

Abstract
Phage display is a widely used technology for the isolation of peptides and proteins with specific binding properties from large libraries of these molecules. A drawback of the common phagemid/helper phage systems is the high infective background of phages that do not display the protein of interest, but are propagated due to non-specific binding to selection targets. This and the enhanced growth rates of bacteria harboring aberrant phagemids not expressing recombinant proteins leads to a serious decrease in selection efficiency. Here we describe a VCSM13-derived helper phage that circumvents this problem, because it lacks the genetic information for the infectivity domains of phage coat protein pIII. Rescue of a library with this novel CT helper phage yields phages that are only infectious when they contain a phagemid-encoded pIII-fusion protein, since phages without a displayed protein carry truncated pIII only and are lost upon re-infection. Importantly, the CT helper phage can be produced in quantities similar to the VCSM13 helper phage. The superiority of CT over VCSM13 during selection was demonstrated by a higher percentage of positive clones isolated from an antibody library after two selection rounds on a complex cellular target. We conclude that the CT helper phage considerably improves the efficiency of selections using phagemid-based protein libraries.
INTRODUCTION
The display of peptide and protein libraries on filamentous phages, in combination with effective selection strategies, has proved a rapid and successful method to obtain proteins with specific binding properties (1,2). A powerful application of phage display is the generation of high-affinity recombinant antibodies with unique specificities for a wide variety of antigens. Although antibody molecules are very large and complex, relatively small scFv antibody fragments can be efficiently displayed on the phage surface. ScFv libraries with a diversity of up to 1011 have been created by genetically fusing scFv repertoires to phage coat protein pIII. These scFv–pIII libraries are encoded by phagemids, which are plasmids containing a phage origin of replication and morphogenetic signal, both required for packaging of the phagemid into phage particles. The pIII protein, present in three to five copies per phage particle, consists of N-terminal N1 and N2 domains involved in phage infectivity and a C-terminal (CT) domain essential for phage assembly. Importantly, fusion of scFv to the N-terminus of N1 does not abolish functional activity of the pIII protein. After transformation with a phagemid library, bacteria need to be infected by helper phage such as VCSM13 that provides all the genes required for phage assembly. Enrichment of scFv libraries for antigen-specific phages is accomplished by incubation with the antigen, removal of non-bound phages and elution of bound phages. Eluted phages are used to infect Escherichia coli bacteria, which are then cultured and superinfected with helper phages to rescue phages that can be used in subsequent selection rounds.
A well-known problem in phage display is the less efficient incorporation into the phage coat of phagemid-encoded scFv–pIII, compared with native pIII encoded by the helper phage genome. As a result the vast majority of phages do not contain scFv in their coat. In addition, phagemid libraries often contain many clones that do not express a scFv–pIII fusion, due to stop codons and frame shifts introduced during library construction. Undesirable enrichment of such aberrant phagemids may result during amplification steps, caused by enhanced growth rates of bacteria not expressing scFv–pIII. As a consequence, the number of phages without a fusion protein has been estimated to be up to 99.5% of the total phage population (3). This large background of ‘bald’ phages reduces selection efficiencies by binding non-specifically to complex antigens and subsequently infecting bacteria during amplification of the selected phages. This holds not only for display of scFv, but for phage display selections using any recombinant protein library.
Here we present a VCSM13-derived mutant helper phage, named CT helper phage, to circumvent the described problems. The CT helper phage genome lacks the infectivity domains N1 and N2 of protein pIII, rendering rescued phages infective only if they display a phagemid-encoded fusion protein of scFv and full-length pIII. We demonstrate that the use of this novel helper phage substantially reduces the background of irrelevant phages, resulting in highly improved selection efficiencies.
MATERIALS AND METHODS
General DNA procedures
All DNA handling and cloning procedures were performed according to standard protocols (4) or protocols provided with DNA isolation kits from Qiagen. Cloning of PCR fragments into pCR4-TOPO and pCR4-BluntII-TOPO was done according to protocols provided with the TOPO TA Cloning Kit for Sequencing and the Zero Blunt TOPO PCR Cloning Kit (Invitrogen). Constructed clones were checked by restriction analysis as well as DNA sequencing using the ABI PRISM BigDye Terminator Cycle Sequencing Kit from Applied Biosystems.
Construction of CT helper phage genome
Primers 5′-TTAGGTTGGTGCCTTCGTAG-3′ and 5′-GGATCCAGCGGAGTGAGAATAGAAAGG-3′ (Invitrogen) were used in a PCR reaction with VCSM13 DNA (Stratagene) as a template to amplify a 400 bp fragment that included the pIII leader sequence with an introduced unique BamHI site downstream. This PCR fragment was cloned into pCR4-TOPO, yielding plasmid pCR4-TOPO-L. Primers 5′-GGATCCTCTGGTTCCGGTGATTTTGATTATG-3′ and 5′-TTGCTTCTGTAAATCGTCGC-3′ were used in a PCR reaction with VCSM13 DNA as a template to amplify a 1779 bp fragment that included the pIII CT domain with an introduced unique BamHI site upstream. Cloning of this fragment into pCR4-TOPO yielded plasmid pCR4-TOPO-CT. The 413 bp NotI–BamHI fragment of pCR-TOPO-L was ligated into the 5717 bp NotI–BamHI fragment of pCR4-TOPO-CT, resulting in plasmid pCR4-TOPO-LCT. The 2117 bp SnaBI–PacI fragment of pCR4-TOPO-LCT was ligated into the 5803 bp SnaBI–PacI fragment of the VCSM13 genome, resulting in the CT helper phage genome which lacks domains N1 and N2 of gene III.
