

Abstract
The positive-strand RNA genomes of caliciviruses are not capped, but are instead covalently linked at their 5′ ends to a viral protein called VPg. The lack of a cap structure typical of eukaryotic mRNA and absence of an internal ribosomal entry site suggest that VPg may function in translation initiation on calicivirus RNA. This hypothesis was tested by analyzing binding of Norwalk virus VPg to translation initiation factors. The eIF3d subunit of eIF3 was identified as a binding partner of VPg by yeast two-hybrid analysis. VPg bound to purified mammalian eIF3 and to eIF3 in mammalian cell lysates. To test the effects of the VPg– eIF3 interaction on translation, VPg was added to cell-free translation reactions programmed with either capped reporter RNA, an RNA containing an EMCV internal ribosomal entry site (IRES) or an RNA with a cricket paralysis virus IRES. VPg inhibited translation of all reporter RNAs in a dose-dependent manner. Together, the data suggest that VPg may play a role in initiating translation on calicivirus RNA through unique protein–protein interactions with the translation machinery.
Keywords: calicivirus/initiation factors/RNA virus/translation initiation/VPg
Introduction
Cap-dependent translation initiates through a complex set of protein–protein and RNA–protein interactions that begin with binding of initiation factor eIF4F to the 5′ terminal 7-methylguanosine (m7G) cap structure on the mRNA (reviewed in Hershey and Merrick, 2000). eIF4F is composed of subunits eIF4E, eIF4GI and eIF4A. Recruitment of the 43S pre-initiation complex, consisting of the Met-tRNAiMet–eIF2–GTP ternary complex and eIF3 bound to the 40S ribosomal subunit, to capped mRNA is mediated primarily through interactions between eIF4GI and eIF3 (Hentze, 1997; Gingras et al., 1999). This 48S complex scans to the AUG in the appropriate context to initiate protein synthesis. Translation initiation also can be cap- and end-independent. This mechanism is exemplified by internal ribosomal entry sites (IRES) of picornavirus RNA genomes (Pestova et al., 2001), although internal initiation on some cellular mRNAs also occurs (Carter et al., 2000; Hellen and Sarnow, 2001). IRES-mediated assembly of initiation complexes occurs through RNA–protein interactions, and protein synthesis begins at initiation codons downstream of the IRES. Thus, ribosome recruitment and translation initiation on IRES-containing mRNAs is independent of the 5′ terminus and an m7G cap.
Positive-strand RNA viruses in the families Picornaviridae, Potyviridae, Luteoviridae, Comoviridae and Caliciviridae lack m7G cap structures. Instead, their RNAs are covalently linked at the 5′ end to a small protein called VPg (viral protein genome linked) (reviewed in Sadowy et al., 2001). VPg of the picornaviruses does not function in initiation of translation on the viral RNA, as initiation is IRES driven (Jang et al., 1990). The potyviral VPg is multifunctional, and binds to eIF(iso)4E (Wittmann et al., 1997), but the consequences of this interaction with respect to translation initiation are not clear, as elements in the 5′ untranslated region (UTR) of the potyviral RNA direct cap-independent translation (Carrington and Freed, 1990). In contrast to these examples, there is significant suggestive evidence of a role for VPg in initiation of protein synthesis on calicivirus RNA. Removal of VPg from calicivirus RNA results in loss of infectivity (Burroughs and Brown, 1978), and dramatically reduces translation of feline calicivirus (FCV) RNA in vitro (Herbert et al., 1997). These observations suggested VPg was important in initiation of protein synthesis, perhaps functioning as a cap analogue, as proposed by Herbert and co-workers (Herbert et al., 1997). However, a putative VPg-directed mechanism must deviate from that displayed by m7G cap-dependent initiation, because addition of m7G cap analogue to translation reactions of VPg-linked FCV RNA had no effect on protein synthesis.
The Caliciviridae include the prototype human calicivirus Norwalk (NV), FCV, rabbit hemorrhagic disease virus (RHDV), vesicular exanthema of swine virus (VESV) and others (Clarke and Lambden, 2000). Calici virus RNA genomes are 7–8 kb in length, positive sense, polyadenylated and covalently linked to a 12–15 kDa VPg at the 5′ ends of both genomic and subgenomic RNAs (Clarke and Lambden, 2000). The genomes of NV, FCV and RHDV have been completely sequenced (Meyers et al., 1991; Carter et al., 1992; Jiang et al., 1993; Hardy, 1999). The first strong context initiation codon in each of the viral genomic RNAs is near the 5′ terminus, at nucleotide 11, 20 and 10, respectively. The N-terminal protein encoded in the first open reading frame is expressed in vitro and in infected cells (Liu et al., 1996; Wirblich et al., 1996; Clarke and Lambden, 2000), suggesting the absence of an IRES that would initiate translation at downstream AUGs. Such features of calicivirus genomes raise interesting questions regarding how translation on calicivirus RNA is initiated in the absence of a cap or an IRES.
NV and other human caliciviruses cause epidemic outbreaks of acute gastroenteritis (Fankhauser et al., 1998; Hardy, 1999). These viruses do not grow in cell culture, and thus the molecular mechanisms by which the proteins encoded in their genomes are expressed are not well understood. We formulated the hypothesis that initiation of protein synthesis on NV RNA proceeds by a unique mechanism that is dependent on interactions between VPg and the cellular translation machinery. In this study, we show that VPg directly binds to initiation factor eIF3. We also show that VPg inhibits cap-dependent and IRES-driven translation of reporter mRNAs in a dose-dependent manner. Based on data described herein, we propose VPg may function to recruit translation initiation complexes to calicivirus mRNA through direct protein–protein interactions with eIF3, and potentially other eIFs as well.
