


Abstract
Ligation-mediated PCR was employed to quantify cyclobutane pyrimidine dimer (CPD) formation at nucleotide resolution along exon 2 of the adenine phosphoribosyltransferase (aprt) locus in Chinese hamster ovary (CHO) cells following irradiation with either UVA (340–400 nm), UVB (295–320 nm), UVC (254 nm) or simulated sunlight (SSL; λ > 295 nm). The resulting DNA damage spectrum for each wavelength region was then aligned with the corresponding mutational spectrum generated previously in the same genetic target. The DNA sequence specificities of CPD formation induced by UVC, UVB or SSL were very similar, i.e., in each case the overall relative proportion of this photoproduct forming at TT, TC, CT and CC sites was ∼28, ∼26, ∼16 and ∼30%, respectively. Furthermore, a clear correspondence was noted between the precise locations of CPD damage hotspots, and of ‘UV signature’ mutational hotspots consisting primarily of C→T and CC→TT transitions within pyrimidine runs. However, following UVA exposure, in strong contrast to the above situation for UVC, UVB or SSL, CPDs were generated much more frequently at TT sites than at TC, CT or CC sites (57% versus 18, 11 and 14%, respectively). This CPD deposition pattern correlates well with the strikingly high proportion of mutations recovered opposite TT dipyrimidines in UVA- irradiated CHO cells. Our results directly implicate the CPD as a major promutagenic DNA photoproduct induced specifically by UVA in rodent cells.
INTRODUCTION
Exposure to sunlight is the major determinant in the multistage development of melanoma and non-melanoma forms of skin cancer (1,2). This can be largely attributed to the induction of highly genotoxic dipyrimidine photoproducts, namely cis–syn cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs), which are formed via direct absorption of solar UV photons by DNA. Indeed, sequencing studies have clearly shown that collections of mutated p53 tumor suppressor genes isolated from human and murine skin tumors consistently bear the hallmark ‘signature’ of causation by dipyrimidine photoproducts, i.e., a striking predominance of C→T transitions at dipyrimidine target sites including some tandem CC→TT events, and an accompanying paucity of mutations at A:T base pairs (3,4). In addition, different lines of evidence strongly support a preeminent role specifically for the CPD, which, relative to the 6-4PP, forms in substantially higher yield after sunlight exposure and is repaired much more slowly (5,6). Nonetheless, at least some genotoxic contribution by the 6-4PP (and/or by its Dewar valence photoisomer) should not be ruled out (7).
Long-wavelength UVB (295–320 nm) and UVA (320– 400 nm) constitute ∼0.3 and ∼5.1% of the terrestrial solar spectrum, respectively. UVC (190–280 nm) and short- wavelength UVB (280–295 nm) are virtually completely blocked by the atmosphere and therefore not biologically relevant. The widely employed model mutagen 254-nm UV (hereafter called UVC), being very close to the absorption maximum of DNA (λ = 260 nm), is the most proficient UV wavelength in the generation of dipyrimidine photoproducts. CPDs and 6-4PPs can also be readily formed through direct absorption of UVB photons by DNA, although (in line with the relative DNA absorption potential of UVB) at a yield ∼20–100-fold lower than UVC (8,9). UVA is absorbed orders of magnitude (105-fold) more weakly by DNA compared with UVC (10), and is therefore proportionately far less effective in the induction of dipyrimidine photoproducts (see below). Nonetheless, purified UVA has been shown to exhibit significant mutagenic and carcinogenic potential at physiological fluencies (11–14). This is of major concern since levels of human exposure to UVA have increased significantly in recent decades due to (i) the (previous) widespread use of UVB-specific sunscreens, allowing greatly increased periods of recreational sun tanning by blocking UVB-induced erythema, but without providing UVA protection, and (ii) the current popularity of high-intensity UVA tanning sources, emitting up to 99.9% and 0.1% of energy in the UVA and UVB ranges, respectively (15,16).
Since UVA is so weakly absorbed by DNA, its mutagenic effect has generally not been attributed to dipyrimidine photoproducts, but rather to excitation of non-DNA chromophores leading to the production of reactive oxygen species which, in turn, attack the double helix to yield oxidized bases and DNA strand breaks (11,17,18). In particular, purified UVA induces photooxidation products of guanine, most notably the highly mutagenic adduct 8-oxo-7,8-dihydroguanine (8-oxoGua), through generation of singlet oxygen or via a type I photosensitization reaction (19–22). It should be mentioned that exposure to UVB or broad-spectrum sunlight (but not to UVC) also significantly alters the cellular redox state (23) and induces some 8-oxoGua (22). In a highly characteristic manner, 8-oxoGua would be expected to cause G→T signature mutations; however, such mutations have been only rarely recovered either at target loci in cultured mammalian cells irradiated with either UVB or UVA (13,24,25), or among mutated p53 tumor suppressor genes derived from sunlight-associated skin tumors (26). This latter revelation, together with studies (cited above) demonstrating the powerful promutagenic potential of CPDs in living cells, indicate a minor role at best for oxidative DNA damage in solar mutagenesis. In summary, compared with the well-characterized situation for cells irradiated with UVC, UVB or broad-spectrum natural sunlight, i.e., where CPDs clearly underlie the vast majority of genetic alterations, the primary promutagenic lesion(s) induced by purified UVA at present remain unclear.