Construction of pIII-complementing plasmid pUC19-g3
Primers 5′-CAAATTCTATTTCAAGGAGACAGTCATAATGAAAAAATTATTATTCGCAATTCCTTTAG-3′ and 5′-GCTAACATACTGCGTAATAAGGAGTCTTAAGAATTCCAGTTCTTT-3′ were used in a PCR reaction with VCSM13 DNA as a template to amplify a 1318 bp fragment consisting of the complete pIII gene and a downstream EcoRI site. Primers 5′-GATTACGCCAAGCTTGCATGCAAATTCTATTTCAAGGAGA-3′ and 5′-GCTAACATACTGCGTAATAAGGAGTCTTAAGAATTCCAGTTCTTT-3′ were used in a PCR reaction with the 1318 bp fragment as a template to elongate this fragment, thereby introducing a unique HindIII site upstream of the pIII gene. This fragment was cloned into pCR4-BluntII-TOPO, yielding plasmid pCR4-BluntII-TOPO-g3. The 1314 bp HindIII–EcoRI fragment of pCR4-BluntII-TOPO-g3 was ligated into the 2635 bp HindIII–EcoRI fragment of pUC19 (New England Biolabs), resulting in plasmid pUC19-g3 which contains the wild-type pIII gene under the control of the lacZ promoter.
Construction of phagemid pUC119-TG1
A 273 bp DNA fragment including restriction sites EcoRI, HindIII, NcoI and NotI was synthesized using a proprietary method developed by BaseClear and cloned into a pGEM-T vector (Promega), resulting in plasmid pGEM-NN. The 267 bp HindIII–EcoRI fragment of pGEM-NN was ligated into the 3111 bp HindIII–EcoRI fragment of phagemid pUC119 (Takara), resulting in phagemid pUC119-NN. The 738 bp NcoI–NotI fragment of phagemid pHEN-TG1 (described below) containing a thyroglobulin-specific scFv gene was ligated into the 3330 bp NcoI–NotI fragment of pUC119-NN, resulting in phagemid pUC119-TG1. This phagemid expresses soluble thyroglobulin-specific scFv fragments.
CT helper phage production
CT phages harboring no full-length pIII but only its CT domain were produced as follows. Tetracycline-resistant XL1-Blue bacteria (Stratagene) were transformed with the CT phage genome and grown in 20 ml 2YT-KT medium (2YT medium with 25 µg/ml kanamycin and 10 µg/ml tetracycline) overnight shaking at 37°C. Bacteria were pelleted by centrifugation and the supernatant was incubated at least 1 h on ice-water with 1/5 vol of PEG/NaCl (20% PEG 6000, 2.5 M NaCl). Precipitated phages were pelleted by centrifugation and supernatants were discarded. Phages were resuspended in PBS/1% BSA, followed by removal of remaining cell debris by centrifugation. The supernatant was sterilized through a 0.45 µm filter and stored at 4°C.
CT helper phages harboring both full-length and truncated pIII were produced as follows. XL1-Blue was transformed with plasmid pUC19-g3 and the transformants were made electrocompetent. The CT genome was transformed into these cells where after the cells were mixed with 1 ml SOC medium containing 100 µg/ml ampicillin, 10 µg/ml tetracycline and 4.5% glucose. After incubating for 1 h shaking at 37°C, 0.5 ml of this mixture was added to 5 ml 2YT-AKT medium (2YT-KT medium with 100 µg/ml ampicillin). This culture was incubated for 6 h shaking at 37°C, bacteria were pelleted and the supernatant discarded. The cells were resuspended in 20 ml 2YT-AKTI medium (2YT-AKT medium with 1 mM IPTG) and incubated overnight shaking at 30°C, followed by isolation of the produced phages via PEG precipitation as above. These CT helper phages were used to produce a larger batch of helper phages as follows. XL1-Blue(pUC19-g3) was grown at 37°C to mid-log phase in 150 ml 2YT-ATG medium (2YT medium with 100 µg/ml ampicillin, 10 µg/ml tetracycline and 4.5% glucose). This culture was infected with 2 × 1011 c.f.u. of CT helper phages (a 10-fold excess of phage over bacteria) for 30 min at 37°C without shaking. The culture was centrifuged and the supernatant discarded. Bacterial cells were resuspended in 1.5 l 2YT-AKTI medium and grown overnight shaking at 30°C. Bacteria were pelleted by centrifugation and phages were precipitated as described above. Phage pellets were resuspended in PBS/1% BSA, filter-sterilized and stored at 4°C.
Phage rescue
Phages were rescued from a previously described scFv library (5,6) and similar libraries as follows. Bacteria were grown at 37°C to mid-log phase in 2YT-AG medium (2YT-ATG medium without tetracycline). The culture was infected with VCSM13 or CT helper phage (using a 10-fold excess of phages over bacteria) for 30 min at 37°C without shaking. Bacterial cells were pelleted by centrifugation and resuspended in 2YT-AK medium (2YT-AKT medium without tetracycline). Phages were produced by growing the culture overnight shaking at 30°C. Bacteria were pelleted by centrifugation and phages were isolated from the culture supernatant by PEG precipitation as described above.
Rescue of phagemids expressing monoclonal phage antibodies was performed essentially as described above. Four clones were amplified that specifically bind either thyroglobulin (pHEN-TG1), CD8α on T lymphocytes (pHEN-CD8) or have other well-defined staining patterns on peripheral blood cells (pHEN-BG and pHEN-BT) (5,6).
