1984) Trisomy in man. Annu. Rev. Genet., 18, 69–97. [DOI] [PubMed] [Google Scholar]
- 2.Epstein C.J. (1995) Epilogue: toward twenty-first century with Down syndrome—a personal view of how far we have come and how far we can reasonably expect to go. Prog. Clin. Biol. Res., 393, 241–246. [PubMed] [Google Scholar]
- 3.Epstein C.J. (1986) The Consequences of Chromosome Imbalance: Principles, Mechanisms and Models. Cambridge University Press, New York.
- 4.Hattori M., Fujiyama,A., Taylor,T.D., Watanabe,H., Yada,T., Parks,H.S., Toyoda,A., Ishii,K., Totoki,Y., Choi,D.K. et al. (2000) The DNA sequence of human chromosome 21. Nature, 405, 311–319. [DOI] [PubMed] [Google Scholar]
- 5.Reymond A., Marigo,V., Yaylaoglu,M.B., Leoni. A., Ucla,C., Scamuffa,N., Cacciopoli,C., Dermitzakis,E.T., Lyle,R., Banfi,S. et al. (2002) Human chromosome 21 gene expression atlas in the mouse. Nature, 420, 582–586. [DOI] [PubMed] [Google Scholar]
- 6.Gitton Y., Dahmane,N., Baik,S., Ruiz i Altaba,A., Neidhardt,L., Scholze,M., Herrmann,B.G., Kahlem,P., Benkahla,A., Schrinner,S. et al. (2002) The HSA21 expression map initiative. A gene expression map of human chromosome 21 orthologues in the mouse. Nature, 420, 586–590. [DOI] [PubMed] [Google Scholar]
- 7.Lisitsyn N., Lisitsyn,N. and Wigler,M. (1993) Cloning the differences between two complex genomes. Science, 259, 946–951. [DOI] [PubMed] [Google Scholar]
- 8.Lisitsyn N.A. (1995) Representational difference analysis: finding the differences between genomes. Trends Genet. 11, 303–307. [DOI] [PubMed] [Google Scholar]
- 9.Hedrick S.M., Cohen,D.I., Nielsen,E.A. and Davis,M.M. (1984) Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature, 308, 149–153. [DOI] [PubMed] [Google Scholar]
- 10.Gurskaya N.G., Diatchenko,L., Chenchik,A., Siebert,P.D., Khaspekov,G.L., Lukyanov,K.A., Vagner,L.L., Ermolaeva,O.D., Lukyanov,S.A. and Sverdlov,E.D. (1996) Equalizing cDNA subtraction based on selective suppression of polymerase chain reaction: cloning of Jurkat cell transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Anal. Biochem., 240, 90–97. [DOI] [PubMed] [Google Scholar]
- 11.Diatchenko L., Lau,Y.-F.C., Chenchik,A., Moqadam,F., Huang,B., Lukyanov,S.A., Lukyanov,K.A., Gurskaya,N.G., Sverdlov,E.D. and Siebert,P.D. (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl Acad. Sci. USA, 93, 6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velculescu V.E., Zhang,L., Vogelstein,B. and Kinzler,K.W. (1995) Serial analysis of gene expression. Science, 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 13.McClelland M., Mathieu-Daude,F. and Welsh,J. (1995) RNA fingerprinting and differential display using arbitrarily primed PCR. Trends Genet., 11, 242–246. [DOI] [PubMed] [Google Scholar]
- 14.Livesey F.J. and Hunt,S.P. (1996) Identifying changes in gene expression in the nervous system: mRNA differential-display. Trends Neurosci., 19, 84–88. [DOI] [PubMed] [Google Scholar]
- 15.Slonim D.K. (2002) From patterns to pathways: gene expression data analysis comes of age. Nature Genet., 32 (suppl.), 502–508. [DOI] [PubMed] [Google Scholar]
- 16.de Haan J., Cristiano,F., Iannello,R.C. and Kola,I. (1995) Cu/Zn-superoxide dismutase and glutathione peroxidase during aging. Biochem. Mol. Biol. Int., 35, 1281–1297. [PubMed] [Google Scholar]