Nonetheless, a number of recent studies have provided new insight into the mechanism of UVA mutagenesis. In particular, the global genomic distributions of various classes of DNA photoproducts were investigated in Chinese hamster ovary (CHO) cells exposed to different rigorously filtered sources emitting UVC, UVB, UVA or broad-spectrum simulated sunlight (SSL). CPDs were by far the major photolesion produced by all types of radiation, including by UVA which was shown to generate levels of CPDs at a yield 105-fold lower than UVC (9,22,27), consistent with a mechanism of UVA-induced CPD formation based on direct absorption of UVA photons by DNA (10). In addition, it was shown that the 6-4PP and its Dewar valence photoisomers were readily produced by UVB and SSL, although no such 6-4PP derivatives could be detected following UVA treatment (9). Also, the yield of CPDs was reported to be ∼3-fold higher than that of 8-oxoGua upon UVA exposure (22). Finally, using a sensitive genomic sequencing method (ligation-mediated PCR; see below), under conditions where extremely high levels of CPDs were induced, it was unexpectedly revealed that both 6-4PPs and oxidized DNA bases are virtually undetectable in SSL-exposed cultured cells (28). Taken together, these data demonstrate significant variations in DNA damage distribution depending on incident wavelength and indicate, at least on quantitative grounds, that CPDs might constitute an important promutagenic lesion induced by purified UVA.
Despite the studies described immediately above, which probed global genomic photoproduct distribution in CHO cells exposed to various components of the solar UV wavelength spectrum, no studies to date have coordinately examined the precise nature of genetic damage induced specifically by UVA in living cells and its relationship to UVA mutagenesis at the DNA sequence level. We therefore employed ligation- mediated PCR (LMPCR) to compare the frequency of CPD formation at nucleotide resolution along the transcribed strand (TS) and non-transcribed strand (NTS) of exon 2 of the adenine phosphoribosyltransferase (aprt) locus in CHO cells irradiated with rigorously-filtered sources emitting either UVA (340–400 nm), UVB (295–320 nm), UVC (254 nm) or broad-spectrum SSL (λ > 295 nm). For each wavelength region, the resulting spectrum of CPD damage was compared to the corresponding mutational spectrum generated previously in the same genetic target (13). Our results shed new light on the mechanism of UVA mutagenesis in a mammalian chromosomal gene.
MATERIALS AND METHODS
Cells lines, irradiation conditions and digestion of genomic DNA
CHO cells (strain UVL-9) were grown to 80–90% confluence in 60-mm Petri dishes and irradiated on ice in PBS buffer with 254-nm UVC, UVB (295–320 nm) or SSL (λ > 295 nm) using, respectively, a 254-nm germicidal UVC lamp (G25T8 germicidal lamp, Sankyo Denki, Japan), a fluorescent polychromatic UVB lamp (a series of six 15 W tubes,Vilber Lourmat, France) or a 2500 W xenon arc lamp (XBO, OSRAM, Rosny-sous-Bois, France) (9,13). For UVA irradiation, a 5000 W Mutzhas SUPERSUN lamp (Münich, Germany) emitting virtually exclusively in the UVA1 range (340–440 nm) was utilized. The spectral outputs of these lamps, and properties of the UV wavelength-cutoff filters (Schott, Mainz, Germany) employed to remove contaminating wavelengths (necessary only in the case of UVB and SSL), have been previously characterized (29) (Results and Discussion). Following treatment of living cells with each of the four light sources, genomic DNA was immediately extracted, quantified and digested with T4 endonuclease V (kindly provided by Dr Stephen Lloyd, The University of Texas Medical Branch, Galveston, USA) to incise the DNA very efficiently and with high specificity at CPD sites. This treatment produces 5′-pyrimidine overhangs precisely at the termini of incised strands, which were then removed by photoreactivation using Escherichia coli CPD photolyase (kindly provided by Dr Tim O’Connor, Beckman Research Institute of the City of Hope, Duarte, USA) in order to generate 5′ ligatable ends (30). Prior to using this T4 endonuclease V/photolyase-digested genomic DNA for DNA sequence level analysis via LMPCR (see immediately below), aliquots were run on an alkaline agarose gel to estimate the global CPD frequency induced by each type of UV treatment (31,32).
LMPCR analysis
The LMPCR technique for analysis of CPD distribution at nucleotide resolution in UV-irradiated cells has been described previously in detail (32). Briefly, following denaturation of the T4 endonuclease V/photolyase-digested DNA, an aprt-specific oligonucleotide was annealed downstream of the region to be analyzed, and a set of genomic cleavage products (terminating precisely at sites where T4 endonuclease V incised the DNA adjacent to CPDs) was generated via primer extension with cloned Pfu polymerase (Stratagene, Cedar Creek, TX). An asymmetric double-stranded oligonucleotide linker was ligated to the phosphate groups at the fragment termini, thus providing a common sequence on the 5′ end of all fragments. The longer oligonucleotide primer of the linker, in conjunction with another aprt-specific primer, was then used in a PCR reaction to amplify the gene-specific cleavage products of interest. These products were subjected to electrophoresis on 8% polyacrylamide gels alongside a Maxam–Gilbert sequencing ladder, transferred to nylon membranes, hybridized to a 32P-labeled gene-specific probe and visualized by autoradiography.
Each band on an autoradiogram represents a nucleotide position where a CPD cleavage has occurred, and the intensity of the band reflects the number of DNA molecules with a cleaved CPD at that position. All bands corresponding to CPDs at dipyrimidine sites, and yielding a measurable signal above background, were quantified using a Fuji BAS 1000 phosphorimager (Fuji Medical System, Stanford, CA) as described immediately below. Each sample was assayed in duplicate. A screening sequencing gel was initially run to ensure that there was no significant variation in band intensity between samples. The duplicates were then pooled and a ‘combined’ gel was run. Each irradiation experiment was performed in triplicate in two different laboratories using similar light sources and filters. Two series of LMPCR protocols for each primer set were applied to each DNA sample issued from the various irradiation experiments. The band patterns as well as the quantifications of cleavage products in gels did not differ significantly between individual experiments or manipulators.
To analyze CPD distribution on the NTS of aprt exon 2 (spanning positions +199 to +287), the following primers were used: primer arp394 (for primer extension), 5′-TATGTCACCCTCAGTCCATC-3′ (Tm = 56.3°C); primer arp377 (for PCR amplification), 5′-GCAGGCTCCCCTCCCTTCCCTTAT-3′ (Tm = 71.4°C); primer arp354 (for PCR probe preparation), 5′-CGTGCTGGTCCCCCACTGTG-3′ (Tm = 70.1°C).