Phage titration
Total phage titers were measured by ELISA. Nunc-Immuno plates were coated with anti-M13 monoclonal antibody (Amersham Biosciences) in PBS. Wells were washed with PBS, blocked with PBS/1% milkpowder (PBSM) and incubated with serial dilutions of phage in PBSM. After washing with PBS/0.1% Tween 20 and PBS, wells were incubated with HRP/Anti-M13 conjugate (Amersham Biosciences). Wells were washed with PBS/0.1% Tween 20 and with PBS, followed by detection of bound phages with FAST OPD (Sigma). The reaction was stopped with 1 M sulfuric acid. Absorbances at 492 nm were used to calculate phage titers in comparison with a VCSM13 reference taken during the procedure.
Infective titers of VCSM13 helper phages were determined by plaque count assay. XL1-Blue grown at 37°C to mid-log phase in 2YT-T medium (2YT-KT medium without kanamycin) was infected for 30 min at 37°C with a serial dilution of the helper phage. Top agar (1:1 mixture of TYE-agar and 2YT medium) was added and spread on TYE-T plates (TYE-agar with 10 µg/ml tetracycline) that were incubated overnight at 37°C. The number of plaques was used to calculate the phage titers.
Infective titers of CT helper phages were determined by colony count assay. XL1-Blue grown at 37°C to mid-log phase in 2YT-T medium was infected for 30 min at 37°C without shaking with varying dilutions of CT helper phage. Bacteria were plated on TYE-KT (TYE-T with 25 µg/ml kanamycin) and grown overnight at 37°C. The number of colonies was used to calculate the phage titers.
For identification and titration of CT helper phages carrying pUC19-g3, the colony count assay was performed as above, except for plating on TYE-ATG (TYE-T with 100 µg/ml ampicillin and 4.5% glucose) and TYE-AKTG (TYE-ATG with 25 µg/ml kanamycin).
Infective titers of phages rescued from the scFv library were determined by the colony count assay as above, except for growing in 2YT-TG medium (2YT-T medium with 4.5% glucose), and plating on TYE-ATG.
Western blot
Monoclonal scFv-displaying phages were analyzed using western blot analysis. Samples containing 1010 phages were incubated in reducing loading buffer for 5 min at 100°C, run on a 10% Bis-Tris gel and transferred to PVDF membrane according to the instructions provided with the NuPAGE system (Invitrogen). The membrane was blocked in PBSM and incubated with anti-pIII antibody (MoBiTec) recognizing the CT domain. After washing in PBS/0.2% Tween, the membrane was incubated with rabbit anti-mouse/HRP (DAKO) and washed in PBS/0.2% Tween 20. Proteins were visualized using ECL substrate (Amersham Biosciences).
ScFv detection
ScFv molecules displayed by rescued monoclonal phages were detected via the Myc tag in the scFv–pIII fusion protein using ELISA essentially as described above for phage titration, except for using anti-c-Myc-peroxidase (Roche) instead of the HRP/Anti-M13 conjugate.
The fraction of phages rescued from a scFv library that is infective and carries a functional scFv gene was determined by a colony lift immunoassay. XL1-Blue grown at 37°C to mid-log phase in 2YT-T medium was infected with phages rescued from the scFv library, for 30 min at 37°C without shaking. Infected bacteria were grown on TYE-ATG overnight at 37°C and lifted from the plate using a nitrocellulose filter (Schleicher and Schuell). The filter was placed on a TYE-ATI plate (TYE-T with 100 µg/ml ampicillin and 1 mM IPTG) and incubated overnight at 30°C, resulting in expression of soluble scFv in addition to scFv–pIII. Subsequently the filter was washed in PBS/0.05% Tween 20 to remove adherent bacteria, blocked in PBSM, washed in PBS/0.05% Tween 20 and incubated with mouse anti-c-Myc (Roche). After washing in PBS/0.05% Tween 20, the filter was incubated with rabbit anti-mouse/HRP (DAKO) and washed in PBS/0.05% Tween 20. The filters were developed by incubation in DAB substrate (Sigma) followed by rinsing with water.
Flow cytometry
Monoclonal scFv-displaying phages rescued with either VCSM13 or CT helper phage were evaluated by flow cytometry essentially as described before (6). In short, 5 × 105 peripheral blood leukocytes were incubated with 5 × 1011 monoclonal phages in PBSM, washed, incubated with mouse anti-M13-biotin (Crucell), washed, and incubated with streptavidin-PE (Caltag). Cells were analyzed using a FACSCalibur cytometer with CellQuestPro software (Becton Dickinson).
Model experiment for phage enrichment
XL1-Blue(pHEN-TG1) was mixed with XL1-Blue(pUC119-TG1) in a ratio of 1:10, 1:100 or 1:1000 based on absorbance at 600 nm of the respective cultures grown in 2YT-ATG. Phages were rescued from the mixture using either VCSM13 or CT as described above. XL1-Blue was infected with the rescued phages and plated, followed by a colony lift immunoassay as described above to identify colonies producing scFv encoded by pHEN-TG1. The identity of clones that were negative in this screening was verified by PCR analysis using a primer combination specific for pUC119-TG1.