Results
NV VPg binds translation initiation factor eIF3
Removal of VPg from calicivirus RNA results in loss of infectivity of transfected RNA (Burroughs and Brown, 1978). However, capped synthetic RNA transcribed from a full-length cDNA clone of FCV is infectious (Sosnovtsev and Green, 1995), indicating that m7G can substitute for VPg. These observations suggest that VPg may function in initiation of translation on viral RNA. To test this hypothesis, we employed NV VPg as bait in a yeast two-hybrid screen of an epithelial cell cDNA library to identify binding partners of VPg that may suggest a role for this protein in translation of viral RNA. Approximately 1.3 × 106 yeast transformants were screened for cDNA clones encoding proteins that interacted with VPg. Eighty colonies were isolated, and the cDNA inserts of activation domain plasmids extracted from 22 colonies were sequenced. BLASTx searches with these cDNA sequences revealed similarities with thymosin β-10, RanBPM and predominately hypothetical proteins predicted from sequencing projects. One cDNA encoded the eIF3d subunit (p66) of translation initiation factor eIF3. The impetus to perform the two-hybrid screen with VPg was to identify candidate binding partners that may point to a role for VPg in translation initiation. Therefore, although eIF3d was recovered only once in our screen, we chose to pursue further characterization of the potential interaction between VPg and eIF3.
eIF3 is a 600 000 Da complex formed by 11 non-identical subunits (Hershey and Merrick, 2000). Among other functions, eIF3 stabilizes binding of the Met-tRNAiMet–eIF2–GTP ternary complex to the 40S ribosomal subunit to form 43S pre-initiation complexes (Hershey and Merrick, 2000). The eIF3d subunit is a 66 000 Da protein that is the major RNA binding subunit of eIF3 (Asano et al., 1997b). The 1.6 kb cDNA encoding eIF3d isolated in the yeast two-hybrid screen lacked only the N-terminal 10 amino acids when compared with the published sequence (Asano et al., 1997b). eIF3d in the activation domain plasmid was unable to activate reporter gene expression to allow growth on nutrient-deficient medium in the absence of VPg (data not shown). Likewise, VPg did not activate reporter gene expression in the absence of eIF3d, or in the presence of an irrelevant protein in the activation domain plasmid.
VPg binds purified mammalian eIF3
A glutathione S-transferase (GST) pull-down assay utilizing a GST–VPg fusion protein and eIF3 purified from HeLa cells was performed to test whether VPg interacted with mammalian eIF3. GST–VPg, but not GST, bound to purified eIF3 (Figure 1A). The subunits pulled down in the eIF3 complex were defined by a banding pattern which was similar to that described in characterization of the eIF3 antibody (Meyer et al., 1982). eIF3 subunits eIF3a, eIF3b and eIF3c were clearly identified, as compared with input purified eIF3. Detectable levels of eIF3f and eIF3j also were apparent on longer exposures. Interestingly, the eIF3j subunit displayed strongly in the input eIF3 was absent in the eluates of GST–VPg. Asano and co-workers have reported that eIF3j is loosely associated with the eIF3 complex (Asano et al., 1997a), yet whether VPg specifically displaces eIF3j, or any of the other subunits, from the holocomplex currently is not known. The interaction of VPg and eIF3d in yeast suggested the eIF3 complex may be bound to VPg through interaction with this subunit. The eIF3d antibody titer in the eIF3 polyclonal antibody is too low to allow detection of eIF3d in the pull-down experiments with cell lysates or with purified eIF3. Therefore, to test whether eIF3 bound to VPg through interactions with eIF3d, radiolabeled eIF3d translated in vitro was used in the pull-down assays. Figure 1B shows that eIF3d bound to GST–VPg, but not to GST, suggesting the eIF3 holocomplex may be bound at least partly through interactions between VPg and eIF3d.
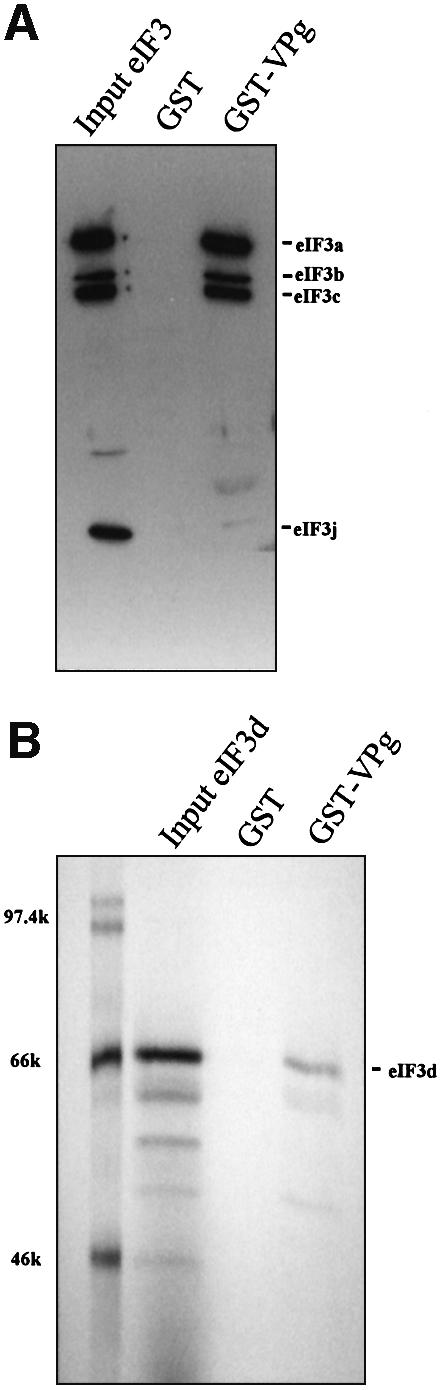
Fig. 1. VPg binds initiation factor eIF3. Pull-down assays were conducted with GST or GST–VPg immobilized on glutathione–Sepharose 4B beads. (A) Pull-down with purified eIF3. The input eIF3 lane represents 10% of the amount of protein used in the pull-down reaction, and the western blot was probed with anti-eIF3 polyclonal antibody. (B) Pull-down with 35S-labeled in vitro translated subunit eIF3d. The input eIF3d lane represents 25% of the amount used in the GST and GST–VPg pull-downs. Pull-down eluates were separated by 10% SDS–PAGE and exposed to film for autoradiography.
The ability of VPg to interact with eIF3 in its native form in mammalian cell lysates was examined by pull-down assay with extracts prepared from CaCo-2 cells. GST–VPg, but not GST, bound eIF3 from CaCo-2 cell lysates (Figure 2A), similar to the results obtained in the assay using purified eIF3. As before, subunits eIF3a, eIF3b and eIF3c were clearly detected. Pre-treating the cell lysates with S7 nuclease prior to the pull-down assay did not disrupt the interaction, indicating that binding of VPg to eIF3 was not mediated by RNA (Figure 2B). eIF3 also was not detected by pull-down with GST fused to an irrelevant protein (data not shown). We cloned and expressed VPg of the Snow Mountain strain of human calicivirus (SMV) to test whether the interaction with eIF3 was unique to VPg of the NV strain. VPg of these two calicivirus strains share 68% amino acid identity (Lochridge and Hardy, 2003). SMV VPg also bound eIF3 in a GST pull-down assay performed with CaCo-2 cell lysates (Figure 2C).

