

Abstract
Mucopolysaccharidosis VII (MPS VII, Sly syndrome) is an autosomal recessive lysosomal storage disease caused by β-glucuronidase (GUS) deficiency. A naturally occurring mouse model of that disease has been very useful for studying experimental approaches to therapy. However, immune responses can complicate evaluation of the long-term benefits of enzyme replacement or gene therapy delivered to adult MPS VII mice. To make this model useful for studying the long-term effectiveness and side effects of experimental therapies delivered to adult mice, we developed a new MPS VII mouse model, which is tolerant to both human and murine GUS. To achieve this, we used homologous recombination to introduce simultaneously a human cDNA transgene expressing inactive human GUS into intron 9 of the murine Gus gene and a targeted active site mutation (E536A) into the adjacent exon 10. When the heterozygote products of germline transmission were bred to homozygosity, the homozygous mice expressed no GUS enzyme activity but expressed inactive human GUS protein highly and were tolerant to immune challenge with human enzyme. Expression of the mutant murine Gus gene was reduced to about 10% of normal levels, but the inactive murine GUS enzyme also conferred tolerance to murine GUS. This MPS VII mouse model should be useful to evaluate therapeutic responses in adult mice receiving repetitive doses of enzyme or mice receiving gene therapy as adults. Heterozygotes expressed only 9.5–26% of wild-type levels of murine GUS instead of the expected 50%, indicating a dominant-negative effect of the mutant enzyme monomers on the activity of GUS tetramers in different tissues. Corrective gene therapy in this model should provide high enough levels of expression of normal GUS monomers to overcome the dominant negative effect of mutant monomers on newly synthesized GUS tetramers in most tissues.
INTRODUCTION
Mucopolysaccharidosis VII (MPS VII or Sly syndrome) is a lysosomal storage disease caused by a deficiency of β-glucuronidase (GUS, EC.3.2.1.31) (1), an enzyme involved in stepwise degradation of glycosaminoglycans (GAGs) (2). The enzyme is a tetrameric glycoprotein acid hydrolase localized primarily in lysosomes and found in virtually all mammalian cells (3). It removes glucuronic acid residues from the non-reducing termini of GAGs. In its absence, chondroitin sulfate, dermatan sulfate and heparan sulfate are only partially degraded and accumulate in the lysosomes of many tissues, eventually leading to cellular and organ dysfunction. Over 45 different mutations have been found in the GUS gene in patients with MPS VII, accounting for the clinical variability among MPS VII patients (4–10). Around 90% of mutations identified in MPS VII patients were point mutations expressing an inactive protein.
Opportunities for experimental therapy for MPSs and related disorders were greatly expanded by the discovery of the MPS VII mouse by Birkenmeier et al. (11–13). The original MPS VII (gusmps/mps) mouse has a 1 bp deletion in exon 10 resulting in a progressive degenerative disease, which reduces lifespan and causes facial dysmorphism, growth retardation, deafness and behavioral defects. Progressive lysosomal accumulation of undegraded GAGs affects the spleen, liver, kidney, eye, brain, heart and bone. These morphologic, genetic and biochemical characteristics closely resemble those of human MPS VII.
The availability of the MPS VII mouse, with known and uniform genetic constitution, made it an attractive model to study multiple experimental therapies for lysosomal storage disorders. Thus, MPS VII (gusmps/mps) mice have been used effectively for evaluating the responses to bone marrow transplantation (14–18), enzyme replacement (ERT) (19–25), and gene therapy with retroviral (26–33), adenoviral (34–38) and adeno-associated viral vectors (39–44). However, cellular and humoral immune responses to repeatedly injected enzymes and to enzymes expressed in some gene therapy vectors have been recognized as impediments to evaluating experimental ERT and gene therapy strategies in adult MPS VII mice and other models. Antibodies to the gene products in MPS I, MPS VI and MPS VII animal models were detected after multiple injections of enzyme (45,46).
To make the MPS VII model suitable for long-term studies of repetitive ERT and gene therapy administered to adult animals, we initially used a traditional transgenic approach to make mice immunotolerant to human GUS enzyme. After identifying residue E540 as the active site nucleophile of human GUS (47,48), we introduced a transgene expressing human GUS E540A on the MPS VII (gusmps/mps) background. The transgenic mouse with human GUS E540A transgene retained the MPS VII phenotype but had the added desirable feature of being immunotolerant to human GUS, even though the human GUS protein was expressed at such a low level that it could not be detected in most tissues by western blot (49).
In this study, we report a novel, second approach to creating a tolerant mouse model of MPS VII that has the advantage of being tolerant to both the human and mouse gene products. The new mouse model lacks GUS activity because of a targeted missense mutation (E536A) in the mouse Gus gene (which corresponds to the active site mutation E540A in the human GUS gene) and which confers tolerance to murine GUS. It is also tolerant to human GUS because a human GUS E540A cDNA transgene was simultaneously introduced into intron 9 of the mouse Gus gene. This approach to making tolerant mouse models of human diseases is potentially generalizable.
RESULTS
Generation of Gustm(hE540A·mE536A)Sly MPS VII mice
To introduce the E536A point mutation in the Gus gene and human GUS cDNA with an E540A mutation into the adjacent intron, we designed a targeting vector with a total of 12.4 kb of homologous mouse genomic sequence flanking the neor cassette and human GUS cDNA (Fig. 1). After electroporation of the construct into embryonic stem (ES) cells and selection with G418 and ganciclovir, doubly resistant clones were screened for homologous recombination by PCR, and by southern blots hybridized with a 3′ external probe. Of 190 clones screened by PCR, three contained the EcoRI uncleaved 3910 bp PCR fragment diagnostic of homologous recombination in one allele, in addition to the 3580 and 330 bp restriction fragments from the wild-type allele. Moreover, two out of three clones contained the E536A point mutation, which was confirmed by BstUI restriction enzyme digestion (Fig. 2). Targeted ES cells containing one mutant allele were injected into C57BL/6 blastocysts and chimeric males were obtained, followed by germ-line transmission of the mutant allele. Heterozygous F1 offspring were independently intercrossed to generate F2 homozygous mice of 129/Sv × C57BL/6 hybrid strain background.
Figure 1.