For analysis of the TS (spanning positions +321 to +211), the following primers were used: primer arp160R (for primer extension), 5′-CAGTCTCGGGGATCTTG-3′ (Tm = 55.3°C); primer arp181R (for PCR amplification), 5′-TTGTGGTCTCCGCCCCCCTTTCCC-3′ (Tm = 77.1°C); primer arp192R (for PCR probe preparation), 5′-TTTCCCCGGCCACCAGC-3′ (Tm = 68.3°C).
Quantification of band intensity at dipyrimidine sites
Using the phosphorimager software package provided by the manufacturer (Image Gauge V3.0), a rectangle was drawn around each band on the LMPCR autoradiogram corresponding to a dipyrimidine site. The band intensity value corresponding directly to the amount of radioactive material was read within each rectangle as counts per band surface. The amount of radioactive material at every position corresponding to a dipyrimidine site was quantified in irradiated samples as well as in the unirradiated (NoUV) control lane. The relative intensity (in percentage) of each band in a particular lane was calculated as the fraction of counts at that precise position in comparison with the total amount of counts at all bands corresponding to all other dipyrimidine sites in that lane, after correction for background by subtracting the corresponding value in the unirradiated control lane. In other words, the corrected number of counts for a specific band was divided by the total number of counts of all bands in that lane. Where irradiation had been performed at various doses, the mean value was calculated for each dipyrimidine site, since only the global frequency of CPDs varies as a function of dose but not their distribution at individual dipyrimidine sites. The quantification of each autoradiogram was performed blindly by two independent investigators.
RESULTS AND DISCUSSION
CHO cells were exposed to photons from defined regions of the solar wavelength spectrum, and the frequency of CPD formation along the TS and NTS of aprt exon 2 was mapped at nucleotide resolution by LMPCR. For each type of radiation, the various doses employed correspond to those which can be obtained during a few hours of natural sunlight exposure in Paris (France) at summer zenith (27). The UVB (295–320 nm) and SSL (λ > 295 nm) sources were purified using 2-mm-thick UV wavelength-cutoff filters (Schott WG305 and WG320, respectively) to rigorously exclude any contaminating UVC and far-UVB. The energy emitted by the filtered SSL lamp was composed of ∼0.3% UVB, ∼6% UVA, ∼47% visible light and ∼47% infrared. The corresponding values for terrestrial sunlight are 0.3, 5.1, 62.7 and 31.9%, respectively (33). We note that the UVB source contained a UVA component that could not be excluded; however, as previously discussed (13), this very minor component would not be expected to exert any significant effect on the end-points (i.e., DNA damage and mutation) studied here (29). The broad-band UVA lamp was employed without any cutoff filters. However, the emission spectrum of this lamp consists almost entirely of UVA1 radiation (340–400 nm) and does not contain any biologically significant UVB or UVC (i.e., wavelengths below 330 nm contributing <10–6 of the total energy output, according to the emission spectrum provided by the manufacturer). Consequently, the UVA-induced CPD frequency distribution described herein can be entirely attributed to the action of UVA photons.
The distributions of CPDs following UVB, SSL and UVC irradiation are quite similar
Figure 1A shows a typical LMPCR autoradiogram reflecting the CPD frequency distribution at nucleotide resolution along the NTS of aprt exon 2 in cells irradiated with 0.03, 0.6 or 3000 kJ/m2 of UVC, UVB or SSL, respectively. Similar raw data (not shown) were obtained for the TS. Relative CPD frequencies at individual nucleotide positions along aprt exon 2 were determined by phosphorimager analysis. In the case of UVC, UVB and SSL, CPDs were detected at all dipyrimidine sites along both strands. It is particularly noteworthy that the LMPCR band patterns and intensities generated by UVC, UVB and SSL were very similar (visual inspection of Fig. 1A). Indeed, as graphically depicted in the upper portion of Figure 2 and in Figure 3 (corresponding to the NTS and TS, respectively), phosphorimager analysis clearly revealed that initial CPD frequencies induced by each type of radiation along aprt exon 2 were generally equivalent when considering individual dipyrimidine target sites. Furthermore, in actually calculating the proportion of CPDs formed at each of the four classes of dipyrimidine sites, it was revealed that, in the case of either UVC, UVB or SSL, the overall proportions of CPDs forming at TT, TC, CT and CC sites were ∼28, ∼26, ∼16 and ∼30%, respectively (Table 1). This relative CPD frequency distribution is atypical, especially when compared with the general situation for UVC-irradiated total genomic DNA, where the proportions of CPDs at TT, TC, CT and CC have been estimated at 68, 16, 13 and 3%, respectively (34,35). In addition, although not apparent in the present case, the ratio of CPDs at C-containing dipyrimidines relative to TT dipyrimidines has been shown to increase significantly following exposure to either UVB or SSL as opposed to exposure to UVC (35,36). The apparently peculiar CPD frequency distribution observed along aprt exon 2 can be at least partially explained by the relatively small size (100 bp) of this genetic target. Most notably in this latter respect, aprt exon 2 is very rich in C-containing dipyrimidines together with a paucity in runs of Ts where CPDs are readily formed after UVC (37,38). Specifically, aprt exon 2 contains 65 potential dipyrimidine targets in total, i.e., 5 TT, 11 TC, 9 CT and 19 CC on the NTS, and 3 TT, 6 TC, 7 CT and 5 CC on the TS.
Figure 1.