Fig. 2. VPg binds eIF3 present in cell lysates. Pull-down assays were conducted with GST, GST–VPg or GST–VPg mutant constructs immobilized on glutathione–Sepharose 4B beads and incubated with CaCo-2 cell extracts. (A) CaCo-2 cell lysates. (B) S7-treated CaCo-2 lysates. (C) GST–SMV-VPg immobilized to glutathione–Sepharose beads. (D) VPg deletion mutants. Western blots were probed with anti-eIF3 polyclonal antibody. Asterisks indicate a protein likely to be eIF4GI.
We constructed two GST fusion proteins that contained either the N-terminal domain (amino acids 1–69, GST–VPg1–69) or the C-terminal domain (amino acids 70–138, GST–VPg70–138) of VPg, in order to identify regions that may be important in mediating the interaction with eIF3. Figure 2D shows that GST–VPg70–138 strongly bound eIF3 in the pull-down assay. The amount of eIF3 recovered in the pull-down eluate was similar to that observed with the full-length VPg. In contrast, GST–VPg1–69 showed only slight reactivity with eIF3. These data suggest the eIF3 binding domain of VPg resides primarily in the C-terminal half of the protein, although some extension of the binding domain towards the N-terminus is possible. Taken together, the data presented above show that VPg binds specifically to eIF3, binding is mediated primarily by the C-terminal half of the protein and the interaction is not dependent on RNA or other protein–protein interactions.
Translation initiation factors present in VPg pull-down eluates
In cap-dependent initiation, the 43S complex is recruited to mRNA through interactions between eIF3 and the eIF4GI component of the cap-binding complex eIF4F (Hentze, 1997; Gingras et al., 1999). A high molecular weight protein was detected in all of the GST–VPg pull-down assays in which CaCo-2 cell lysates were used (Figure 2, asterisks). We could not relate this protein to the standard eIF3 banding pattern, and it was never detected in pull-down assays that employed purified eIF3. Previous characterization of the anti-eIF3 serum demonstrated reactivity to eIF4G (p220) (Etchison et al., 1982). Based on the estimated molecular weight of the protein, characteristics of the eIF3 antibody and known eIF3 binding proteins, we tested whether this unknown protein might be eIF4GI. GST–VPg pull-down eluates were probed in a western immunoblot with an anti-eIF4GI antibody. The data shown in Figure 3A indicate that eIF4GI was present in eluates of GST–VPg, but not in eluates of GST, suggesting that the unknown band seen in the blots with the anti-eIF3 antiserum may be eIF4GI. Similar analyses of eluates from GST–VPg deletion constructs showed that eIF4GI was strongly present in eluates from GST– VPg70–138, and significantly less so in the eluates of the N-terminal VPg domain GST–VPg1–69. The excess of eIF4GI detected in eluates from GST–VPg70–138 may reflect the stronger interaction of this construct with eIF3. It should be noted that the purified eIF3 preparation also was probed for the presence of contaminating eIF4GI, and the result was negative. We currently do not know whether there is a direct interaction between VPg and eIF4GI, or whether eIF4GI was pulled down through its association with eIF3 in cell lysates, as yeast two-hybrid assays to address this question yielded inconclusive results (data not shown).
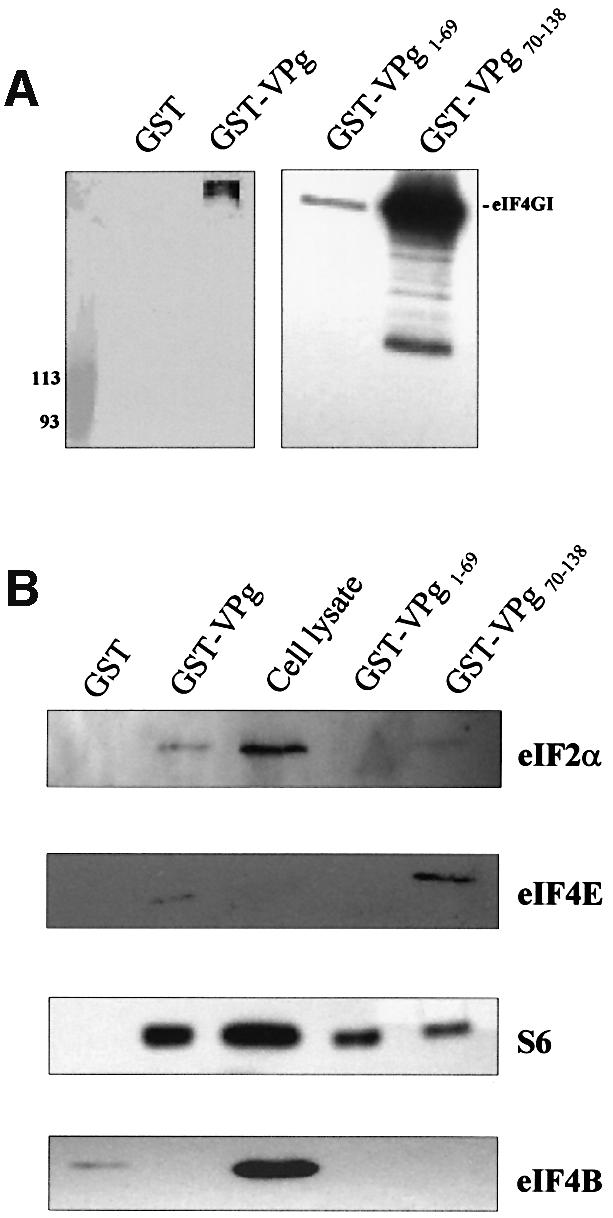
Fig. 3. VPg interactions with eIF4GI and other initiation factors. Pull-down assays were conducted with GST, GST–VPg or GST–VPg mutant constructs immobilized on glutathione–Sepharose 4B beads and incubated with CaCo-2 cell extracts. Western blots were probed with (A) anti-eIF4GI, or (B) anti-eIF2α, anti-eIF4E, anti-S6 or anti-eIF4B.