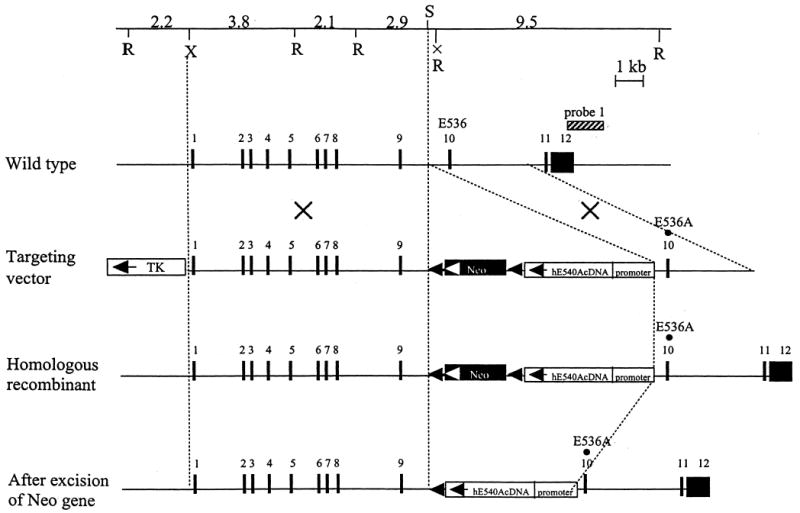
Targeted mutagenesis of the Gus gene. The structure of the endogenous gene, the targeting construct, the homologous recombinant allele, and the neo-excised allele are presented schematically on successive lines. Filled rectangles represent exons and neor, whereas two open rectangles indicate TK and human GUS cDNA, respectively. The striped bar over the wild-type allele represents the probe used for Southern blots. Abbreviations for restriction enzymes are R, EcoRI; S, SalI; X, XhoI. The EcoRI site in intron 9 (X) was lost during the construction of the targeting vector by in vitro mutagenesis without any effect on the consensus splicing sequences. The homologous E to A amino acid change was introduced in both the mouse Gus gene (E536) and the human GUS cDNA (E540).
Figure 2.
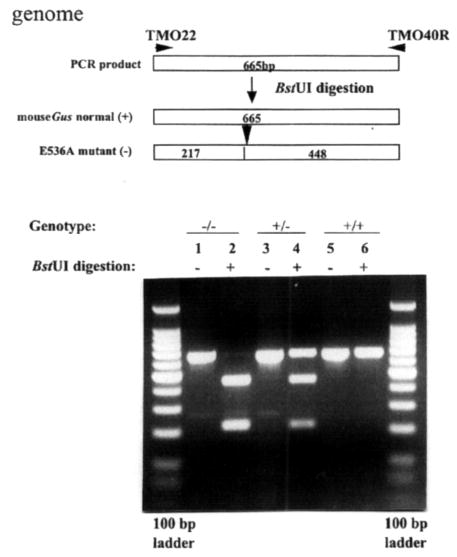
Detection of E536A point mutation in the murine Gus gene by genomic PCR amplification and subsequent BstUI digestion. The E536A mutation (an A→C transversion) creates a new restriction site, BstUI. Restriction enzyme analysis of the mutation introduced at codon 536, E536A, was performed using DNA from Gustm(hE540A·mE536A)Sly (−/−), Gustm(hE540A·mE536A)Sly/+ (+/−), and Gus+/+ (+/+) mice. Unnumbered lanes at each end are DNA ladders of 100 bp markers. Lane 1, undigested amplified PCR product (665 bp) from an E536A homozygote (−/−); lane 2, DNA from an E536A homozygote digested with BstUI (217 and 448 bp); lane 3, undigested amplified PCR product from a heterozygote (+/−); lane 4, DNA from a heterozygote digested with BstUI (217, 448 and 665 bp); lane 5, undigested amplified PCR product from a wild-type control (+/+); lane 6, DNA from a wild-type control digested with BstUI.
Phenotype of Gustm(hE540A·mE536A)Sly mice
Homozygous E536A MPS VII mice carrying the human GUS E540A cDNA, herein referred to as Gustm(hE540A·mE536A)Sly, were not distinguishable from heterozygous Gustm(hE540A·mE536A)Sly/+ and Gus+/+ littermates at birth without genotyping, but could easily be identified visually by the time of weaning from their shortened faces and slightly smaller size. As they aged, their growth retardation, shortened extremities and facial dysmorphism became more prominent. Figure 3 shows the difference in phenotype between wild-type and mutant mice at 4 months of age. By this age, radiographic analysis of the axial and appendicular skeleton of Gustm(hE540A·mE536A)Sly mice demonstrated marked dysplasia with a narrow thorax, sclerosis of the calvarium, and shortened, broad, sclerotic long bones (Fig. 3B). Typically, the mutant mice became progressively less active, stopped eating and underwent a sharp drop in body weight in the few days before death. Other aspects of the MPS VII mutant phenotype (which include deafness, failure to reproduce and shortened survival) were quite similar to the clinical phenotype described for the original MPS VII (gusmps/mps) mice (5,6). Combined data from crosses between F1 heterozygous progeny showed a distribution of 28% Gus+/+, 57% Gustm(hE540A·mE536A)Sly/+ and 15% Gustm(hE540A·mE536A)Sly in 150 offspring analyzed at weaning, suggesting that homozygous MPS VII offspring have reduced survival in the neonatal period, as had been noted for the original MPS VII (gusmps/mps) mice (24).
Figure 3.
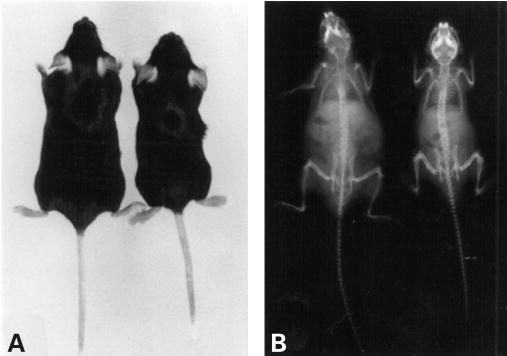
Phenotype of MPS VII doubly tolerant mouse. (A) A 4-month-old MPS VII doubly tolerant female mouse (right) compared with a 4-month-old normal female (left). The MPS VII mouse is smaller than the normal mouse and has shortened limbs, a hobbled gait, and a dysmorphic face with a blunted nose. (B) The skeleton of a 4-month-old MPS VII doubly tolerant male mouse (right) shows sclerosis of the cranial bones, a broad zygomatic arch, shortened limb bones, and a narrow rib cage, compared with the skeleton of a normal 4-month-old male mouse (left).
The colony of heterozygous Gustm(hE540A·mE536A)Sly/+ mice, which were phenotypically normal, was maintained by brother–sister matings, genotyped by PCR analysis of genomic DNA, and by enzymatic analysis of tail sample extracts for GUS activity. Homozygous and heterozygous offspring from this colony were analyzed for morphologic, biochemical and histopathologic phenotypes and tested for tolerance to immune challenge with human and mouse GUSs.