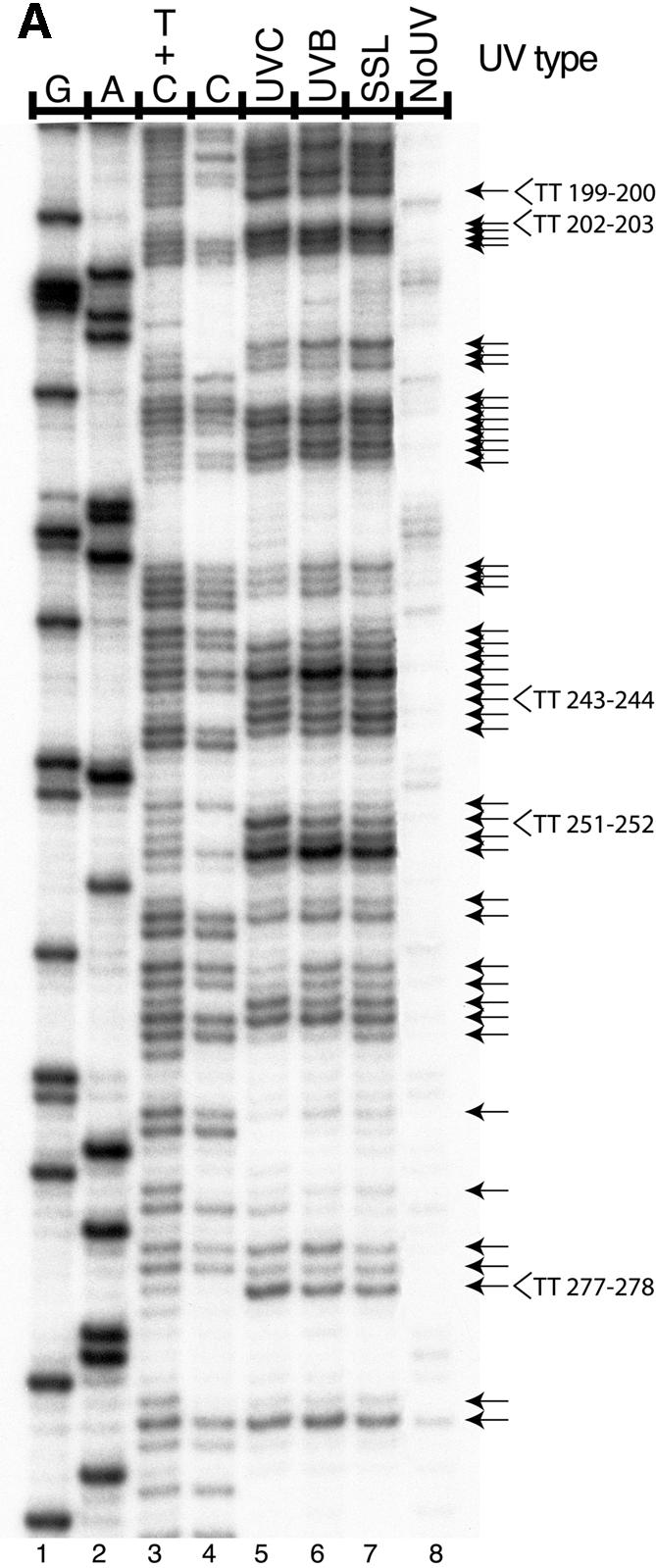

Induction of CPDs by UVA, UVB, UVC radiation and SSL, at nucleotide resolution, in exon 2 of the aprt gene of CHO cells. Cells were either unirradiated (NoUV) or irradiated as indicated and CPD formation was analyzed by LMPCR along the NTS of aprt gene exon 2. The doses for UVB, SSL and UVC induced approximately equal global CPD frequencies as determined by alkaline denaturing agarose gel electrophoresis of T4 endonuclease V-treated genomic DNA (data not shown). The arrows on the right side of the autoradiograms indicate dipyrimidine sites quantified with phosphorimager. The first four lanes from the left on each autoradiogram exhibit LMPCR of DNA treated in standard Maxam–Gilbert cleavage reactions. (A) The three lanes to the right of the Maxam–Gilbert sequencing ladder represent LMPCR of DNA isolated from cells irradiated with 0.03 kJ/m2 of UVC, 0.6 kJ/m2 of UVB or 3000 kJ/m2 of SSL, respectively. (B) The nine lanes to the right of the Maxam–Gilbert sequencing ladder represent LMPCR of DNA isolated from cells irradiated with (from left to right, respectively): 4500, 3000, 1500 kJ/m2 of UVA; 3.75, 1.20, 0.60 kJ/m2 of UVB; 7500, 5000, 2500 kJ/m2 of SSL.
Figure 2.

Distribution of CPDs and mutations induced by UVA, UVB, UVC radiation and SSL in exon 2 of the aprt gene of CHO cells. CPDs were mapped along the NTS of exon 2. Quantification of CPDs was performed as described in Materials and Methods. The upper part of the figure represents the relative frequencies of CPD formation at every dipyrimidine site. In the lower part of the figure, mutations as reported by Drobetsky et al. (13), and mapped along the sequence independently of the strand where the premutagenic lesions at the origin of the base changes are expected. The relative frequency of mutations at each site was calculated as the ratio of the number of mutants recovered at a particular site to the total number of mutants occurring along exon 2 of the aprt gene after irradiation. All events were considered in the calculation and double mutations were counted as one change at each of the two positions. The underlined nucleotides correspond to positions 238–246.
Figure 3.

Distribution of CPDs induced by UVA, UVB, UVC radiation and SSL in exon 2 of the aprt gene of CHO cells. CPDs were mapped along the TS. Quantification of CPDs was performed as described in Materials and Methods.
Table 1. The relative proportionsa of CPDs formed at the four classes of dipyrimidine sites, along both strands of exon 2 of the aprt gene in CHO cells exposed to UVA, UVB, UVC radiation and SSL.
| UVA | UVB | SSL | UVC | |
|---|---|---|---|---|
| TT | 57% | 27% | 29% | 26% |
| TC | 18% | 26% | 28% | 24% |
| CT | 11% | 17% | 17% | 15% |
| CC | 14% | 30% | 26% | 35% |
aFollowing quantification of autoradiograms, the frequency of CPDs formed at a particular class of dipyrimidine sites was divided by the number of dipyrimidine sites in this class contained in exon 2 of the aprt gene, including transcribed and non-transcribed strands (8 TT, 17 TC, 16 CT and 24 CC sites). The relative proportion of CPDs formed at the four different classes of dipyrimidine sites was then normalized to 100%.