Since eIF4GI was strongly present in the pull-down eluates of full-length VPg and its C-terminal domain, we probed for additional components of translation initiation complexes including eIF2α, eIF4B, eIF4E and the S6 protein of the 40S subunit (Figure 3B). Small amounts of eIF2α were detected in eluates from full-length VPg and GST–VPg70–138. Similar results were observed for eIF4E. The S6 protein was detected in eluates from all three constructs, consistent with an affinity of VPg for 40S (unpublished data). eIF4B was not detected in any of the eluates, with the exception of slight reactivity with GST alone. Taken together, the binding data suggest that several translation initiation factors are present in VPg-interacting complexes. The extent to which these interactions are direct with VPg, or mediated through eIF3, is not yet known.
VPg inhibits translation of reporter RNAs in rabbit reticulocyte lysate
NV and other human caliciviruses do not grow in cell culture, making functional studies difficult. Therefore, to begin to understand the functional consequences of the interaction between VPg and eIF3, we investigated the effects of the presence of VPg on cap-dependent translation in vitro. A capped reporter RNA (m7G-Luc) was translated in rabbit reticulocyte lysate (RRL) in the presence of increasing concentrations of GST–VPg or GST. Translation of m7G-Luc RNA was inhibited in the presence of GST–VPg, but not GST, in a dose-dependent manner (Figure 4A).

Fig. 4. VPg inhibits translation of reporter RNAs. (A) m7G-Luc RNA, (B) IRES-Luc RNA, and (C) CrPV IGR-IRES RNA were translated in Flexi RRL without or with RNA (lanes 1 and 2) and with increasing concentrations of GST–VPg (lanes 3, 4 and 5 = 12, 24 and 48 pmol, respectively), or increasing concentrations of GST (lanes 6, 7 and 8 = 19, 38 and 76 pmol, respectively). 35S-labeled proteins were separated by SDS–10% PAGE and visualized by autoradiography. (D) Average OD of luciferase synthesized in the presence of GST–VPg in the translation reactions shown in (A–C). m7G-Luc RNA (squares), IRES-Luc (triangles) and CrPV IGR-IRES (crosses). A control reaction where GST was used as the inhibitor in the IRES-Luc translation reactions is shown as a representative (diamonds). Data are normalized to 1.
Pestova and co-workers reported that canonical initiation factors were utilized in both cap-dependent initiation and translation initiation mediated by an IRES (Pestova et al., 1996). The role of the eIF4F complex in translation of an EMCV IRES-containing mRNA was different from its role in m7G-dependent translation, as assembly of functional initiation complexes did not require eIF4E binding to a 5′ terminal cap. We reasoned that elucidating potential mechanisms of translation inhibition would aid in understanding the function of VPg, and thus asked whether VPg could inhibit translation of an mRNA under the control of an EMCV IRES (IRES-Luc). Increasing concentrations of GST–VPg or GST were added to IRES-Luc mRNA translation reactions. GST–VPg eliminated translation of the IRES-Luc RNA, as it did for the m7G-Luc RNA (Figure 4B), also in a dose-dependent manner.
The RNA genome of cricket paralysis virus (CrPV) is a functionally dicistronic mRNA (Wilson et al., 2000b). Translation of the virus structural proteins encoded in the second open reading frame is mediated by an intergenic region (IGR) that functions as an IRES. The IGR-IRES of CrPV is unique in that 80S ribosomes can assemble in the absence of the canonical eIFs (Wilson et al., 2000a). We tested whether VPg could inhibit translation of the CrPV IGR-IRES that does not require eIF3. As in the previous experiments, GST–VPg inhibited translation from this RNA in a dose-dependent manner, whereas GST alone had no effect (Figure 4C). Complete inhibition required approximately twice as much VPg as did the EMCV IRES, and although reproducible, the physiological relevance of this difference currently is not known. One interpretation of these data is that the interaction between VPg and eIF3 does not play a role in inhibiting translation. An extension of this prediction is that VPg inhibits translation of all three RNAs by the same mechanism, and the least common denominator for which we have shown an interaction with VPg is the 40S subunit. Although canonical eIFs are not required for assembly of 80S ribosomes on the CrPV IGR-IRES, these factors are still present in the lysates used to translate the RNA, and the effects of these endogenous eIFs on translation from the IGR-IRES are not known. An alternative explanation is that the interactions between VPg and eIF3, and VPg and 40S, are not mutually exclusive, and that binding of VPg to either eIF3 or 40S subunits (or both) could inhibit translation of any of the reporter RNAs analyzed here. The mechanism of inhibition then would be dependent on the specific factor requirements of each RNA. The ability of VPg to inhibit translation of capped mRNA and mRNAs that contain IRES sequences with different eIF requirements suggest that VPg may be inhibiting translation through protein interactions common to cap-dependent and internal initiation.
Discussion
Mechanisms of eukaryotic ribosome recruitment to mRNA are distinguishable by their requirements for initiation factors and 5′ end dependence (Jackson, 2000; Pestova et al., 2001). The mechanism utilized by the majority of cellular and viral mRNAs is 5′ end and m7G cap dependent, and the 43S pre-initiation complex is recruited to the mRNA through its interactions with the eIF4GI component of the cap-binding complex eIF4F. 5′ terminus-independent internal initiation driven by an IRES, typified by those present in some positive strand RNA virus genomes, has different initiation factor requirements depending on the IRES analyzed (Pestova et al., 1996, 1998; Jackson, 2000). We put forth the hypothesis that translation initiation on calicivirus RNA may proceed by a unique mechanism that involves protein–protein interactions between VPg and the cellular translation machinery. In this study, we have shown that recombinant VPg binds to purified eIF3, and to native eIF3 in mammalian cell lysates; the C-terminal half of VPg is the primary mediator of the interaction; and eIF4GI and other translation initiation factors are present in VPg-interacting complexes. These data, interpreted in the context of those previously reported for translation of the FCV genomic RNA, suggest that VPg may function as a cap analogue through protein–protein interactions distinct from those thus far described as important in m7G cap-dependent or IRES-mediated translation initiation. Herbert and co-workers have postulated a role for VPg in direct ribosome recruitment (Herbert et al., 1997), and our data so far support this model. Future studies will address the current hypothesis that the interactions described herein function as an end-dependent mechanism of ribosome recruitment to calicivirus RNA, mediated by interactions between a 5′ terminus-linked viral protein VPg and host cell translation initiation complexes.