- 17.de Haan J., Wolvetang,E.J., Cristiano,F., Iannello,R., Bladier,C., Kelner,M.J. and Kola,I. (1997) Reactive oxygen species and their contribution to pathology in Down syndrome. Adv. Pharmacol., 38, 379–402. [DOI] [PubMed] [Google Scholar]
- 18.Oyama F., Cairns,N.J., Shimada,H., Oyama,R., Titani,K. and Ihara,Y. (1994) Down’s Syndrome: up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J. Neurochem., 62, 1062–1066. [DOI] [PubMed] [Google Scholar]
- 19.Kitzmueller E., Labudova,O., Rink,H., Cairns,N. and Lubec,G. (1999) Altered gene expression in fetal Down syndrome brain as revealed by the gene hunting technique of subtractive hybridization. J. Neural Transm. Suppl., 57, 99–124. [DOI] [PubMed] [Google Scholar]
- 20.Chrast R., Scott,H.S., Papasavvas,M.P., Rossier,C., Antonarakis,E.S., Barras,C., Davisson,M.T., Schmidt,C., Estivill,X., Dierssen,M. et al. (2000) The mouse brain transcriptome by SAGE: differences in gene expression between P30 brains of the partial trisomy 16 mouse model of Down syndrome (Ts65Dn) and normals. Genome Res., 10, 2006–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng L., Haten,M.E. and Heintz,N. (1994) Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron, 12, 895–908. [DOI] [PubMed] [Google Scholar]
- 22.Kurtz A., Zimmer,A., Schnütgen,F., Brüning,G., Spener,F. and Müller,T. (1994) The expression of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development, 120, 2637–2649. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu F., Watanabe,T.K., Shinomiya,H., Nakamura,Y. and Fujiwara,T. (1997) Isolation and expression of a cDNA for human brain fatty acid-binding protein (B-FABP). Biochim. Biophys. Acta, 1354, 24–28. [DOI] [PubMed] [Google Scholar]
- 24.Godbout R., Bisgrove,D.A., Shkolny,D. and Day,R.S. (1998) Correlation of B-FABP and GFAP expression in malignant glioma. Oncogene, 16, 1955–1962. [DOI] [PubMed] [Google Scholar]
- 25.Bass N.M. (1993) Cellular binding proteins for fatty acids and retinoids—similar or specialized functions. Mol. Cell. Biochem., 123, 191–202. [DOI] [PubMed] [Google Scholar]
- 26.Veerkamp J.H. and Maatman,R.G. (1995) Cytoplasmic fatty acid-binding proteins: their structure and genes. Prog. Lipid Res., 34, 17–52. [DOI] [PubMed] [Google Scholar]
- 27.Schnütgen F., Börchers,T., Müller,T. and Spener,F. (1996) Heterologous expression and characterisation of mouse brain fatty acid binding protein. Biol. Chem. Hoppe-Seyler, 377, 211–215. [DOI] [PubMed] [Google Scholar]
- 28.Hartfuss E., Galli,R., Heins,N. and Götz,M. (2001) Characterization of CNS precursor subtypes and radial glia. Dev. Biol., 229, 15–30. [DOI] [PubMed] [Google Scholar]
- 29.Xu L.X., Sánchez,R., Sali,A. and Heintz,N. (1996) Ligand specificity of brain lipid-binding protein. J. Biol. Chem., 271, 24711–24719. [DOI] [PubMed] [Google Scholar]
- 30.Innis S.M. (1991) Essential fatty acids in growth and development. Prog. Lipid Res., 30, 39–103. [DOI] [PubMed] [Google Scholar]
- 31.Martínez M. (1996) Docosahexaenoic acid therapy in docosahexaenoic acid-deficient patients with disorders of peroxisomal biogenesis. Lipids, 31, 145–152. [DOI] [PubMed] [Google Scholar]
- 32.Feng L. and Heintz,N. (1995) Differentiating neurons activate transcription of the brain lipid-binding protein gene in radial glia through a novel regulatory element. Development, 121, 1719–1730. [DOI] [PubMed] [Google Scholar]