Histopathology of the Gustm(hE540A·mE536A)Sly mouse
Multiple tissues from eight homozygous mice from 1 to 8 months of age, five heterozygote mice at 4–13 months of age, and four wild-type control mice at 4–6 months of age were studied morphologically as described (12). Tissues were evaluated for the extent of lysosomal storage and alterations were compared with those described in the murine MPS VII (gusmps/mps) model (12). In the homozygote mice, widespread lysosomal storage was seen throughout the fixed tissue macrophage system whereas heterozygotes had no storage apparent in the fixed tissue macrophages (Fig. 4A–P). In the liver (Fig. 4A) and spleen (Fig. 4C) of homozygous mice, lysosomal storage was marked in the sinus lining cells. Kupffer cells were distended with variably sized lysosomes. Hepatocytes contained a few small vacuoles, primarily in a pericanalicular location. In the homozygote kidney, the glomerular visceral epithelial cells, the cortical tubular epithelial cells, and the interstitial cells were distended with enlarged lysosomes (data not shown). Heart valve stromal cells and interstitial cells in the myocardium also had lysosomal distention in the homozygotes. The homozygote brain had lysosomal storage in neurons and glial cells (Fig. 4E) in the neocortex, hippocampus and cerebellum as well as in meningeal cells (Fig. 4G). The cerebellar Purkinje cells had more opaque storage in their cytoplasm than that seen in neurons elsewhere in the central nervous system, similar to that seen in the previously described MPS VII mice. The cerebellar Purkinje cells in the heterozygote mice contained complex material stored in lysosomes that was similar to the stored material seen in the homozygous animals (Fig. 5). Corneal stromal fibrocytes (Fig. 4I) and the retinal pigment epithelium (Fig. 4K) both had storage in homozygotes. The bone storage in homozygotes was marked, with lysosomal distention in osteoblasts and osteocytes lining the cortical and trabecular bone, in chondrocytes, and in the sinus lining cells in the bone marrow (Fig. 4M). There were clusters of foamy macrophages in the homozygote marrow, indicating lysosomal storage. The bone and joints had altered architecture with synovial thickening and vacuolated cells in the synovium. The articular chondrocytes were distended with storage (Fig. 4O). The heterozygotes had normal bone.
Figure 4.
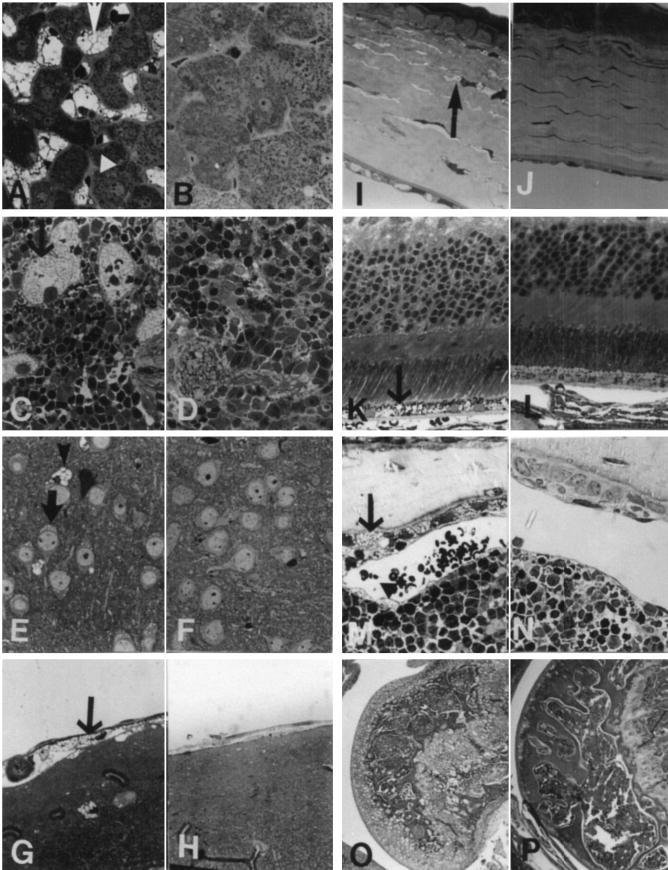
Morphological alteration in the MPS VII doubly tolerant Gustm(hE540 A·mE536A)Sly mice. (A) Liver from an 8-month-old Gustm(hE540A·mE536A)Sly mouse has Kupffer cells that are distended with lysosomal storage (arrow). The hepatocytes have only a small amount of cytoplasmic storage (arrowhead). (B) Neither hepatocytes nor Kupffer cells were altered in an adult heterozygote Gustm(hE540A.mE536A)Sly/+ mouse. (C) Spleen from a 7-month-old Gustm(hE540A·mE536A)Sly mouse has prominent lysosomal storage in the sinus lining cells. (D) No storage is apparent in the spleen of a Gustm(hE540A·mE536A)Sly/+ mouse. (E) The neocortical neurons (arrow) and glial cells (arrowhead) in a 1-month-old Gustm(hE540A.mE536A)Sly mouse have lysosomal distention. (F) The neocortex in a Gustm(hE540A·mE536A)Sly/+ mouse has no evidence of lysosomal storage in either neurons or in glial cells. (G) The meninges covering the brain contain cells distended with lysosomal storage (arrow) in a 1-month-old Gustm(hE540A·mE536A)Sly mouse. (H) In the meninges of a Gustm(hE540A·mE536A)Sly/+ mouse there is no evidence of lysosomal storage. (I) The cornea from a 1-month-old Gustm(hE540A·mE536A)Sly mouse has stromal fibrocytes (arrow) with a moderate amount of lysosomal distention. (J) The Gustm(hE540A·mE536A)Sly/+ mouse has no storage in the corneal fibrocytes or epithelium. (K) The retinal pigment epithelium at the base of the retina in a 1-month-old Gustm(hE540A·mE536A)Sly mouse is distended with storage (arrow). Other layers of the retina have no lysosomal storage accumulation apparent at the light microscopic level. (L) The retina from a Gustm(hE540A·mE536A)Sly/+ mouse has no morphological abnormality. (M) Bone from the rib of a 2-month-old Gustm(hE540A·mE536A)Sly mouse shows distended osteoblasts lining the cortical bone (arrow) and osteocytes within the bone with a moderate amount of lysosomal distention. The sinus lining cells in bone marrow (arrowhead) also contain a small amount of storage. (N) Neither the bone marrow nor the bone had a morphological alteration in the Gustm(hE540A·mE536A)Sly/+ mice. (O) A stifle joint from the limb of an 8-month-old Gustm(hE540A·mE536A)Sly mouse shows the distortion of the bone architecture. There is storage and structural alteration of both the articular and epiphyseal cartilage plate chondrocytes. (P) A similar joint from an adult Gustm(hE540A·mE536A)Sly/+ mouse has no structural alteration. (A–N, toluidine blue, 1 cm=27 μm; O,P hematoxylin and eosin, 1 cm=425 μm).