Phage selection on tumor cells
A selection for binding to molecules expressed on U937 cells was performed using a phagemid-based scFv library (5) rescued using either the VCSM13 or CT helper phage. Briefly, 106 U937 cells were incubated at 4°C with 0.5 ml of the amplified libraries (1013 phages), pelleted by centrifugation and washed three times in RPMI 1640 medium (Invitrogen) to remove unbound phages. Bound phages were eluted using 50 mM glycine, pH 2.2, neutralized using 1 M Tris–HCl, pH 7.5 and used to infect XL1-Blue. After overnight growth, colonies were scraped from plates and grown to mid-log phase, followed by rescue using VCSM13 or CT helper phage as above. Undesired phages were depleted from the phage mixture using incubation with a large excess of peripheral blood leukocytes. Remaining phages were used in a second round of selection on U937 cells. Phages of individual clones were prepared and screened for binding to U937 cells by flow cytometry as described above. DNA of positive clones was sequenced to establish the selected scFv gene diversity.
RESULTS
Strategy
Rescue of phagemids encoding scFv–pIII fusions using helper phages like VCSM13 yields phage particles displaying scFv, as depicted schematically in Figure 1A. However, the majority of rescued phages do not display scFv (Fig. 1B), as phagemid-encoded scFv–pIII is incorporated less efficiently into the phage coat than pIII encoded by the helper phage. These ‘bald’ phages may bind non-specifically to selection targets, however, and will infect bacteria during amplification, thereby reducing the efficiency of selections. We reasoned that this problem could be solved by deleting the infectivity domains N1 and N2 of pIII from the genome of helper phage VCSM13, while leaving the CT domain intact. Rescue of phagemids with this CT helper phage would yield a population containing both truncated pIII and scFv–pIII (Fig. 1C), in addition to a large fraction harboring truncated pIII only (Fig. 1D). Since the phages without a scFv also lack the infectivity domains, this background of irrelevant phages would be lost upon re-infection of bacteria.
Figure 1.

Schematic representation of phages rescued with VCSM13 or CT helper phage. The phage coat is depicted as a black rectangle, domains N1, N2 and CT of coat protein pIII as labeled circles, and scFv as a labeled square. (A and B) Phages rescued with VCSM13 helper phage. A minor population displays scFv (A), while the majority contains only wild-type pIII encoded by VCSM13 helper phage (B). Both forms are infective due to the presence of N1 and N2. (C and D) Phages rescued with CT helper phage. A minor population displays scFv (C), while the majority contains only the CT domain of pIII encoded by CT helper phage (D). Phages with displayed protein are infective due to the presence of N1 and N2, while phages without displayed protein are not infective since they lack N1 and N2.
CT helper phage production
The CT helper phage genome was constructed by removing the region encoding the infectivity domains N1 and N2 of the pIII protein from the VCSM13 genome (Fig. 2). Expression of this genome in XL1-Blue yielded a stock containing 1011 phage particles per milliliter, as determined by ELISA. To verify that these phages are not infective, XL1-Blue was incubated with 108 CT phages (carrying kanamycin resistance) and grown overnight in the presence of kanamycin. No growth was observed, while bacteria infected with VCSM13 grew normally.
Figure 2.

Schematic representation of pIII and pIII-derived proteins encoded by the various DNA constructs used in this study. Anti-thyroglobulin scFv and domains N1, N2 and CT of coat protein pIII are depicted as labeled open boxes, Myc tag is drawn as a dashed box, and linker sequences are shown as black boxes. An amber stop codon is located between the Myc tag and the N1 domain in pHEN-TG1. ScFv–pIII fusion proteins encoded by pHEN-BG, pHEN-CD8 and pHEN-BT have the same structure as depicted for pHEN-TG1. N-terminal leader sequences present in the precursors of the five mature proteins are not depicted here.
CT helper phage needs to be infective in order to transfer its genome into bacteria carrying a phagemid; therefore, the complementing plasmid pUC19-g3 was constructed to provide full-length pIII during helper phage production (Fig. 2). XL1-Blue was transformed with pUC19-g3, followed by transformation of CT DNA into these bacteria. After overnight culture 1012 phage particles were obtained. All these CT helper phages were infective, indicating incorporation of full-length pIII. Upscaling of this protocol proved impractical, therefore CT helper phages from this batch were used to transfer CT DNA to XL1-Blue(pUC19-g3) by infection rather than transformation. This procedure yielded 5 × 1014 CT helper phages from 1.5 l culture, similar to yields routinely obtained when producing VCSM13.
Plasmids sometimes incorporate into phage particles, even though they are not single stranded and lack the packaging signal (7). To check for packaging of pUC19-g3 in CT helper phage, XL1-Blue was infected with CT helper phage and grown on plates containing appropriate antibiotics. According to the number of colonies, 1 in every 5 × 105 CT helper phages may contain pUC19-g3, while approximately 1 in every 5 × 106 may carry both the pUC19-g3 plasmid and the CT genome. A plaque count assay was performed to detect infective CT helper phages that give rise to subsequent generations of phages that remain infective. Small plaques were observed for 1 in every 105 phages, indicating that this fraction contains a source of full-length pIII. Therefore, in agreement with the colony count assay, 0.001% of CT helper phages may contain the pUC19-g3 plasmid and thus a native pIII gene.