The organization of calicivirus genomic and subgenomic RNA, specifically a 5′-genome-linked protein and short 5′ UTRs, led to the hypothesis that VPg may function in translation initiation. Generally, short 5′ UTRs confer an inefficiency of translation on the mRNAs in which they are present (Kozak, 1987, 1991a,b, 1994). The 5′ UTRs of all the calicivirus genomes sequenced to date are less than 20 nucleotides long. In addition, an equally short 5′ UTR is present on the subgenomic RNA encoding the calicivirus capsid protein (Clarke and Lambden, 2000). It is possible then, that a direct interaction between VPg and eIF3 bound to 40S subunits positions the ribosome precisely at the initiating AUG, and thus these viral RNAs would not be subject to the diminutive effects of a short 5′ leader sequence on translational efficiency. Such a direct recruitment has been proposed for the HCV IRES, where 40S subunits and eIF3 directly bind the IRES in the absence of other initiation factors (Pestova et al., 1998). In this study, VPg bound directly to purified eIF3. Furthermore, the ribosomal subunit protein S6 was detected in pull-down eluates of full-length VPg, and both the C- and N-terminal VPg domains. It may be that binding to 40S subunits is mediated through the N-terminal domain of VPg. It is tempting to draw a parallel with the HCV IRES with respect to these interactions functioning to position the ribosome at or near the initiator codon. Still, the mechanisms and initiation factor requirements must be different between the two virus systems, as the HCV IRES–eIF3– 40S interaction is RNA–protein, whereas the VPg interactions are protein–protein.
The presence of additional components of translation initiation complexes, such as eIF4GI, eIF2α and eIF4E, when binding assays were performed with cell extracts instead of purified eIF3 suggests one alternative mechanism that involves VPg binding to multiple initiation factors (including 40S subunits) to assemble an initiation complex that more closely resembles that recruited by an m7G cap. Asano and co-workers reported the presence of a multifactor intermediate complex composed of eIF3, eIF1, eIF5 and eIF2–GTP–Met-tRNAiMet in cell extracts that exist free of 40S subunits (Asano et al., 2000). It is conceivable that the presence of one or more of the additional eIFs detected by immunoblot in the pull-down assays reflects their associations with eIF3 instead of direct interactions with VPg. Still, detection of components of eIF4F, including eIF4GI and eIF4E, point to the possibility of a complex set of interactions that finally would result in assembly of a functional initiation complex on VPg-linked RNA.
The effects of VPg on translation of reporter RNA in vitro were tested to investigate the functional consequences of the eIF3 interactions. VPg inhibited translation of a capped reporter RNA and a reporter RNA containing an EMCV IRES. We considered the possibility that the mechanism of inhibition of translation of these two RNAs may involve disruption of the interaction between eIF4GI and eIF3. eIF4GI was detected in GST–VPg pull-down eluates, but we were unable to show, or exclude, a direct interaction through a two-hybrid analysis of eIF4GI and VPg. These two proteins may bind the same sites on eIF3, leading to an inhibition of translation in the presence of VPg. The binding site(s) of eIF4GI on eIF3 is not known, but if the mechanism of translation inhibition by VPg includes preventing the eIF4GI–eIF3 interaction, the eIF3d subunit of eIF3 may play a role. VPg may sequester eIF3, and potentially other initiation factors bound to eIF3, making them unavailable to form functional initiation complexes. Obvious candidates for inhibition targets include those required for cap-dependent translation, such as components of eIF4F. An alternative and equally plausible hypothesis is provided by the ability of VPg to inhibit translation mediated by the CrPV IGR-IRES, which can assemble 80S ribosomes from purified 40S and 60S subunits in the absence of any other eIF (Wilson et al., 2000a). These data suggest that inhibition of translation from this mRNA (and perhaps the others) by VPg is mediated through the 40S subunit. However, the presence of endogenous eIFs in the reticulocyte lysates used for these experiments complicates this straightforward interpretation. Thus the mechanism by which VPg is able to inhibit translation of reporter RNAs that have different factor requirements remains unclear at this point. Studies that add purified initiation factors to VPg-inhibited translation reactions to rescue protein synthesis are ongoing in order to define the inhibitory mechanism reported here.
VPg of positive-strand RNA viruses in families other than the Caliciviridae has been ascribed a number of functions (Sadowy et al., 2001). The picornavirus VPg is comparatively small (2–4 kDa), functions in protein-primed RNA synthesis (Paul et al., 1998) and is not necessary for infectivity of transfected viral RNA. The function of VPg linked to potyvirus genomes appears more complex. Potyviral VPg is larger than the calicivirus VPg (24–26 kDa). In addition to its role in RNA synthesis (Hong et al., 1995), VPg has been found in the nucleus (Restrepo et al., 1990), functions in long-distance cell–cell movement (Schaad et al., 1997) and binds eIF(iso)4E (Wittmann et al., 1997; Leonard et al., 2000). The role of potyvirus VPg binding to eIF(iso)4E with respect to protein synthesis is not known, yet a recent study has confirmed the importance of this interaction by showing that mutant Arabidopsis thaliana resistant to potyviral infection do not express eIF(iso)4E (Lellis et al., 2002). Whether potyviral VPg functions in translation initiation remains to be established, as it is clear that sequences in the 5′ UTR of potyviral RNA direct cap-independent translation enhancement (Carrington and Freed, 1990). Thus far, there is no evidence that NV RNA contains an IRES to direct cap- or 5′ terminus-independent translation. Assays to address directly if and how the NV VPg may function in ribosome recruitment are challenging. This virus does not grow in cell culture and, consequently, there is no source of NV VPg-linked RNA to perform recruitment studies. Efforts to establish a system with a cultivatable calicivirus from which native VPg-linked RNA can be obtained are in progress. Such a system will allow us to address directly translation initiation complex recruitment through functional assays.
Materials and methods
Yeast plasmids and transformations
Construction of yeast plasmids and transformations. Matchmaker yeast two-hybrid vectors were purchased from Clontech. The activation domain vector pGADT7 carries the LEU2 nutritional marker and the DNA-binding domain vector pGBKT7 carries the TRP1 nutritional marker for selection in yeast. The oligo(dT)-primed MA104 cell cDNA library was cloned into pGADT7 and is described elsewhere (Graff et al., 2002). The sequence encoding VPg was amplified from a full-length NV cDNA clone (Hardy et al., 2002) by PCR with two primers, VPg EcoRI(+) 5′-ccggaattcggaaagaacaaaggcaagacc-3′ and VPg-BamHI(–) 5′-cgcggatccttcaaaattgatcttttcattataat-3′. Restriction enzyme sites are underlined. Amplification conditions consisted of 30 cycles of 94°C for 1 min, 50°C for 30 s and 72°C for 30 s. The resulting 400 bp fragment was cloned into pGBKT7 to generate pGBK-VPg. Binding domain plasmids were transformed into AH109 yeast cells by the lithium acetate/PEG method as described by Gietz and Woods (1998).