Figure 5.
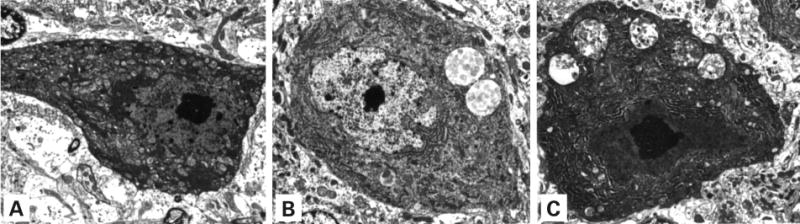
Storage in cerebellum of control and MPS mice. (A) A cerebellar Purkinje cell in a wild-type control mouse has normal cytoplasm with no evidence of storage. (B) A Purkinje cell from a homozygous mouse contains several large membrane-bound accumulations of flocculent fibrillar material, identical to the storage material seen in the previously described Birkenmeier MPS VII murine model. (C) A Purkinje cell from a heterozygous mouse contains membrane-bound stored material that is similar, although slightly more complex ultrastructurally, than that seen in the homozygous mutant mouse (A, B and C, uranyl acetate–lead citrate; 2080×).
Storage was well established in the fixed tissue macrophage system in the bone, brain and eye, even in 1-month-old homozygote mice.
Biochemical phenotype of the Gustm(hE540A·mE536A)Sly mice
Table 1 summarizes data comparing the tissue levels of GUS in Gustm(hE540A·mE536A)Sly, Gustm(hE540A·mE536A)Sly/+ heterozygotes and Gus+/+ mice. The homozygous Gustm(hE540A·mE536A)Sly mice showed profound deficiency of GUS comparable to that of the original MPS VII mice (11,49), which is consistent with the targeted allele having an active site mutation. Heterozygotes had less than the expected normal GUS activity (Table 1). Instead of 50% normal levels in each tissue, the values were 9.5–26.2% in different tissues. Thus, although the absolute level of enzyme varied in different tissues, the evidence for a dominant negative effect of the mutant allele in the heterozygote was seen in all tissues tested.
Table 1.
Tissue levels of GUS in wild-type, homozygous doubly tolerant MPS VII mice and heterozygous littermates
| Tissue | Wild-type (n = 4) | Gustm(hE540A·mE536A)Sly (homozygotes, n = 8) | Gustm(hE540A·mE536A)Sly/+ (heterozygotes, n = 12) | Gustm(hE540A·mE536A)Sly/+ (heterozygotes, percentage of WT) |
|---|---|---|---|---|
| Brain | 16.69 ± 2.0 | 0.13 ± 0.01 | 1.92 ± 0.2 | 11.5 |
| Liver | 185 ± 11.9 | 0.064 ± 0.03 | 40.5 ± 5.5 | 21.9 |
| Spleen | 301 ± 26.6 | 0.057 ± 0.005 | 32.5 ± 4.8 | 10.8 |
| Kidney | 108 ± 7.5 | 0.106 ± 0.03 | 17.9 ± 3.6 | 16.6 |
| Heart | 20.8 ± 12.5 | 0.163 ± 0.140 | 1.97 ± 0.5 | 9.5 |
| Lung | 88.8 ± 1.8 | 0.103 ± 0.115 | 10.63 ± 3.5 | 12.0 |
| Muscle | 7.32 ± 1.6 | 0.127 ± 0.105 | 1.92 ± 0.3 | 26.2 |
| Serum (u/ml) | 37.6 ± 12.0 | 0.063 ± 0.052 | 5.82 ± 2.3 | 15.5 |
Gus levels expressed as units/mg cell protein.
Two other lysosomal enzymes, β-galactosidase and β-hexosaminidase, were elevated in tissues of 3–5-month-old mice (Fig. 6). These ‘secondary elevations’ are similar to those reported in the original MPS VII mice (14,15,20). Elevations of enzyme activities in tissues of 3-month-old mice were the same as those in tissues of 5-month-old mice (data not shown). Table 2 presents data demonstrating the significant elevation in urinary GAG in the adult Gustm(hE540A·mE536A)Sly mice compared with wild-type B6 mice.
Figure 6.
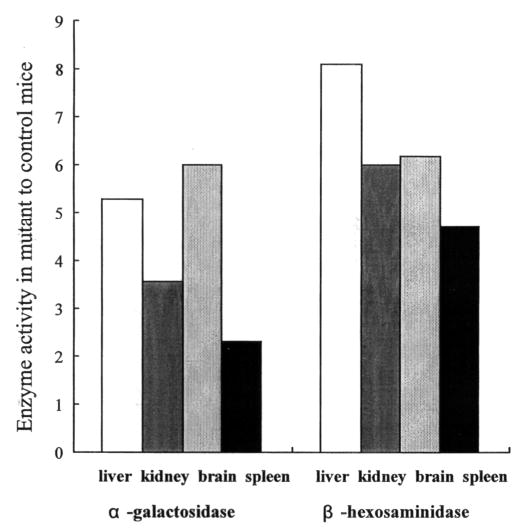
Secondary elevation of α-galactosidase and β-hexosaminidase. Levels of α-galactosidase and β-hexosaminidase in tissues of Gustm(hE540A·mE536A)Sly mice, expressed as fold increase over levels found in B6 control mice. Normal B6 control mean α-galactosidase levels in liver, kidney, brain and spleen are 45, 28, 19 and 67 units/μg protein, respectively. Normal B6 control mean β-hexosaminidase levels in liver, kidney, brain and spleen are 442, 781, 876 and 2525 units/μg protein, respectively.
Table 2.
Urinary GAG excretion in Gustm(hE540A·mE536A)Sly mice
| Genotype (n = 8) | mg GAG/g creatinine |
|---|---|
| Gustm(hE540A.mE536A)Sly | 893.5 (823–1050)* |
| Wild-type | 213.5 (0–321.8) |
Doubly tolerant MPS VII mice and wild-type littermates were 3–6 months of age. GAG values represent the means and (ranges).
P < 0.0001.
Murine Gus and human GUS mRNA transcript levels
To assess the expression of the GUS (or Gus) gene product in Gustm(hE540A·mE536A)Sly mice, we performed northern blot analyses on total RNA isolated from liver, kidney and spleen of Gustm(hE540A·mE536A)Sly (−/−), Gustm(hE540A·mE536A)Sly/+ heterozygote (+/−), and Gus+/+ wild-type (+/+) littermates. The result from the liver is shown in Figure 7A. A murine Gus transcript of 2.3 kb, present in multiple tissues from +/+ mice, was present in reduced amounts in −/−mice. However, the easily detectable transcript in this mutant contrasts with that in the classical Birkenmeier mutant, where the murine message was estimated to be 200-fold lower than the level of the normal transcript (11). A human GUS transcript of 2.3 kb was also abundant in −/− and +/− mice. RT–PCR amplified both Gus and GUS transcripts from RNA from Gustm(hE540A·mE536A)Sly mice. Sequencing of the PCR products showed no alterations except the E536A mutation (GAG to GCG) in the murine transcript and the E540A mutation in the human transcript. Digestion with BstUI could be used to verify the E540A mutation in the human transcript (Fig. 7B) and distinguished the mutant murine allele from the normal murine allele (Fig. 7C).