Phage rescue and analysis
To validate the CT helper phage, four monoclonal phagemids selected previously from a scFv library were rescued. One of these clones specifically binds to thyroglobulin, while three clones recognize different defined cell populations within a mixture of peripheral blood leukocytes [see (5,6) and Materials and Methods]. Total and infective phage titers were determined by ELISA and colony count assays, respectively (Fig. 3). Average total titers of CT- and VCSM13-rescued phages were similar, the latter being moderately higher. For VCSM13, the infective titer of rescued phages was comparable with the total titer, the small difference representing some variability between the two assays. In contrast, the infective titer of CT-rescued phages was 300-fold lower than the total titer. These results support the prediction that ∼99% of rescued phages do not display scFv, this population being infective when VCSM13 is used, while non-infective when CT is used.
Figure 3.

Titers of VCSM13- and CT-rescued phages. Total titers (white bars) and infective titers (gray bars) of four monoclonal scFv-displaying phages rescued with either VCSM13 or CT were determined by ELISA and colony counting, respectively, as described in Materials and Methods. Average titers of rescued monoclonal phages pHEN-TG1, pHEN-BG, pHEN-CD8 and pHEN-BT are shown for VCSM13 and CT. Error bars indicate standard errors of average values.
To investigate the relative amount of scFv–pIII incorporated into the monoclonal phages, an ELISA was performed using an antibody recognizing the Myc tag present in the scFv–pIII fusion protein. Absorbances were corrected for differences in total phage titer. Some variation in observed signals was apparent among the four clones, but for all CT-rescued clones the values were higher (8-fold on average) than for the corresponding VCSM13-rescued clones (Fig. 4). Western blot analysis using anti-pIII antibody confirmed the incorporation of scFv–pIII in rescued phages. Comparable amounts of scFv–pIII were detected as a band of ∼85 kDa in samples containing 1010 VCSM13- or CT-rescued phages of clone pHEN-CD8 (Fig. 5). For phages rescued with VCSM13, a second protein of ∼60 kDa indicated the incorporation of full-length pIII, while a band of ∼17 kDa demonstrated the presence of truncated pIII in CT-rescued phages.
Figure 4.

ScFv display in VCSM13- and CT-rescued phages. Incorporation of scFv in the coat of four monoclonal scFv-displaying phages rescued with either VCSM13 or CT was determined by ELISA as described in Materials and Methods. Values in arbitrary units (a.u.) were obtained by subtraction of a blank measurement (no phages added) and division by the number of phages in each sample.
Figure 5.
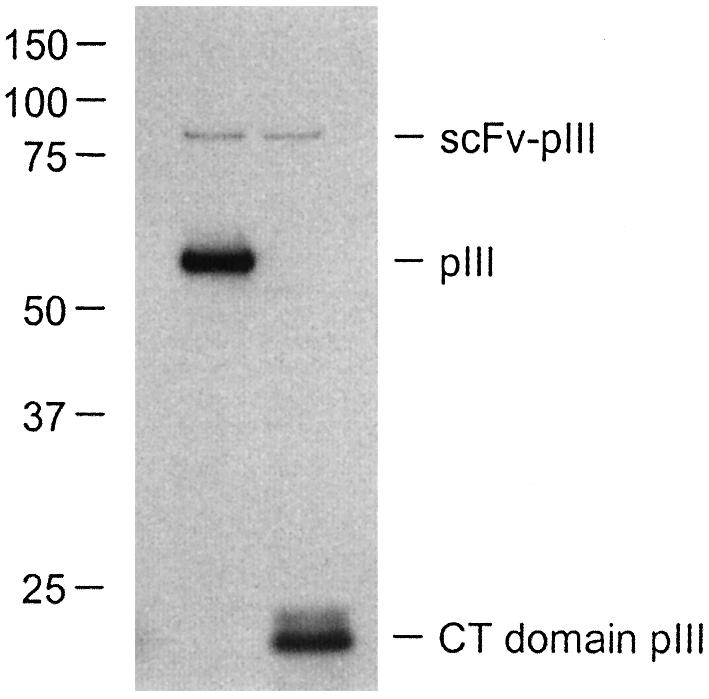
SDS–PAGE and western blot analysis of monoclonal scFv- displaying phages using anti-pIII antibody. Samples containing 1010 phages of clone pHEN-CD8 rescued using VCSM13 (left) or CT helper phages (right) were analyzed as described in Materials and Methods. Molecular masses (kDa) of marker proteins are indicated.
To compare the antigen-binding properties of the monoclonal phages, they were incubated with peripheral blood leukocytes and detected using flow cytometry. Dot plots for clones pHEN-TG1 and pHEN-CD8 are shown in Figure 6. Clone pHEN-TG1 does not bind to leukocytes (Fig. 6B and C) while clone pHEN-CD8 binds to a subpopulation of lymphocytes only (Fig. 6D and E), independent of the helper phage used for rescue. The mean fluorescent intensity (MFI) of the positive population obtained using CT-rescued pHEN-CD8 (MFI 1725) was reproducibly (data not shown) higher than when using VCSM13-rescued pHEN-CD8 (MFI 1247). Similar results were obtained using CT- and VCSM13-rescued clone pHEN-BG, specific for B lymphocytes and granulocytes (data not shown).
Figure 6.

Analysis by flow cytometry of VCSM13- and CT-rescued scFv-displaying phages binding to B- and T-lymphocytes (top) and granulocytes (bottom). Peripheral blood leukocytes were incubated with phages, followed by detection using biotin-labeled phage-specific antibodies and PE-labeled streptavidin, as described in Materials and Methods. Values of MFI for the positive populations are indicated in each plot. (A) Negative control without phages. (B) VCSM13-rescued pHEN-TG1 (specific for thyroglobulin, not present on leukocytes). (C) CT-rescued pHEN-TG1. (D) VCSM13-rescued pHEN-CD8 (specific for T lymphocytes expressing CD8α). (E) CT-rescued pHEN-CD8.