Two-hybrid interactions were scored by the ability of yeast to grow on SC –L–W medium and to activate reporter genes HIS3, ADE2 and MEL1. Activation of reporter gene expression was indicated by growth in the absence of histidine (H) and adenine (A), and by the ability to metabolize the chromogenic substrate X-α-gal (ICN). Approximately 6 × 109 pGBK-VPg yeast cells were transformed with 120 µg of the MA104 cDNA library by the lithium acetate/PEG procedure. Transformations were plated on SC –L–W–H–A medium and cultured for 2–4 days at 30°C. Colonies then were re-streaked on the same selective medium with the addition of 400 µg X-α-gal.
Isolation of plasmid DNA from yeast and identification of cDNA. Plasmid DNA was isolated from yeast as described previously (Gietz and Woods, 1998). The activation domain plasmids with cDNAs encoding potential interactors were recovered by electroporation into DH10B cells and culture on LB agar containing 50 µg/ml ampicillin. Small-scale plasmid purifications were performed with Eppendorf Perfect Prep. DNA was sequenced on an ABI 310 Genetic Analyzer with BigDye Terminator® chemistry.
Expression and purification of GST–VPg and GST–VPg deletion mutants
To construct vectors expressing GST fusion proteins, sequences encoding NV VPg were amplified by PCR from the full-length NV cDNA clone with two primers, VPg-EcoRI(+) and VPg-XhoI(–) 5′-ccgctcgagttcaaaattgatcttttcattataat-3′ under the same conditions as described above. Sequences encoding VPg of SMV were amplified by PCR from a SMV cDNA clone (Lochridge and Hardy, 2003) with two primers SMV-VPg (+) 5′-ccggaattcagtgacatcacgcctgaaggc-3′ and SMV-VPg (–) 5′-acgcgtcgacctcaaaactgagtttctcatt-3′. Amplification conditions were 25 cycles of 94°C for 15 s, 55°C for 15 s and 72°C for 1 min. The N-terminus of NV VPg (amino acids 1–69) was amplified with VPg-EcoRI(+) and VPg-XhoI(–)N term 5′-ccgctcgagaccatcaccacctgcctgtacctc-3′ to generate GST–VPg1–69. The C-terminus of NV VPg (amino acids 70–138) was amplified with VPg-EcoRI(+)C term 5′-ccggaattcggcataggagaaactgaaatgg-3′ and VPg-XhoI(–) to generate GST–VPg70–138. Amplification conditions were 30 cycles of 94°C for 1 min, 55°C for 1 min and 72°C for 2 min, followed by a 5 min extension at 72°C.
Each PCR fragment was cloned into pGEX-4T-1 (Amersham Pharmacia) and expressed as a GST fusion protein in BL21(DE3) cells. Bacteria were cultured to an OD600 of 0.6 and recombinant protein expression was induced with 1 mM IPTG for 2 h. Bacteria were collected by centrifugation and suspended in buffer containing 50 mM Tris–HCl pH 8, 2 mM EDTA, 1% Triton X-100 and 100 µg/ml lysozyme. This suspension was incubated for 15 min at 30°C and then sonicated three times on ice for 10-s pulses. Soluble and insoluble proteins were separated by centrifugation for 10 min at 12 000 g. The supernatant containing soluble protein was retained for the purification of GST–VPg. GST was expressed as a control and purified under the same conditions described below.
A 50% slurry of glutathione–Sepharose 4B beads (Amersham Pharmacia) was prepared following instructions provided by the manufacturer. One hundred microliters of prepared beads were mixed with 3 ml soluble bacterial cell lysate from either 50, 15 or 3 ml GST–VPg-induced culture (volume dependent on the experiment), and rocked for 10 min at room temperature. Beads were washed twice with cold phosphate buffered saline (PBS), and collected by centrifugation for 5 min at 500 g. GST–VPg was eluted from the beads by three 10 min room temperature incubations in elution buffer containing 10 mM reduced glutathione/50 mM Tris–HCl pH 8. Beads were removed by centrifugation for 5 min at 500 g. The eluates were combined, and eluted proteins were evaluated for purity by SDS–PAGE. GST–VPg and GST were quantified with the Bio-Rad protein assay with bovine serum albumin as the assay standard.
GST pull-down assay
GST–VPg pull-down assays employed purified eIF3, in vitro translated eIF3d and native eIF3 in mammalian cell (CaCo-2) lysates. Purified eIF3 was obtained from HeLa cells by previously described methods (Falvey and Staehelin, 1970; Meyer et al., 1982). 35S-labeled eIF3d was translated in vitro as described below for luciferase RNA. Translation reactions were treated with 0.4 µg/µl RNAse A for 30 min at 37°C before use in pull-down assays. CaCo-2 colon adenocarcinoma cells were grown to confluency in 60 mm dishes in MEM (Invitrogen) containing 15% fetal bovine serum (Atlanta Biologicals). CaCo-2 cells were chosen for these experiments because they are intestinal in origin and are relevant to our enteric virus model system. The cells were washed twice with PBS, scraped from the plate and collected in 100 µl of lysis buffer containing 50 mM Tris–HCl pH 8, 15 mM NaCl, 140 mM KCl, 2% NP-40, 6 µg/ml aprotinin, 6 µg/ml pepstatin A and 6 µg/ml leupeptin. The lysate was brought to a final volume of 1 ml with wash buffer (20 mM Tris–HCl pH 7.5, 15 mM NaCl, 140 mM KCl and 0.1% NP-40), mixed with the prepared glutathione–Sepharose 4B beads bound to GST–VPg or GST alone, or the indicated mutant, and rotated end-over-end for 4 h at 4°C. In some experiments, the lysates were pretreated with 75 U/ml S7 micrococcal nuclease (Roche Biochemicals) in the presence of 1 mM CaCl2 for 15 min at 20°C. Nuclease digestions were terminated by addition of EGTA to a final concentration of 2 mM. The beads were collected by centrifugation for 5 min at 500 g at 4°C and washed three times with wash buffer. GST–VPg and interactors were eluted from the beads by three 10 min incubations in elution buffer as before.