Figure 7.
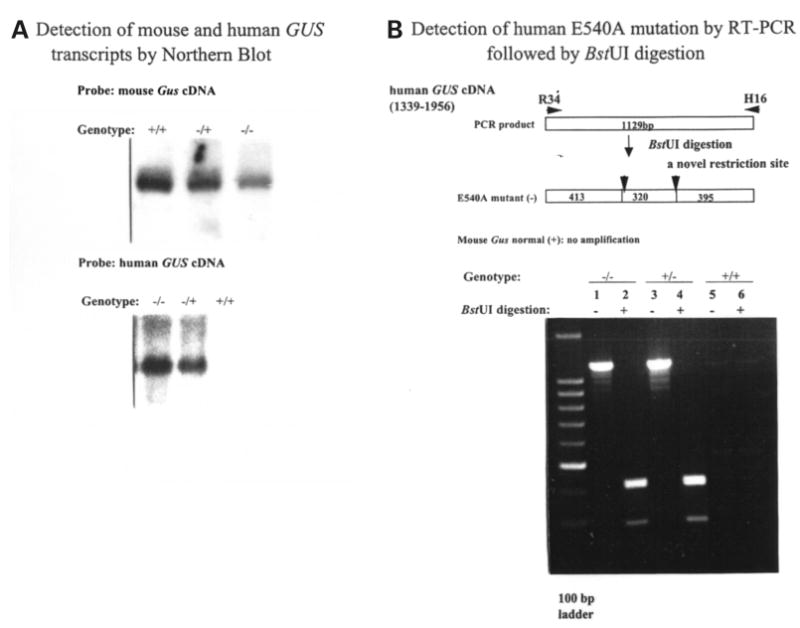
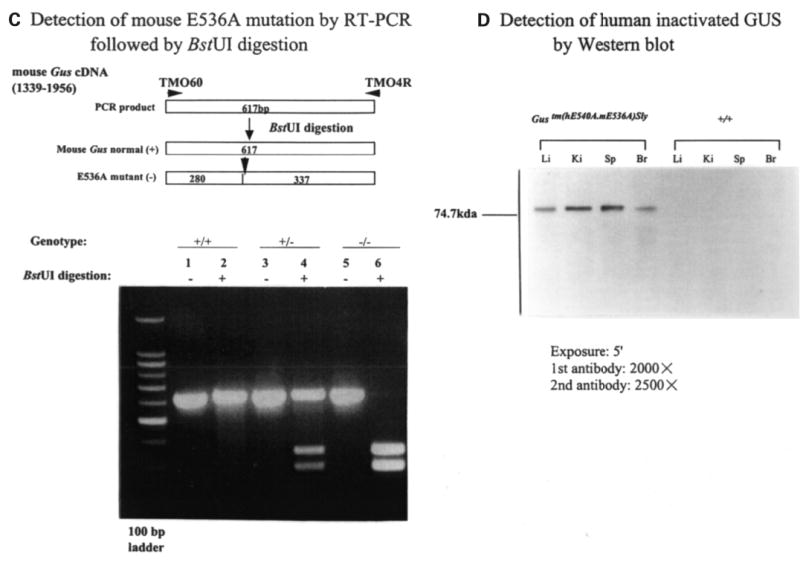
Expression of murine Gus (or human GUS) mRNA and GUS protein. (A) Northern blot analysis of murine Gus (upper panel) or human GUS (lower panel) mRNA from the livers of wild-type (+/+), heterozygote (+/−) and Gustm(hE540A·mE536A)Sly homozygote (−/−) mice. The 2.3 kb murine Gus mRNA transcript present in homozygous Gustm(hE536A·mE536A)Sly liver (upper panel, lane 3) was reduced in comparison to wild-type (+/+) and heterozygote (+/−) mice (upper panel, lanes 1 and 2). The murine probe does not cross hybridize with the human transcript under these conditions (data not shown). The human GUS mRNA was expressed only in the transgene containing Gustm(hE540A·mE536A)Sly homozygote and heterozygote tissues (lower panel, lanes 1 and 2). (B) BstUI analysis of human E540A mutation in hGUS transgene using RT–PCR. Fragments of 1129 bp were amplified from Gustm(hE540A·mE536A)Sly and heterozygote mice (lanes 1 and 3) using primers R34 and H16 (and visualized on a 2% agarose gel). No amplification from the wild-type mouse DNA (lanes 5 and 6) were seen. The 1129 bp fragments amplified from the mutant human transgene were cleaved into 413, 320 and 395 bp fragments by digestion (lanes 2 and 4). Note that the 413 and 395 bp fragments do not separate, so a doublet of these two and a single 320 bp band are seen. (C) BstUI analysis of mouse genomic DNA for the E536A mutation using RT–PCR. The 617 bp fragments were amplified using primers TMO60 and TMO4R from the homozygous Gustm(hE540A·mE536A)Sly (lanes 5 and 6), wild-type (lanes 1 and 2), and heterozygous Gustm(hE540A·mE536A)Sly/+ (lanes 3 and 4) mice, respectively. BstUI digestion cleaves the products from the mutant allele into 280 and 337 bp fragments (lanes 4 and 6). The 617 bp product of the normal allele is not cleaved by BstUI (lane 2). (D) Western blot for human GUS protein in extracts of tissues from Gustm(hE540A·mE536A)Sly and wild-type mice. The hGUS proteins from liver, kidney, spleen and brain tissues of the Gustm(hE540A·mE536A)Sly mouse were identified on western blot by anti-human GUS antibody (lanes 1–4). No band was observed in any tissue from the control wild-type mouse.
Expression of human GUS
Liver, kidney, spleen and brain tissues from Gustm(hE540A·mE536A)Sly and Gus+/+ mice were homogenized to analyze the expression of the hGUS protein. Western blots of these tissues are shown in Figure 7D. A single band with the expected Mr of the hGUS protein (75 kDa) was detected by the anti-human GUS antibody in all tissues of Gustm(hE540A·mE536A)Sly mice. No signal for hGUS protein was found in the tissues from the Gus+/+ control mice. The mGUS protein was also detectable in each tissue from Gustm(hE540A·mE536A)Sly mice, although the amount is substantially reduced compared with that of normal mice (data not shown).