The UVA-induced CPD distribution differs from that induced by UVB, SSL and UVC
The LMPCR autoradiogram in Figure 1B depicts the CPD distribution along the NTS of aprt exon 2 following irradiation with increasing doses of UVA, UVB or SSL. In the case of UVB and SSL, CPD frequencies increased as a function of dose at all possible dipyrimidine target sites on both strands (visual inspection of Fig. 1B; autoradiogram for the TS not shown). However, the site specificity for DNA damage formation following exposure to rigorously purified UVA was strikingly different relative to UVB or SSL. Indeed, CPDs were detected at all potential dipyrimidine sites along both strands of aprt exon 2 in UVA-exposed cells only at the highest dose, i.e., 4.5 kJ/m2 (upper portions of Figs 2 and 3). The UVA doses also generated a lower global CPD frequency than the doses of UVB and SSL, as judged by denaturing agarose gel electrophoresis (data not shown).
UVA-induced CPDs are predominantly formed at thymine–thymine dipyrimidine sites
The proportion of CPDs formed at each of the four classes of dipyrimidine sites (TT, CC, CT and TC) have been calculated independently of the number of each of these sites in the analyzed sequence (Table 1). Assuming the same number of dipyrimidine sites for each class, the proportion of CPDs formed at TT sites following UVA radiation would be 57%, at least in the sequence context of aprt exon 2. Moreover, the increased proportion of CPDs generated at TT sites following irradiation with UVA versus either UVB or SSL is significantly different (P < 0.0004 and <0.00002; Fisher’s exact test). Using a sensitive HPLC–MS/MS assay, a large majority of CPDs were recovered at TT sites in bulk genomic DNA from CHO cells exposed to UVA, but not to UVB or SSL (88% versus 42 and 48%, respectively; T.Douki, J.Cadet and E.Sage, manuscript submitted).
Although DNA absorbs UVA extremely weakly relative to UVC or UVB (10), it was nonetheless previously shown that the global yield of CPDs in genomic DNA isolated from CHO cells treated with UVA is consistent with a mechanism of formation based on direct absorption of UVA photons (330–360 nm) by DNA (9,27). Moreover, based on action spectra and relative spectral effectiveness for CPD induction, it has been demonstrated that photons of longer UVA wavelength can also be implicated in the production of CPDs (19,22). Furthermore, the distribution of CPDs formed by direct absorption of UVA photons is not expected to differ from that of CPDs produced by UVB or SSL. This latter notion, coupled with the strikingly high frequency of CPDs observed here at TT sites uniquely after UVA treatment, strongly suggest that UVA-induced CPDs are formed through means other than (or in addition to) direct absorption by DNA. In particular, triplet photosensitization would be expected to produce CPDs predominantly at TT sites, to a lesser extent at TC and CT sites and not at all at CC sites, due to the lower energy of the triplet state of thymine in comparison with cytosine. The preferential formation of CPDs at TT sites via photosensitization after UV exposure has been well documented in vitro (39–43). Various non-DNA chromophores are able to absorb UVA photons and presumably transfer energy or electrons to other cellular macromolecules (11); however, potential photosensitizers that could be implicated in preferential CPD formation at TT sites in UVA-exposed cells remain to be investigated.
Induction of CPDs at TT sites by UVA radiation correlates with sites of mutation along exon 2 of the CHO aprt gene
It is now well established that the probability of mutation fixation at a given nucleotide position in UV-irradiated cells is strongly dependent upon the initial frequency of CPDs and their subsequent rate of removal via nucleotide excision repair (44,45). Therefore, in order to shed further light on the mechanism of solar UV mutagenesis, and of UVA-induced mutagenesis in particular, the CPD damage spectrum generated here for each of UVC, UVB, UVA and SSL was aligned and compared with the corresponding mutational spectrum characterized previously in the same genetic target (13,46,47). These prior studies showed that the vast majority (>90%) of aprt– mutations induced by either UVC or UVB at the CHO aprt locus was of the UV signature type, i.e., C→T transitions at dipyrimidine sites including some tandem CC→TT events, and correspondingly very few mutations at A:T base pairs. UV signature mutations also comprised a majority of genetic alterations induced by SSL at the aprt locus, although T→G transversions at TT sites were induced at significantly higher levels (25% of the SSL-induced mutation spectrum) compared with either UVC (5%) or UVB (9%). The relatively high frequency of this unusual type of base substitution in SSL-exposed cells could be attributed to the UVA component of the SSL source, since it was also revealed in the same study that T→G transversions at TT dipyrimidines were represented in strikingly high proportion (37%) at the CHO aprt locus following exposure to rigorously purified UVA. It was originally postulated, based on the above study, that UVA-induced mutations might be attributed to CPDs, since the vast majority of these mutations, including T→G, were targeted to dipyrimidine sites. Furthermore, three highly characteristic CC→TT UV signature tandem mutations, in addition to a significant proportion of C→T transitions at dipyrimidine sites (5 and 22%, respectively), were recovered after UVA treatment.
Figure 2 illustrates the alignment of damage and mutation spectra along the NTS of exon 2 at the CHO aprt locus, where >60% of all aprt– mutations induced by UVC, UVB, UVA or SSL were recovered in the previous study described immediately above. The pyrimidine run spanning nucleotides 238–246 (especially site 241, but also 242–244; nucleotides 238–246 are underlined in Fig. 2), and to a lesser extent the one spanning nucleotides 250–254 (only position 253), constitute the hottest sites for mutation induction following exposure to UVC, UVB or SSL. Furthermore, these pyrimidine runs are clearly the sites that are most frequently damaged following exposure to any of the three types of radiation. CPDs were also very frequently formed at positions 202–206; however, this pyrimidine run is located within an intron, i.e., where mutations may not yield a selectable aprt– phenotype. At positions 241–242 and 253, following UVA irradiation, GC→AT and tandem CC→TT mutations were also recovered where CPDs formed, but to a much lesser extent relative to UVC, UVB or SSL.