Enrichment of phages displaying scFv in a model experiment
A model experiment was performed to demonstrate efficient elimination of phages without scFv–pIII. Phagemid pHEN-TG1 encodes a thyroglobulin-specific scFv fused via a Myc tag to full-length pIII (Fig. 2). The related phagemid pUC119-TG1 (both are derivatives of pUC119) encodes the same scFv, but lacks the Myc tag and pIII (Fig. 2). Phages can incorporate pUC119-TG1, but the encoded scFv cannot be displayed. Therefore, these phages mimic the background of phages without a displayed scFv that are usually formed after rescue of phagemids encoding scFv–pIII. We reasoned that rescue of a mixture of pHEN-TG1 and pUC119-TG1 with CT helper phage would enrich for clones containing pHEN-TG1 while such enrichment would not occur when using VCSM13. XL1-Blue bacteria transformed with phagemid pHEN-TG1 or pUC119-TG1 were mixed in a 1:10 ratio, followed by rescue of phages from the mixture with either VCSM13 or CT helper phage. Rescued phages were used to infect XL1-Blue, followed by growth on plate, colony lifting and identification of the pHEN-TG1 phagemid using the Myc tag expressed from this phagemid only. Using VCSM13, 1 of 182 counted colonies was positive for the Myc tag (Table 1). The fraction of scFv–pIII producing clones was apparently not maintained at 10%, and had even decreased to ∼0.5%. In contrast, 448 of 622 colonies (72%) were positive for the Myc tag when CT helper phage was used (Table 1). The fraction of positive clones was thus two orders of magnitude higher when CT was used instead of VCSM13, indicating a considerable enrichment for pHEN-TG1 that encodes scFv–pIII. Analysis of 16 negative colonies confirmed that all contained the pUC119-TG1 phagemid. Comparable results were obtained when the initial ratios of XL1-Blue(pHEN-TG1) and XL1-Blue(pUC119-TG1) were 1:100 and 1:1000 (Table 1).
Table 1. Rescue of phages from a mixture of phagemids leads to selective amplification of phagemids encoding a functional scFv–pIII fusion.
| Helper phage | Input fraction scFv–pIII gene | Output fraction scFv–pIII gene | Enrichment factor |
|---|---|---|---|
| VCSM13 | 0.1 | 0.005 (1 of 182) | 0.05 |
| 0.01 | <0.005 (0 of 190) | <0.5 | |
| 0.001 | <0.004 (0 of 245) | <4 | |
| CT | 0.1 | 0.72 (448 of 622) | 7 |
| 0.01 | 0.51 (144 of 280) | 51 | |
| 0.001 | 0.11 (57 of 516) | 110 |
XL1-Blue(pHEN-TG1) expressing scFv–pIII was mixed with a 10-, 100- or 1000-fold excess of XL1-Blue(pUC119-TG1) expressing scFv without pIII, followed by rescue of phages from the mixture with VCSM13 or CT helper phage. XL1-Blue was infected with these rescued phages, colony lifted and used to determine the fraction of phages expressing scFv–pIII. See Materials and Methods for experimental details.
Enrichment of phages displaying scFv from a library
Next we examined whether CT helper phage could enrich for phagemids expressing functional scFv–pIII from a partially randomized scFv library, which inherently contains a substantial fraction of clones without a functional scFv–pIII gene. The complete scFv library (5) was rescued with either CT or VCSM13. XL1-Blue was infected with a small portion of the rescued libraries and grown on plate. Colonies were filter-lifted and clones producing scFv were detected using Myc tag specific antibody. As shown in Table 2, 28% of the clones infected with VCSM13-rescued phages produced scFv, whereas 84% of CT-rescued phages produced scFv.
Table 2. Rescue of a large semi-synthetic scFv library with CT helper phage leads to selective amplification of phages displaying scFv.
| Helper phage | Positive clones in experiment A | Positive clones in experiment B | Positive clones in experiment C | Total positive clones |
|---|---|---|---|---|
| VCSM13 | 32% (48 of 148) | 21% (21 of 102) | 27% (20 of 73) | 28% (89 of 323) |
| CT | 82% (108 of 132) | 87% (79 of 91) | 84% (37 of 44) | 84% (224 of 267) |
XL1-Blue bacteria were infected with phages from the VCSM13- or CT-rescued scFv library, followed by colony lifting and detection of scFv-producing clones as described in Materials and Methods. Percentages of scFv-positive clones were calculated for three individual experiments.
Phage selection on tumor cells
To test whether the improved helper phage results in more efficient selections, a selection was performed on tumor cell line U937 using the scFv phagemid library rescued with either VCSM13 or CT helper phages. Two selection rounds were performed, using the corresponding helper phage to rescue phages after the first round. The number of colonies obtained after the first and second round of selection shows that 10 times less colonies were obtained when CT helper phage was used instead of VCSM13 (Table 3). Phages were rescued from 93 individual colonies from both selections and screened for binding to U937 cells using flow cytometry. When VCSM13 was used throughout, none of the screened clones was positive. In contrast, 32 clones (34%) were positive when CT helper phage was used. DNA sequencing of 10 randomly picked positive clones revealed 7 different scFv genes (data not shown).