Tolerance of the Gustm(hE540A·mE536A)Sly mice to immune challenge with human and mouse β-glucuronidase
We next tested the hypothesis that the gene products expressed for the human E540A cDNA in intron 9 and the endogenous Gus gene containing the E536A mutation in exon 10 would confer tolerance to human and murine GUSs. To provide a maximum immunogenic challenge, we used i.p. injection of either human or mouse GUS in complete Freund’s adjuvant as the initial challenge, followed by two boosts with the respective antigen in incomplete Freund’s adjuvant at 28 and 42 days, as described previously (49). As a control, we used homozygous B6 MPS VII (gusmps/mps) mice, which do not express human and mouse GUSs and received the same immunogens on the same schedule. At the first bleed (12 days after the first boost), both of the MPS VII (gusmps/mps) controls, but none of the Gustm(hE540A·mE536A)Sly mice, showed anti-human GUS antibodies by ELISA (data not shown). Figure 8A shows the ELISA plate assay on blood taken 12 days following the second boost with hGUS (i.e. 54 days after the initial challenge). None of the Gustm(hE540A·mE536A)Sly mice showed any response. Both MPS VII control mice had titers of 105 or greater. The same was true for the murine GUS immunogenic challenge, where Gustm(hE540A·mE536A)Sly mice showed no reaction but MPS VII (gusmps/mps) mice had a substantial reaction (Fig. 8B). These data demonstrate two important points: (i) the MPS VII (gusmps/mps) mice that do not express human and mouse GUSs are capable of mounting a strong antibody response to human and mouse GUSs when challenged in this manner; and (ii) the Gustm(hE540A·mE536A)Sly alleles conferred tolerance to both human and mouse GUSs, even when each was provided in an extreme immunogenic challenge.
Figure 8.
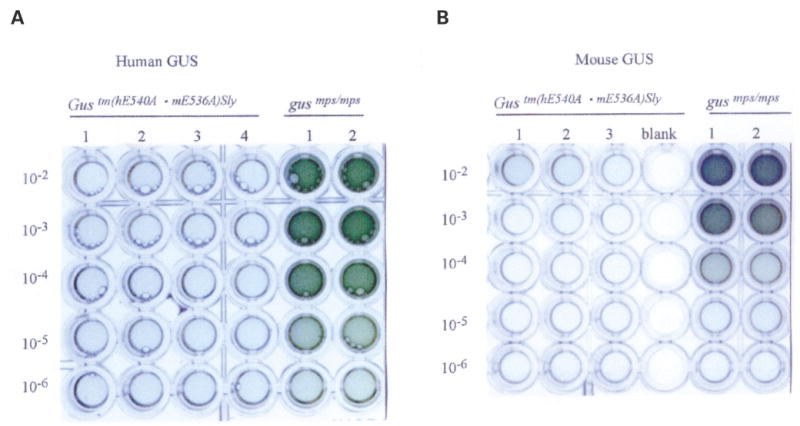
Humoral immune tolerance of Gustm(hE540A·mE536A)Sly mice to human and murine GUSs. (A) ELISA plate assay of antibodies to hGUS in serum of Gustm(hE540A·mE536A)Sly mice (left, lanes 1–4) and control MPS VII (gusmps/mps) mice (right, lanes 1 and 2) following primary immunization with human GUS in complete Freund’s adjuvant and two boosts with human GUS in incomplete Freund’s adjuvant. Gustm(hE540A·mE536A)Sly mice show no antibody response whereas control mice have antibodies detectable at 105 dilutions or greater. (B) ELISA plate assay of antibodies to mGUS in serum of Gustm(hE540A·mE536A)Sly mice (left, lanes 1–3) and control MPS VII (gusmps/mps) mice (right, lanes 1 and 2) following primary immunization with mouse GUS in complete Freund’s adjuvant and two boosts with mouse GUS in incomplete Freund’s adjuvant.
DISCUSSION
The original MPS VII (gusmps/mps) mouse generates an immune response to both human and murine GUSs when administered repetitively to adult mice (49,50). Antibodies to the infused enzymes can change the targeting and fate of the infused enzymes and may limit the response to therapy (45,51,52). Expression of human GUS in experimental gene therapy protocols can also lead to antibody production to human GUS (27,28). Cellular responses to cells expressing foreign genes following gene therapy have been noted to be important factors limiting persistence of gene expression in other models (46,53). To eliminate these immunological obstacles to evaluating enzyme and gene therapy in the adult MPS VII mouse model, we initially used a simple transgenic approach to create tolerance to human GUS, by introducing a transgene expressing inactive human GUS on the MPS VII (gusmps/mps) background (49).
In the present study, we developed a novel strategy to produce mice tolerant both the human and murine gene products. In this approach, we introduced a construct into ES cells that carried not only an active site mutation (E536A) in the endogenous Gus gene, but also the human E540A cDNA inserted in the adjacent intron. The homologous recombinant ES cells, which contained both the targeted mutation (E536A) and the cDNA, were used to generate mutant mice that expressed inactive but stable forms of both human and mouse enzymes. The Gustm(hE540A·mE536A)Sly mice retained the full MPS VII phenotype. However, the stable inactive enzymes they expressed induced tolerance to immune challenge by each of the respective wild-type enzymes.
The deficiency of one lysosomal enzyme involved in the degradation of MPS is often accompanied by secondary increases in others (50). This had been found in MPS I, II, III (54) and VII (50), and is true in this new mouse model of MPS VII. Decreases in these secondary elevations have been shown to provide a convenient biochemical marker of correction by enzyme or gene therapy (14,20). The presence of storage in the cerebellar Purkinje cells of the heterozygote mice may indicate greater expression of the mutant human transgene in these cells than in others and a correspondingly larger dominant negative effect on these cells, or that these cells require more β-glucuronidase than most cells to prevent storage. These cells have been more resistant than other cell types to treatment in previous therapeutic trials in homozygous MPS VII mice (20). Why these cells might require more β-glucuronidase activity than other cells requires further investigation.
Production of a tolerant MPS VII model by this approach has several advantages. First, since gene-targeted mutagenesis is used to introduce the mutated human GUS cDNA and to inactivate the endogenous mouse Gus at the same time, one does not require a natural mouse mutant or a gene-targeted knock-out mouse to begin. Second, unlike the original transgenic tolerant mouse model, where the transgene is present on a different chromosome than the gusmps allele, the human E540 cDNA and the E536-targeted mutation are very closely linked on the same chromosome and are simultaneously transmitted to all offspring in crosses to other strains. Third, the Gustm(hE540A·mE536A)Sly mouse produces enough stable, catalytically inactive human GUS, detectable by immunoblot to allow the evaluation of the level of expression of the inactive gene in various tissues. Fourth, by controlling the localization and copy number of the human GUS cDNA in the mouse genome, we reduced the chances that changes in the expression of other genes by the transgene might influence the phenotype. The standard method of producing transgenic mice does not control the insertion site.