All UVA-induced CPD damage hotspots occur at TT sites and correlate well with UVA-induced mutation hotspots. For example, the dipyrimidine site TT 243–244, where the majority of UVA-induced T→G mutations were recovered, represents a very strong hotspot for CPD formation specifically after UVA exposure. In addition, TT 276–277, which was more frequently damaged by UVA than by other types of radiation, was also more frequently mutated by UVA and SSL, and only T→G mutations were recovered (13). Although the UVA-induced CPD hotspot at TT 199–200 is located in an intron, one tandem TT→GG mutation was observed after UVA, strongly suggesting a CPD as the premutagenic lesion. In contrast, the TT site 251–252, which is a hotspot for UVA-induced CPDs, is not mutated by UVA. However, a T→G base substitution at the 5′ position of the TT would not modify the amino acid residue, making this an unselectable mutation. Such a change at the 3′ position of the TT would change a serine into alanine, but may not lead to a mutated phenotype. In any case, among hundreds of mutations at the aprt locus generated by exposure to diverse DNA damaging agents, no mutations have ever been recovered at TT 251–252 (48).
The overall data provide strong support for the increasingly well-established notion that CPDs underlie the vast majority of mutations generated by natural sunlight exposure, and directly implicate CPDs as a preeminent premutagenic photoproduct induced by rigorously purified UVA, at least in CHO cells. In all likelihood, 6-4PPs do not play a significant role in UVA-induced mutagenesis, because neither the 6-4PP nor its Dewar valence photoisomer are detectable following UVA exposure (9). Also, investigators have shown that among the plethora of oxidized DNA bases characterized to date, UVA generates mainly 8-oxoGua and very few oxidized pyrimidines (19,22,49). The highly mutagenic 8-oxoGua usually induces G→T and not T→G transversions (50). Very few, if any, G→T transversions have been recovered after UVA irradiation (13,24). Finally, since reactive oxygen species are produced by UVA, and consequently by sunlight (11), a role for the highly mutagenic oxidative intermediate 8-oxodGTP in the generation of T→G transversions cannot be discounted. However, incorporation of 8-oxodGTP is not expected to be sequence specific, nor to be responsible for the induction of tandem TT→GG mutations as we have observed at the CHO aprt locus. Further support for the notion that CPDs at TT sites can induce T→G transversions comes from the fact that UVC irradiation has been shown to frequently generate such transversions at the hypoxanthine-guanine phosphoribosyltransferase (hprt) locus in hamster cells (51).
Although the relatively high proportion of CPDs at TT sites appears to provide a clear basis for the striking predominance of T→G transversions at the CHO aprt locus following irradiation with UVA, we emphasize that this would not appear to underlie the significant increase in T→G mutations also observed after SSL irradiation. Indeed, in the current study, unlike the striking situation for UVA, no difference in levels of TT CPDs were observed for cells exposed to SSL versus UVC or UVB. At present, we have no explanation for this apparent discrepancy. We speculate that, in addition to influencing initial CPD formation, UVA wavelengths may also differentially affect other determinants of mutation fixation relative to the case for UVC or UVB, e.g., by decreasing or increasing the rate of repair and/or of translesion synthesis, respectively, particularly at TT sites.
In conclusion, our data strongly support the notion of the CPD as a major premutagenic lesion in UVA-induced mutagenesis. We would like to emphasize that UVA-induced CPDs at TT sites, presumably formed by photosensitization, could be responsible for T→G transversions in rodent cells. Assuming that these observations can be applied to human skin cancer, our data provide strong molecular impetus for advising the use of sunscreens that contain UVA- as well as UVB-blocking agents. Furthermore, extensive exposure to high intensity UVA tanning lamps could represent a risk for skin cancer through significant TT CPD formation.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr R. Stephen Lloyd and Dr Tim O’Connor for supplying T4 endonuclease V and photolyase, respectively. This work was supported by grants (held separately by E.A.D. and R.D.) from the National Cancer Institute of Canada (NCIC) (with funds from the Canadian Cancer Society and the Terry Fox Run), and from the Institut Curie (Genotoxicology Program) and by Centre National de la Recherche Scientifique (held by E.S.). E.A.D. and R.D. are scholars of the Fonds de la Recherche en Santé du Québec (FRSQ). J.-P.T. is the recipient of a postdoctoral fellowship from the NCIC. P.J.R. holds student fellowships from the Hôpital St-François d‘Assise Research Center and from the Canadian Institute for Health Research (CIHR). D.P. is the recipient of a PhD fellowship from the Ministère de l’Éducation Nationale de l’Enseignement Supérieur et de la Recherche and from the Fondation pour la Recherche Médicale.
REFERENCES
- 1.Setlow R.B. (1974) The wavelengths in sunlight effective in producing skin cancer: a theoretical analysis. Proc. Natl Acad. Sci. USA, 71, 3363–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black H.S., deGruijl,F.R., Forbes,P.D., Cleaver,J.E., Ananthaswamy,H.N., deFabo,E.C., Ullrich,S.E. and Tyrrell,R.M. (1997) Photocarcinogenesis: an overview. J. Photochem. Photobiol. B., 40, 29–47. [DOI] [PubMed] [Google Scholar]
- 3.Brash D.E., Rudolph,J.A., Simon,J.A., Lin,A., McKenna,G.J., Baden,H.P., Halperin,A.J. and Ponten,J. (1991) A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl Acad. Sci. USA, 88, 10124–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ananthaswamy H.N., Fourtanier,A., Evans,R.L., Tison,S., Medaisko,C., Ullrich,S.E. and Kripke,M.L. (1998) p53 Mutations in hairless SKH-hr1 mouse skin tumors induced by a solar simulator. Photochem. Photobiol., 67, 227–232. [DOI] [PubMed] [Google Scholar]
- 5.Mullenders L.H., Hazekamp-van Dokkum,A.M., Kalle,W.H., Vrieling,H., Zdzienicka,M.Z. and van Zeeland,A.A. (1993) UV-induced photolesions, their repair and mutations. Mutat. Res., 299, 271–276. [DOI] [PubMed] [Google Scholar]
- 6.You Y.H., Lee,D.H., Yoon,J.H., Nakajima,S., Yasui,A. and Pfeifer,G.P. (2001) Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J. Biol. Chem., 276, 44688–44694. [DOI] [PubMed] [Google Scholar]
- 7.Gibbs P., Horsfall,M., Borden,A., Kibley,B.J. and Lawrence,C.W. (1993) Understanding spectra of UV-induced mutations: studies with individual photoproducts. In Shima,A.e.a. (ed.), Frontiers of Photobiology. Elsevier Science Publishers, Netherlands, pp. 357–362.