- 33.Josephson R., Müller,T., Pickel,J., Okabe,S., Reynolds,K., Turner,P.A., Zimmer,A. and McKay,R.D.G. (1998) POU transcription factors control expression of CNS stem cell-specific genes. Development, 125, 3087–3100. [DOI] [PubMed] [Google Scholar]
- 34.Knoepfler P.S., Calvo,K.R., Chen,H. and Antonarakis,S.E. (1997) Meis1 and Pknox1 bind DNA cooperatively with Pbx1 utilizing an interaction surface disrupted in oncoprotein E2a-Pbx1. Proc. Natl Acad. Sci. USA, 94, 14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berthelsen J., Zappavigna,V., Mavilio,F. and Blasi,F. (1998) Prep1, a novel functional partner of Pbx proteins. EMBO J., 17, 1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berthelsen J., Zappavigna,V., Ferretti,E., Mavilio,F. and Blasi,F. (1998) The novel homeoprotein Prep1 modulates Pbx-Hox protein cooperativity. EMBO J., 17, 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goudet G., Delhalle,S., Biemar,F., Martial,J.A. and Peers,B. (1999) Functional and cooperative interactions between the homeodomain PDX1, Pbx and Prep1 factors on the somatostatin promoter. J. Biol. Chem., 274, 4067–4073. [DOI] [PubMed] [Google Scholar]
- 38.Ferretti E., Schulz,H., Talarico,D., Blasi,F. and Berthelsen,J. (1999) The PBX-regulating protein PREP1 is present in different PBX-complexed forms in mouse. Mech. Dev., 83, 53–64. [DOI] [PubMed] [Google Scholar]
- 39.Ferretti E., Marshall,H., Popperl,H., Maconochie,M., Krumlauf,R. and Blasi,F. (2000) Segmental expression of Hoxb2 in r4 requires two separate sites that integrate cooperative interactions between Prep1, Pbx and Hox proteins. Development, 127, 155–166. [DOI] [PubMed] [Google Scholar]
- 40.Herzig S., Füzesi,L. and Knepel,W. (2000) Heterodimeric Pbx-Prep1 homeodomain protein binding to the glucagon gene restricting transcription in a cell type-dependent manner. J. Biol. Chem., 275, 27989–27999. [DOI] [PubMed] [Google Scholar]
- 41.Penkov D., Tanaka,S., Di,R.G., Berthelsen,J., Blasi,F. and Ramirez,F. (2000) Cooperative interactions between PBX, PREP and HOX proteins modulate the activity of the alpha 2(V) collagen (COL5A2) promoter. J. Biol. Chem., 275, 16681–16689. [DOI] [PubMed] [Google Scholar]
- 42.Berthelsen J., Viggiano,L., Schulz,H., Ferretti,E., Consalez,G.G., Rocchi,M. and Blasi,F. (1998) PKNOX1, a gene encoding PREP1, a new regulator of PBX activity, maps on human chromosome 21q22.3 and murine chromosome 17B/C. Genomics, 47, 323–324. [DOI] [PubMed] [Google Scholar]
- 43.Chen H., Rossier,C., Nakamura,Y., Lynn,A., Chakravarti,A. and Antonarakis,S.E. (1997) Cloning of a novel homeobox-containing gene, PKNOX1 and mapping to human chromosome 21q22.3. Genomics, 41, 193–200. [DOI] [PubMed] [Google Scholar]
- 44.de la Monte S.M. (1999) Molecular abnormalities of the brain in Down syndrome: relevance to Alzheimer’s neurodegeneration. J. Neural Transm. Suppl., 57, 1–19. [DOI] [PubMed] [Google Scholar]
- 45.Chomczynski P. and Sacchi,N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem., 162, 156–169. [DOI] [PubMed] [Google Scholar]
- 46.Averof M. (1997) Arthropod evolution: same Hox genes, different body plans. Curr. Biol., 7, 634–636. [DOI] [PubMed] [Google Scholar]
- 47.Schreiber E., Mathias,P., Muller,M.M. and Schaffner,W. (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res., 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]