There’s an important caveat to use of this model in evaluating experimental gene therapy. The normal GUS gene product has to be expressed at a high enough level in the homozygous double tolerant MPS VII mice to overcome the dominant negative effect of two alleles expressing the mutant monomers, which readily combine with the normal monomers to form stable hybrid tetramers (7). Some of these hybrid tetramers must be inactive; the fact that the highly expressed mutant E540A human gene product has such a striking dominant negative effect argues that at least two active GUS monomers per tetramer are required for activity. However, the high levels of gene expression seen with most gene therapy vectors should overcome this dominant negative effect, as only a small amount of normal GUS activity is required to correct (or prevent) storage.
This caveat does not apply to experimental enzyme therapy in this model. Monomers of mature GUS do not exchange. Thus, inactive tetramers would not reduce the activity or the corrective potency of normal enzyme provided by ERT. All of the reported experiments involving ERT in murine MPS VII have used the murine enzyme. Since the mouse is tolerant to both mouse and human GUS, the new model could be used to evaluate the response to ERT with either gene product. Studies of the response to human GUS are likely to be important preclinical experiments preceding ERT with the human gene product in MPS patients.
Tolerant mouse models like the one described here should be versatile for evaluating the long-term benefits of ERT or gene therapy whether the murine or human gene product is used. Given the growing interest in producing animal models of human diseases, this method of producing tolerant mouse models of human diseases may have broad applications.
Materials and Methods
Construction of the GUS targeting vector
The mouse Gus gene was cloned from a 129/Sv mouse genomic library (Stratagene, La Jolla, CA, USA). The E536 residue in exon 10 was selected for making a point mutation because the corresponding residue in the human GUS gene (E540) was identified as the active site nucleophile by X-ray crystallography, in vitro mutagenesis, and biochemical studies (47,48,55). The presence of a mutation at this site in a human MPS VII patient produces a severe phenotype. The E536A point mutation introduced in exon 9 of the Gus gene created a new BstUI restriction site.
The pBS vector containing a 3′ PGK neor cassette and a 5′-thymidine kinase (TK) cassette was the starting point. The human GUS E540A cDNA and chicken β-actin promoter were introduced at the 5′ end of the neor gene (Fig. 1). Then the TK-neor-cDNA cassette was introduced into a plasmid containing the 3.9-kb fragment of genomic DNA upstream of the SalI site in intron 9, in which the E536A substitution in exon 10 of the Gus gene had been introduced by in vitro mutagenesis. Next, the 8.5 kb fragment containing exons 1–9 of the Gus gene was added between the TK and neor genes to create the complete targeting vector. The loxp sequences are positioned at both ends of the neor gene. This lox–neo–lox cassette can be eliminated by mating the heterozygotes with transgenic mice expressing the Cre recombinase enzyme. A cassette containing the Herpes simplex virus TK gene under control of its own promoter was cloned into the vector upstream of the Gus gene (Fig. 1). The final construct contained 8.5 and 3.9 kb of 5′ and 3′ homology to the Gus gene, respectively, and human GUS cDNA with an E540A mutation.
Gene targeting in ES cells and generation of mutant mice
The targeting vector (25 μg) was linearized with NotI and introduced into the 129/Sv-derived ES cell line RW4 (Incyte Genome Systems, St Louis, MO, USA; 1 × 107 cells) by electroporation (230 V and 500 μF) in a Bio-Rad gene pulser. After 24 h, the cells were placed under selection with 200 μg/ml G418 (Gibco BRL, Rockville, MD, USA) and 2 μM ganciclovir (Syntex Chemicals, Boulder, CO, USA) for 6 days. Genomic DNAs of resistant ES clones were screened by PCR for the homologous recombinant allele. This method utilizes a forward primer in intron 9 (Sc1: 5′-GACTGACTGCTACGAGCTGCA-GATTGAACCTGG-3′) and a reverse primer in intron 10 outside the targeting sequences (Sc12R: 5′-TCATCAAGGT-GGCCCATGTCTTTAATCCAAGAG-3′), which produces a 3910 bp fragment and retains an EcoRI restriction site in the normal allele but not in the mutant allele. Following cleavage with EcoRI, the 3580 and 330 bp restriction fragments present in the normal allele are distinguished from the uncleaved 3910 bp PCR fragment from the mutant allele on a 1.2% agarose gel. The presence of the E536A point mutation in exon 10 of the Gus gene was detected by PCR of the same ES cell DNA using a forward primer in intron 9 (TMO22: 5′-CCTGTGTCATT-TGCATGTGACTATT-3′) and a reverse primer in intron 10 (TMO40R: 5′-TGTGGGTGCTGGGAAC-CAGACT-GAG-3′), which amplified a 665 bp fragment of the mouse Gus gene. Following digestion with BstUI, the 217 and 448 bp restriction fragments present in the mutant allele are distinguished from the uncleaved 665 bp PCR fragment from the normal allele on a 1.2% agarose gel (Fig. 2). In addition, genomic DNA of resistant clones was digested with EcoRI, followed by southern blot and hybridization with the 5′ 0.8 kb external probe (Fig. 1). The hybridizing fragment is larger in the mutant allele than in the wild-type allele (11 vs 9.5 kb; data not shown).
Two independent targeted ES clones were used for injection into the blastocysts of C57BL/6J mice and transferred into pseudopregnant female mice. Chimeric male offspring were bred to C57BL/6J females and the agouti F1 offspring were tested for transmission of the mutant allele. The F1 mice were crossed with mice expressing Cre enzyme to remove the neor gene and the resultant neo-excised heterozygous mice were mated to produce homozygous mutant mice. The removal of neor was diagnosed by PCR of tail DNA using a forward primer in intron 9 (BGCre2: 5′-TTTGCCTGCATGTGTGA-GGGTGTCGATCC-3′) and a reverse primer in human GUS cDNA (BGO5: 5′-GATGGTGATCGCTCACCAAATC-3′), which amplified a 650 bp fragment while the non-neor-excised allele did not reveal any fragment. Heterozygotes were either intercrossed for experimental use or backcrossed to C57BL/6 mice to put the mutation on a congenic background. Genotyping was performed by PCR analysis of DNA obtained by tail biopsies at 10 days, and confirmed by assay of GUS activity in these tails. The resultant homozygous mice with the E536A mutation on the mouse Gus gene and E540A human cDNA in adjacent intron 9 were named Gustm(hE540A·mE5·6A)Sly (or Gustm(hE540A··mE536A)Sly/tm(hE540A··mE536A)Sly).