- 8.Mitchell D.L., Jen,J. and Cleaver,J.E. (1991) Relative induction of cyclobutane dimers and cytosine photohydrates in DNA irradiated in vitro and in vivo with ultraviolet-C and ultraviolet- B light. Photochem. Photobiol., 54, 741–746. [DOI] [PubMed] [Google Scholar]
- 9.Perdiz D., Grof,P., Mezzina,M., Nikaido,O., Moustacchi,E. and Sage,E. (2000) Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells. Possible role of Dewar photoproducts in solar mutagenesis. J. Biol. Chem., 275, 26732–26742. [DOI] [PubMed] [Google Scholar]
- 10.Sutherland J.C. and Griffin,K.P. (1981) Absorption spectrum of DNA for wavelengths greater than 300 nm. Radiat. Res., 86, 399–409. [PubMed] [Google Scholar]
- 11.Tyrrell R.M. and Keyse,S.M. (1990) New trends in photobiology. The interaction of UVA radiation with cultured cells. J. Photochem. Photobiol. B., 4, 349–361. [DOI] [PubMed] [Google Scholar]
- 12.van Weelden H., van der Putte,S.C., Toonstra,J. and van der Leun,J.C. (1990) UVA-induced tumours in pigmented hairless mice and the carcinogenic risks of tanning with UVA. Arch. Dermatol. Res., 282, 289–294. [DOI] [PubMed] [Google Scholar]
- 13.Drobetsky E.A., Turcotte,J. and Chateauneuf,A. (1995) A role for ultraviolet A in solar mutagenesis. Proc. Natl Acad. Sci. USA, 92, 2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Setlow R.B. (1999) Spectral regions contributing to melanoma: a personal view. J. Investig. Dermatol. Symp. Proc., 4, 46–49. [DOI] [PubMed] [Google Scholar]
- 15.Urbach F. (1993) Environmental risk factors for skin cancer. Recent Results Cancer Res., 128, 243–262. [DOI] [PubMed] [Google Scholar]
- 16.Gasparro F.P. (2000) Sunscreens, skin photobiology, and skin cancer: the need for UVA protection and evaluation of efficacy. Environ. Health Perspect., 108 Suppl 1, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peak M.J. and Peak,J.G. (1990) Hydroxyl radical quenching agents protect against DNA breakage caused by both 365-nm UVA and by gamma radiation. Photochem. Photobiol., 51, 649–652. [PubMed] [Google Scholar]
- 18.Ravanat J.L., Douki,T. and Cadet,J. (2001) Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B., 63, 88–102. [DOI] [PubMed] [Google Scholar]
- 19.Kielbassa C., Roza,L. and Epe,B. (1997) Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis, 18, 811–816. [DOI] [PubMed] [Google Scholar]
- 20.Kvam E. and Tyrrell,R.M. (1997) Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis, 18, 2379–2384. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X., Rosenstein,B.S., Wang,Y., Lebwohl,M., Mitchell,D.M. and Wei,H. (1997) Induction of 8-oxo-7,8-dihydro-2′-deoxyguanosine by ultraviolet radiation in calf thymus DNA and HeLa cells. Photochem. Photobiol., 65, 119–124. [DOI] [PubMed] [Google Scholar]
- 22.Douki T., Perdiz,D., Grof,P., Kuluncsics,Z., Moustacchi,E., Cadet,J. and Sage,E. (1999) Oxidation of guanine in cellular DNA by solar UV radiation: biological role. Photochem. Photobiol., 70, 184–190. [PubMed] [Google Scholar]
- 23.Tyrrell R.M. (1996) Oxidant, antioxidant status and photocarcinogenesis: the role of gene activation. Photochem. Photobiol., 63, 380–383. [DOI] [PubMed] [Google Scholar]
- 24.Robert C., Muel,B., Benoit,A., Dubertret,L., Sarasin,A. and Stary,A. (1996) Cell survival and shuttle vector mutagenesis induced by ultraviolet A and ultraviolet B radiation in a human cell line. J. Invest. Dermatol., 106, 721–728. [DOI] [PubMed] [Google Scholar]
- 25.Sage E., Lamolet,B., Brulay,E., Moustacchi,E., Chateauneuf,A. and Drobetsky,E.A. (1996) Mutagenic specificity of solar UV light in nucleotide excision repair-deficient rodent cells. Proc. Natl Acad. Sci. USA, 93, 176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dumaz N., Stary,A., Soussi,T., Daya-Grosjean,L. and Sarasin,A. (1994) Can we predict solar ultraviolet radiation as the causal event in human tumours by analysing the mutation spectra of the p53 gene? Mutat. Res., 307, 375–386. [DOI] [PubMed] [Google Scholar]
- 27.Kuluncsics Z., Perdiz,D., Brulay,E., Muel,B. and Sage,E. (1999) Wavelength dependence of ultraviolet-induced DNA damage distribution: involvement of direct or indirect mechanisms and possible artefacts. J. Photochem. Photobiol. B., 49, 71–80. [DOI] [PubMed] [Google Scholar]
- 28.Yoon J.H., Lee,C.S., O‘Connor,T.R., Yasui,A. and Pfeifer,G.P. (2000) The DNA damage spectrum produced by simulated sunlight. J. Mol. Biol., 299, 681–693. [DOI] [PubMed] [Google Scholar]
- 29.Grof P., Ronto,G. and Sage,E. (2002) A computational study of physical and biological characterization of common UV sources and filters, and their relevance for substituting sunlight. J. Photochem. Photobiol. B., 68, 53–59. [DOI] [PubMed] [Google Scholar]
- 30.Pfeifer G.P., Drouin,R., Riggs,A.D. and Holmquist,G.P. (1992) Binding of transcription factors creates hot spots for UV photoproducts in vivo. Mol. Cell. Biol., 12, 1798–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drouin R., Gao,S. and Holmquist,G.P. (1996) Agarose gel electrophoresis for DNA damage analysis. In Pfeifer, G. P. (ed.), Technologies for Detection of DNA Damage and Mutations. Plenum Press, New York, NY, pp. 37–43.