Table 3. Selection of phages from a scFv library using CT instead of VCSM13 helper phage results in more positive clones after two rounds of selection.
| Helper phage | Total clones after first round | Total clones after second round | Positive clones after second round |
|---|---|---|---|
| VCSM13 | 4 × 106 | 1 × 106 | <1% (0 of 93) |
| CT | 4 × 105 | 9 × 104 | 34% (32 of 93) |
VCSM13- and CT-rescued scFv libraries were used to isolate clones specifically binding to U937 cells, using the corresponding helper phage to rescue clones after each selection round. Screening for positive clones after the second round was done using flow cytometry. See Materials and Methods for experimental details.
DISCUSSION
Rescue of phages from a phagemid-based phage display library using conventional helper phages like VCSM13 generally results in a high frequency (up to 99.5%) of phages not displaying the protein of interest (3). These irrelevant phages are nevertheless infective and thus form a significant background during selection procedures. In a previous attempt to circumvent this problem, the complete pIII gene was deleted from the helper phage genome, leaving scFv–pIII encoded by the phagemid as the only source of pIII (7–10). This results in the display of multiple copies of scFv, leading to avidity effects that may be undesirable when selections are aimed at finding high-affinity binders. Furthermore, production yields for these helper phages are considerably lower than for VCSM13-like helper phages. Another solution to the problem of infective phages without scFv was provided by helper phages that contain amber stop codons within the pIII gene (11). Production of these helper phages in amber suppressing bacteria leads to the incorporation of native pIII. When the resulting so-called Ex-phages are used to rescue phages in non-suppressing bacteria, no full-length pIII is translated from the helper phage genome. This also leads to multivalent scFv display, which is of limited use as explained above. Moreover, many commonly used phagemids contain an amber codon between the scFv and pIII, preventing the use of Ex-phages with these phagemids. Yet another solution to the background problem is the use of a helper phage that harbors a proteolytic cleavage site between the N2 and CT domains (12). Since phagemid-encoded scFv–pIII does not have a cleavage site between these domains, incubation of selected phages with protease selectively destroys the infectivity of phages without scFv–pIII. The use of this helper phage is obviously restricted to display of protease-resistant proteins.
None of the three mutant helper phages described above proved satisfactory in the sense of universal applicability. Therefore, we designed a new helper phage for monovalent display that does not rely on specific phagemids, bacteria or protease treatments. This CT helper phage encodes a truncated pIII protein lacking the infectivity domains N1 and N2. We reckoned that phages rescued with CT helper phage would be infective only if they display phagemid-encoded scFv, which is fused to a full-length pIII.
A highly efficient protocol for the production of CT helper phage yielded titers comparable with those of VCSM13. We used a pIII-complementing plasmid, a strategy that has been applied in analogous fashion by others for producing helper phages lacking the complete pIII gene (7–9). Low yields were reported for the production of these pIII-less helper phages. This is likely due to insufficient expression of native pIII from engineered expression plasmids, as the CT domain of pIII is essential for completion and release of stable phage particles. Our helper phage expresses the CT domain directly from its genome, in a natural regulatory context. This results in the expression of similar amounts of CT domain compared with complete pIII in VCSM13. Assembly of CT helper phages is therefore less dependent on integration of plasmid-encoded pIII than assembly of pIII-less helper phages, resulting in more efficient phage packaging and consequently high yields of functional phages and phage antibodies. A previously recognized drawback of using a pIII-complementing plasmid is its incorporation into phage particles, despite the fact that it is not single stranded and lacks a packaging signal (7). We found that 1 in every 105 CT helper phages may carry the pUC19-g3 plasmid. An equivalent frequency of 1 in 3 × 105 was reported for the incorporation of pIII-complementing plasmid in VCSM13-derived pIII-less helper phages (7). Incorporated pUC19-g3 can lead to the assembly of phages that are infective while lacking a scFv–pIII fusion. The background of infective phages that do not display a scFv is therefore not completely abolished using CT helper phage, yet drastically reduced since only 0.001% of CT helper phages carry a native pIII gene compared to >99% when VCSM13 is used.
Total phage titers were similar for VCSM13- and CT-rescued monoclonal phagemids, demonstrating the functionality of CT helper phage in phage rescue. All VCSM13-rescued phages were infective, while ∼1% of rescued phages was infective when CT was used, supporting the hypothesis that CT-rescued phages are infective only if they display scFv. ELISA analysis of scFv incorporation in rescued phages showed ∼8-fold higher absorbance when CT helper phage was used instead of VCSM13. Consistent with these results, flow cytometric analysis of phage binding to defined subpopulations of leukocytes demonstrated ∼1.5-fold higher MFI for CT-rescued monoclonal phages. These observations may be explained by improved exposure of the displayed scFv to detecting antibodies and cell surface antigens, respectively, in the environment of truncated pIIIs (157 amino acids) when compared with full-length pIIIs (406 residues). Alternatively, a larger fraction of rescued phages may display scFv when CT helper phage is used, caused by less efficient incorporation of truncated pIII from CT helper phage as compared with wild-type pIII from VCSM13, and hence a relatively more efficient incorporation of scFv–pIII. Interestingly, western blot experiments with equal amounts of VCSM13- and CT-rescued monoclonal phages demonstrated a comparable scFv–pIII content. Due to limited accuracy of the ELISA used for determining the total phage titers of the western blot samples, up to 2-fold differences in scFv–pIII incorporation may have been masked. Nevertheless, these data suggest that improved accessibility of displayed scFv in CT-rescued phages is more probable than the existence of a larger population of phages displaying scFv.