Pull-down eluates were electrophoresed on SDS-10% or 8% polyacrylamide gels, and proteins were transferred to nitrocellulose for western immunoblotting. The membranes were blocked for 1 h with 10% BLOTTO (10% non-fat dry milk in PBS), and then incubated overnight at room temperature with goat anti-rabbit eIF3 (Meyer et al., 1982) diluted 1:2000 in 0.5% BLOTTO. The reactivity of the anti-eIF3 antibody has been described previously (Brown-Luedi et al., 1982; Meyer et al., 1982). This antiserum recognizes all of the subunits of eIF3, in addition to eIF4GI (Etchison et al., 1982). The titer of antibody to the eIF3d subunit is low. Additional antibodies employed in western blots include rabbit anti-eIF4GI diluted 1:2000, rabbit anti-eIF4B diluted 1:1000, rabbit anti-eIF2α diluted 1:1000 and anti-S6 diluted 1:1000. All commercial antibodies were purchased from Cell Signaling Technologies. The membranes were washed three times in 0.5% BLOTTO, then incubated for 2 h at room temperature with horseradish peroxidase-conjugated rabbit anti-goat, or goat anti-rabbit, IgG diluted 1:3000 (Jackson Immunoresearch Laboratories) in 0.5% BLOTTO. Proteins that bound antibodies were detected by enhanced chemiluminescence (Amersham Pharmacia).
In vitro translations in RRLs
The Luciferase T7 DNA (Promega) served as template for synthesis of capped mRNA (m7G-Luc). The luciferase gene was cloned into the pCITE4a+ vector (Novagen) downstream of the EMCV IRES to generate a template for synthesis of IRES-containing mRNA (IRES-Luc). The plasmid pEJ4 that contains the IGR-IRES of CrPV upstream of the firefly luciferase reporter was kindly provided by Dr P.Sarnow (Stanford University Medical School). Templates were linearized with XmnI (T7 DNA), PvuII (pCITE-Luc) or BamHI (pEJ4) for 2 h at 37°C, phenol/chloroform extracted and precipitated with 450 mM NH4OAc and ethanol. m7G-Luc mRNA was transcribed with the Ambion mMessage mMachine system, and IRES-Luc mRNA was transcribed with the Ambion T7 Megascript system following the provided protocols. Plasmid pETp66N was the template for synthesis of eIF3d RNA (Asano et al., 1997b). Linearized DNA was transcribed using the Ambion T7 Megascript system following provided protocols.
RNA was heated to 65°C for 3 min and quenched on ice before use. Translation reactions were performed in Flexi-RRL (Promega) as recommended by the manufacturer. Two hundred and fifty nanograms of reporter RNA were translated in 25 µl reactions containing 16.5 µl of lysate, 4 µCi [35S]methionine (1000 Ci/mmol; Amersham Pharmacia), 20 µM amino acid solution minus methionine, 70 mM KCl, and GST or GST–VPg as inhibitor. Translation reactions were incubated for 1 h at 30°C. Translation products were resolved by SDS–PAGE and visualized by autoradiography. Luciferase bands were quantified by densitometric analysis on a Bio-Rad Molecular Imager FX. Translation inhibition experiments were performed a minimum of three times.
Acknowledgments
Acknowledgements
We extend appreciation to Michael White and Jay Radke for help and advice on cDNA library construction, and to Peter Sarnow for sharing the CrPV IGR-IRES plasmid. This work was supported by Public Health Service grant AI-43450 to M.E.H. and the MAES (2002-43), and GM-22135 to J.W.B.H. K.F.D. is a recipient of a USDA National Needs Fellowship.
References
- Asano K., Kinzy,T.G., Merrick,W.C. and Hershey,J.W. (1997a) Conservation and diversity of eukaryotic translation initiation factor eIF3. J. Biol. Chem., 272, 1101–1109. [DOI] [PubMed] [Google Scholar]
- Asano K., Vornlocher,H.P., Richter-Cook,N.J., Merrick,W.C., Hinnebusch,A.G. and Hershey,J.W. (1997b) Structure of cDNAs encoding human eukaryotic initiation factor 3 subunits. Possible roles in RNA binding and macromolecular assembly. J. Biol. Chem., 272, 27042–27052. [DOI] [PubMed] [Google Scholar]
- Asano K., Clayton,J., Shalev,A. and Hinnebusch,A.G. (2000) A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5 and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev., 14, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Luedi M.L., Meyer,L.J., Milburn,S.C., Yau,P.M., Corbett,S. and Hershey,J.W. (1982) Protein synthesis initiation factors from human HeLa cells and rabbit reticulocytes are similar: comparison of protein structure, activities and immunochemical properties. Biochemistry, 21, 4202–4206. [DOI] [PubMed] [Google Scholar]
- Burroughs J.N. and Brown,F. (1978) Presence of a covalently linked protein on calicivirus RNA. J. Gen. Virol., 41, 443–446. [DOI] [PubMed] [Google Scholar]
- Carrington J.C. and Freed,D.D. (1990) Cap-independent enhancement of translation by a plant potyvirus 5′ nontranslated region. J. Virol., 64, 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter M.J., Milton,I.D., Meanger,J., Bennett,M., Gaskell,R.M. and Turner,P.C. (1992) The complete nucleotide sequence of a feline calicivirus. Virology, 190, 443–448. [DOI] [PubMed] [Google Scholar]
- Carter M.S., Kuhn,K.M. and Sarnow,P. (2000) Cellular internal ribosome entry site elements and use of cDNA microarrays in their investigation. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 615–635.
- Clarke I.N. and Lambden,P.R. (2000) Organization and expression of calicivirus genes. J. Infect. Dis., 181, S309–S316. [DOI] [PubMed] [Google Scholar]
- Etchison D., Milburn,S.C., Edery,I., Sonenberg,N. and Hershey,J.W. (1982) Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem., 257, 14806–14810. [PubMed] [Google Scholar]
- Falvey A.K. and Staehelin,T. (1970) Structure and function of mammalian ribosomes. I. Isolation and characterization of active liver ribosomal subunits. J. Mol. Biol., 53, 1–19. [DOI] [PubMed] [Google Scholar]
- Fankhauser R.L., Noel,J.S., Monroe,S.S., Ando,T. and Glass,R.I. (1998) Molecular epidemiology of ‘Norwalk-like viruses’ in outbreaks of gastroenteritis in the United States. J. Infect. Dis., 178, 1571–1578. [DOI] [PubMed] [Google Scholar]
- Gietz R.D. and Woods,R.A. (1998) Transformation of yeast by the lithium acetate/single-stranded carrier DNA/PEG method. In Brown,A. and Tuite,M. (eds), Methods in Microbiology. Vol. 26. Academic Press, San Diego, CA, pp. 53–66.