- 32.Drouin R., Therrien,J.P., Angers,M. and Ouellet,S. (2001) In vivo DNA analysis. Methods Mol. Biol., 148, 175–219. [DOI] [PubMed] [Google Scholar]
- 33.Piver W.T. (1991) Global atmospheric changes. Environ. Health Perspect., 96, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Setlow R.B. and Carrier,W.L. (1966) Pyrimidine dimers in ultraviolet-irradiated DNAs. J. Mol. Biol., 17, 237–254. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell D.L., Jen,J. and Cleaver,J.E. (1992) Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res., 20, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sage E. (1999) DNA damage and mutations induced by solar UV radiation. In Baumstark-Khan,C., Kozubek,S. and Horneck,G. (eds), Fundamentals for the Assessment of Risks from Environmental Radiation. Kluwer Academic Publishers, Netherlands, pp. 115–126.
- 37.Sage E., Cramb,E. and Glickman,B.W. (1992) The distribution of UV damage in the lacI gene of Escherichia coli: correlation with mutation spectrum. Mutat. Res., 269, 285–299. [DOI] [PubMed] [Google Scholar]
- 38.Sage E. (1993) Distribution and repair of photolesions in DNA: genetic consequences and the role of sequence context. Photochem. Photobiol., 57, 163–174. [DOI] [PubMed] [Google Scholar]
- 39.Lamola A.A. (1968) Excited state precursors of thymine photodimers. Photochem. Photobiol., 8, 619–632. [Google Scholar]
- 40.Charlier M. and Helene,C. (1972) Photochemical reactions of aromatic ketones with nucleic acids and their components. 3. Chain breakage and thymine dimerization in benzophenone photosensitized DNA. Photochem. Photobiol., 15, 527–536. [DOI] [PubMed] [Google Scholar]
- 41.Moysan A., Viari,A., Vigny,P., Voituriez,L., Cadet,J., Moustacchi,E. and Sage,E. (1991) Formation of cyclobutane thymine dimers photosensitized by pyridopsoralens: quantitative and qualitative distribution within DNA. Biochemistry, 30, 7080–7088. [DOI] [PubMed] [Google Scholar]
- 42.Chouini-Lalanne N., Defais,M. and Paillous,N. (1998) Nonsteroidal antiinflammatory drug-photosensitized formation of pyrimidine dimer in DNA. Biochem. Pharmacol., 55, 441–446. [DOI] [PubMed] [Google Scholar]
- 43.Sauvaigo S., Douki,T., Odin,F., Caillat,S., Ravanat,J.L. and Cadet,J. (2001) Analysis of fluoroquinolone-mediated photosensitization of 2′-deoxyguanosine, calf thymus and cellular DNA: determination of type-I, type-II and triplet-triplet energy transfer mechanism contribution. Photochem. Photobiol., 73, 230–237. [DOI] [PubMed] [Google Scholar]
- 44.Holmquist G.P. and Gao,S. (1997) Somatic mutation theory, DNA repair rates, and the molecular epidemiology of p53 mutations. Mutat. Res., 386, 69–101. [DOI] [PubMed] [Google Scholar]
- 45.Holmquist G.P. and Drouin,R. (1998) Cyclobutane dimer frequency and their repair rates drive tumor mutation frequency in p53 genes. In Hönigsmann,H., Knobler,R.M., Trautigner,F. and Jori,G. (eds), Landmarks in Photobiology. OEMF spa, Milan, Italy, pp. 396–398.
- 46.Drobetsky E.A., Grosovsky,A.J. and Glickman,B.W. (1987) The specificity of UV-induced mutations at an endogenous locus in mammalian cells. Proc. Natl Acad. Sci. USA, 84, 9103–9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drobetsky E.A., Moustacchi,E., Glickman,B.W. and Sage,E. (1994) The mutational specificity of simulated sunlight at the aprt locus in rodent cells. Carcinogenesis, 15, 1577–1583. [DOI] [PubMed] [Google Scholar]
- 48.de Boer J.G., Drobetsky,E.A., Grosovsky,A.J., Mazur,M. and Glickman,B.W. (1989) The Chinese hamster aprt gene as a mutational target. Its sequence and an analysis of direct and inverted repeats. Mutat. Res., 226, 239–244. [DOI] [PubMed] [Google Scholar]
- 49.Pouget J.P., Douki,T., Richard,M.J. and Cadet,J. (2000) DNA damage induced in cells by gamma and UVA radiation as measured by HPLC/GC-MS and HPLC-EC and Comet assay. Chem. Res. Toxicol., 13, 541–549. [DOI] [PubMed] [Google Scholar]
- 50.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 51.Menichini P., Vrieling,H. and van Zeeland,A.A. (1991) Strand-specific mutation spectra in repair-proficient and repair-deficient hamster cells. Mutat. Res., 251, 143–155. [DOI] [PubMed] [Google Scholar]