A model experiment involving a mixture of two different phagemids confirmed our hypothesis that the use of CT helper phage leads to loss of phages that do not display a scFv–pIII fusion. In this experiment, we showed that using the CT helper phage very efficiently enriches for phagemids expressing the complete scFv–pIII fusion. Further evidence for the functionality of CT helper phage was provided by the rescue and analysis of a phagemid library containing a partially randomized scFv sequence. A substantial fraction of the clones in such a library may not express scFv, due to the introduction of frame shifts and stop codons during library construction (13). The ability of the CT helper phage to efficiently enrich for those clones that do express a functional scFv–pIII fusion, was demonstrated by a 3-fold higher fraction of scFv-producing clones after rescue with CT instead of VCSM13.
The final test for the CT helper phage comprised a relevant selection procedure aimed at the isolation of clones specifically binding to a tumor cell line. The comparatively low number of clones obtained after the first and second selection rounds using CT helper phage reflected the loss of phages not expressing a scFv–pIII fusion. Screening of 93 individual clones after the second round resulted in 32 positive clones with various sequences from the CT-rescued library, while no positive clones were obtained when VCSM13 was used. This side-by-side comparison demonstrates that use of CT helper phage enables efficient selection of different clones after only two rounds of selection on a complex selection target.
In conclusion, we have constructed a novel helper phage for phage display that is clearly superior over commonly used helper phages. This CT helper phage was validated for the display of scFv antibody fragments, but it should be applicable to phage display selections of any protein. In a basic selection protocol, phages are selected for the binding properties of the displayed protein fragments. However, due to the high numbers of phages added to selection targets, many phages not displaying pIII-fusions will bind to the targets non-specifically. These phages may contain a phagemid encoding a non-relevant pIII-fusion, or a phagemid that does not express fusion protein at all. Such background phages will get rescued as they contain helper phage-derived pIII proteins that allow for infection of the bacteria used in amplification steps. In contrast, phages rescued using the CT helper phage should meet two additional criteria if they are to survive a selection protocol. Not only should they bind to the selection target, the phages should also contain a phagemid expressing a full-length pIII-fusion and this pIII-fusion protein should be displayed on the phage to allow for infection of the E.coli bacteria. Thus, we expect the CT helper phage to represent a major improvement for the rapid isolation of specific clones from phage display libraries.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to acknowledge Erwin Houtzager and Joeke van der Velde for their contribution to this study.
REFERENCES
- 1.Hoogenboom H.R. (2002) Overview of antibody phage-display technology and its applications. Methods Mol. Biol., 178, 1–37. [DOI] [PubMed] [Google Scholar]
- 2.Smothers J.F., Henikoff,S. and Carter,P. (2002) Tech.Sight. Phage display. Affinity selection from biological libraries. Science, 298, 621–622. [DOI] [PubMed] [Google Scholar]
- 3.Azzazy H.M. and Highsmith,W.E. (2002) Phage display technology: clinical applications and recent innovations. Clin. Biochem., 35, 425–445. [DOI] [PubMed] [Google Scholar]
- 4.Sambrook J. and Russell,D.W. (2001) Molecular Cloning. A Laboratory Manual, 3rd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 5.de Kruif J., Boel,E. and Logtenberg,T. (1995) Selection and application of human single chain Fv antibody fragments from a semi-synthetic phage antibody display library with designed CDR3 regions. J. Mol. Biol., 248, 97–105. [DOI] [PubMed] [Google Scholar]
- 6.de Kruif J., Terstappen,L., Boel,E. and Logtenberg,T. (1995) Rapid selection of cell subpopulation-specific human monoclonal antibodies from a synthetic phage antibody library. Proc. Natl Acad. Sci. USA, 92, 3938–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rakonjac J., Jovanovic,G. and Model,P. (1997) Filamentous phage infection-mediated gene expression: construction and propagation of the gIII deletion mutant helper phage R408d3. Gene, 198, 99–103. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths A.D., Malmqvist,M., Marks,J.D., Bye,J.M., Embleton,M.J., McCafferty,J., Baier,M., Holliger,K.P., Gorick,B.D., Hughes-Jones,N.C., Hoogenboom,H.R. and Winter,G. (1993) Human anti-self antibodies with high specificity from phage display libraries. EMBO J., 12, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duenas M. and Borrebaeck,C.A.K. (1995) Novel helper phage design: intergenic region affects the assembly of bacteriophages and the size of antibody libraries. FEMS Microbiol. Lett., 125, 317–322. [DOI] [PubMed] [Google Scholar]
- 10.Rondot S., Koch,J., Breitling,F. and Dubel,S. (2001) A helper phage to improve single-chain antibody presentation in phage display. Nat. Biotechnol., 19, 75–78. [DOI] [PubMed] [Google Scholar]
- 11.Baek H., Suk,K., Kim,Y. and Cha,S. (2002) An improved helper phage system for efficient isolation of specific antibody molecules in phage display. Nucleic Acids Res., 30, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kristensen P. and Winter,G. (1998) Proteolytic selection for protein folding using filamentous bacteriophages. Fold. Des., 3, 321–328. [DOI] [PubMed] [Google Scholar]
- 13.Carcamo J., Ravera,M.W., Brissette,R., Dedova,O., Beasley,J.R., Alam-Moghe,A., Wan,C., Blume,A. and Mandecki,W. (1998) Unexpected frameshifts from gene to expressed protein in a phage-displayed peptide library. Proc. Natl Acad. Sci. USA, 95, 11146–11151. [DOI] [PMC free article] [PubMed] [Google Scholar]