- Gingras A.C., Raught,B. and Sonenberg,N. (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem., 68, 913–963. [DOI] [PubMed] [Google Scholar]
- Graff J.W., Mitzel,D.N., Weisend,C.M., Flenniken,M.F. and Hardy,M.E. (2002) Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J. Virol., 76, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy M.E. (1999) Norwalk and ‘Norwalk-like viruses’ in epidemic gastroenteritis. Clin. Lab. Med., 19, 675–690. [PubMed] [Google Scholar]
- Hardy M.E., Crone,T.J., Brower,J.E. and Ettayebi,K. (2002) Substrate specificity of the Norwalk virus 3C-like proteinase. Virus Res., 89, 29–39. [DOI] [PubMed] [Google Scholar]
- Hellen C.U. and Sarnow,P. (2001) Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev., 15, 1593–1612. [DOI] [PubMed] [Google Scholar]
- Hentze M.W. (1997) eIF4G: a multipurpose ribosome adapter? Science, 275, 500–501. [DOI] [PubMed] [Google Scholar]
- Herbert T.P., Brierley,I. and Brown,T.D. (1997) Identification of a protein linked to the genomic and subgenomic mRNAs of feline calicivirus and its role in translation. J. Gen. Virol., 78, 1033–1040. [DOI] [PubMed] [Google Scholar]
- Hershey J.W.B. and Merrick,W.C. (2000) The pathway and mechanism of initiation of protein synthesis. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 33–88.
- Hong Y., Levay,K., Murphy,J.F., Klein,P.G., Shaw,J.G. and Hunt,A.G. (1995) A potyvirus polymerase interacts with the viral coat protein and VPg in yeast cells. Virology, 214, 159–166. [DOI] [PubMed] [Google Scholar]
- Jackson R.J. (2000) A comparative view of initiation site selection mechanisms. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 127–183.
- Jang S.K., Pestova,T.V., Hellen,C.U., Witherell,G.W. and Wimmer,E. (1990) Cap-independent translation of picornavirus RNAs: structure and function of the internal ribosomal entry site. Enzyme, 44, 292–309. [DOI] [PubMed] [Google Scholar]
- Jiang X., Wang,M., Wang,K. and Estes,M.K. (1993) Sequence and genomic organization of Norwalk virus. Virology, 195, 51–61. [DOI] [PubMed] [Google Scholar]
- Kozak M. (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res., 15, 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (1991a) A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expr., 1, 111–115. [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (1991b) Effects of long 5′ leader sequences on initiation by eukaryotic ribosomes in vitro. Gene Expr., 1, 117–125. [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (1994) Features in the 5′ non-coding sequences of rabbit alpha and beta-globin mRNAs that affect translational efficiency. J. Mol. Biol., 235, 95–110. [DOI] [PubMed] [Google Scholar]
- Lellis A.D., Kasschau,K.D., Whitham,S.A. and Carrington,J.C. (2002) Loss-of-susceptibility mutants of Arabidopsis thaliana reveal an essential role for eIF(iso)4E during potyvirus infection. Curr. Biol., 12, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Leonard S., Plante,D., Wittmann,S., Daigneault,N., Fortin,M.G. and Laliberte,J.F. (2000) Complex formation between potyvirus VPg and translation eukaryotic initiation factor 4E correlates with virus infectivity. J. Virol., 74, 7730–7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Clarke,I.N. and Lambden,P.R. (1996) Polyprotein processing in Southampton virus: identification of 3C-like protease cleavage sites by in vitro mutagenesis. J. Virol., 70, 2605–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochridge V.P. and Hardy,M.E. (2003) Snow mountain virus genome sequence and virus-like particle assembly. Virus Genes, 26, 71–82. [DOI] [PubMed] [Google Scholar]
- Meyer L.J., Milburn,S.C. and Hershey,J.W. (1982) Immunochemical characterization of mammalian protein synthesis initiation factors. Biochemistry, 21, 4206–4212. [DOI] [PubMed] [Google Scholar]
- Meyers G., Wirblich,C. and Thiel,H.J. (1991) Rabbit hemorrhagic disease virus—molecular cloning and nucleotide sequencing of a calicivirus genome. Virology, 184, 664–676. [DOI] [PubMed] [Google Scholar]
- Paul A.V., van Boom,J.H., Filippov,D. and Wimmer,E. (1998) Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature, 393, 280–284. [DOI] [PubMed] [Google Scholar]
- Pestova T.V., Hellen,C.U. and Shatsky,I.N. (1996) Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol., 16, 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova T.V., Shatsky,I.N., Fletcher,S.P., Jackson,R.J. and Hellen,C.U. (1998) A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev., 12, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova T.V., Kolupaeva,V.G., Lomakin,I.B., Pilipenko,E.V., Shatsky, I.N., Agol,V.I. and Hellen,C.U. (2001) Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl Acad. Sci. USA, 98, 7029–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo M.A., Freed,D.D. and Carrington,J.C. (1990) Nuclear transport of plant potyviral proteins. Plant Cell, 2, 987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowy E., Milner,M. and Haenni,A.L. (2001) Proteins attached to viral genomes are multifunctional. Adv. Virus Res., 57, 185–262. [DOI] [PubMed] [Google Scholar]
- Schaad M.C., Lellis,A.D. and Carrington,J.C. (1997) VPg of tobacco etch potyvirus is a host genotype-specific determinant for long-distance movement. J. Virol., 71, 8624–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosnovtsev S. and Green,K.Y. (1995) RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VpG for infectivity. Virology, 210, 383–390. [DOI] [PubMed] [Google Scholar]
- Wilson J.E., Pestova,T.V., Hellen,C.U. and Sarnow,P. (2000a) Initiation of protein synthesis from the A site of the ribosome. Cell, 102, 511–520. [DOI] [PubMed] [Google Scholar]
- Wilson J.E., Powell,M.J., Hoover,S.E. and Sarnow,P. (2000b) Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol. Cell. Biol., 20, 4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirblich C., Thiel,H.J. and Meyers,G. (1996) Genetic map of the calicivirus rabbit hemorrhagic disease virus as deduced from in vitro translation studies. J. Virol., 70, 7974–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann S., Chatel,H., Fortin,M.G. and Laliberte,J.F. (1997) Interaction of the viral protein genome linked of turnip mosaic potyvirus with the translational eukaryotic initiation factor (iso) 4E of Arabidopsis thaliana using the yeast two-hybrid system. Virology, 234, 84–92. [DOI] [PubMed] [Google Scholar]