Northern blot analysis and RT–PCR
Total cellular RNA was isolated from tissues of homozygous MPS VII (Gustm(hE540A·mE536A)Sly), heterozygous (Gustm(hE540A·mE536A)Sly/+), and wild-type mice using a guanidinium–phenol solution (RNA-Stat60, Tel-Test, Friendswood, TX, USA). Twenty micrograms of RNA from each source were denatured in formaldehyde buffer and electrophoresed in 1% agarose, 2.2 M formaldehyde gels. Equivalent loading of intact RNA was assured by visualization of ethidium bromide stained 28S and 18S ribosomal RNA bands. The RNA was transferred to nylon membranes (Amersham Biosciences, Little Chalfont, UK), immobilized by UV crosslinking, and prehybridized at 65°C. Blots were hybridized overnight at 65°C with 32P-labeled human GUS and mouse Gus cDNA probes, respectively. To investigate the message of human GUS and mouse Gus, RT–PCR was performed. Five micrograms of total RNA were mixed with oligo dT primer in a total volume of 20 μl, including RNase inhibitor, dNTPs, reverse transcriptase, DTT and reverse transcriptase buffer according to the instruction manual (GIBCO BRL). Reaction mixtures were incubated at 42°C for 50 min to make the first cDNA, followed by inactivation of the enzyme at 70°C for 15 min. The remainder of total RNA was eliminated by RNase at 37°C for 20 min. Two microliters of the products of one reverse transcriptase reaction were annealed to 50 pmol each of sense and antisense primers. After heating the reaction mixtures for 5 min at 94°C, PCR amplification was carried out for 35 cycles as follows: 45 s denaturation at 94°C, 40 s annealing at 63°C and 1.5 min extension at 72°C. The PCR products were directly sequenced. To amplify human GUS cDNA, a forward primer (R34: 5′-GTGCAGCTGACTGCACAGACG-3′) and a reverse primer in human GUS cDNA (H16: 5′-GCCGTGAACAG-TCCAGGAGGCACTTGTTGA-3′) were used. This procedure on the mutant allele amplifies an 1129 bp fragment with the E540A mutation, while there is no amplification product on the normal allele. After digestion with BstUI to detect the E540A mutation, the 413, 320 and 395 bp restriction fragments present in the mutant allele are distinguished on a 2.0% agarose gel. Similarly, to amplify mouse Gus cDNA, a forward primer (TMO60: 5′-TCTGTGGCCAATGAGCC-TTCCTCTG-3′) and a reverse primer in the murine Gus cDNA (TMO4R: 5′-GAACGTGTGAACGGTCTGCTTCCG-3′) were used, resulting in amplification of a 617 bp fragment. Digestion with BstUI revealed the 280 and 337 bp restriction fragments in the E536A mutant allele and the uncleaved 617-bp PCR fragment in the normal allele.
Western blot analysis
Tissues were dissected and homogenized immediately (by Brinkmann Polytron homogenizer for 30 s at 4°C) in 5 vols of homogenization buffer (25 mM Tris–HCl, pH 7.2, 140 mM NaCl, 1 mM PMSF). Samples containing 20 μg of protein were analyzed by SDS–PAGE under reducing conditions. The polypeptides were electronically transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). After transblotting, the polypeptides were immunostained using polyclonal goat anti-human GUS antibody and polyclonal rabbit anti-mouse GUS antibody, respectively, followed by incubation with rabbit anti-goat IgG (Sigma-Aldrich, St Louis, MO, USA) coupled with peroxidase or sheep anti-mouse IgG (Sigma-Aldrich) coupled with peroxidase. The peroxidase activity was visualized using a chemiluminescent substrate.
Lysosomal enzyme assays
Lysosomal enzymes were assayed fluorometrically using 4-methylumbelliferyl substrates, as described (56–58). Tissues were dissected and homogenized immediately (by Brinkmann Polytron homogenizer for 30 s at 4°C) in 5 vols of homogenization buffer (25 mM Tris–HCl, pH 7.2, 140 mM NaCl, 1 mM PMSF). GUS levels were assayed on dilutions of wild-type total homogenates for 30 min and MPS VII (Gustm(hE540A·mE536A)Sly) homogenates for 24 h. For α-galactosidase and β-hexosaminidase assays, homogenates were centrifuged at 12 000 rpm for 15 min at 4°C in a microcentrifuge. The supernatant was diluted appropriately for assay in PBS, with the final dilution in an equal volume of citrate phosphate buffer, pH 4.4, containing 0.075 M NaCl, 1.0 mg/ml human serum albumin, and 0.001% Triton X-100. Assays of α-galactosidase and β-hexosaminidase were for 1 h. Units were nmol hydrolyzed per hour, and activity was expressed as u/mg protein, as determined by micro-Lowry assay.
Analysis of GAG
To determine the μg of GAGs per mg of urinary creatinine, we measured urine with 1,9-dimethylmethylene blue (17,59,60). Creatinine was measured by mixing 10 μl of a 10-fold diluted urine sample with 50 μl of saturated picric acid (Sigma) and 50 μl of 0.2 M NaOH. Absorbance at 490 nm was read after 20 min and compared to a standard.
Pathology
Multiple tissues, including limbs, liver, spleen, kidney, heart, rib, eye and brain from eight MPS VII doubly tolerant homozygous −/− mice from 1 to 8 months of age, five heterozygous mice from 4 to 13 months of age, and four wild-type mice from 4 to 6 months of age were examined for morphological evidence of lysosomal storage as previously described. Tissues were evaluated for the extent of lysosomal storage and for comparison with the amount of storage seen in the previously described MPS VII mouse model. The skeletons of a 4-month-old MPS VII doubly tolerant mouse and an unaffected littermate were radiographed as previously described.
Immunization method and analysis of sera from immunized mice by ELISA
Four MPS VII (Gustm(hE540A·mE536A)Sly) and four MPS VII (gusmps/mps) mice were immunized with purified human GUS beginning at two months of age. Each mouse received 50 μg human GUS in 0.2 ml complete Freund’s adjuvant intraperi-toneally as an initial challenge, and two subsequent boosts with 50 μg human GUS in 0.2 ml incomplete Freund’s adjuvant intraperitoneally (the first boost at 28 days and the other at 42 days after the initial challenge). Blood was collected by eye bleed to measure antibodies to human GUS by ELISA 12 days after each boost.
Analysis of sera from immunized mice was done by ELISA assay on microtiter aliquots. The wells of 96-well microtiter plates were coated overnight at 4°C with 10 μg/ml purified recombinant human β-glucuronidase in 15 mM Na2CO3, 35 mM NaHCO3, 0.02% NaN3, pH 9.6. The wells were washed three times with TBST (10 mM Tris, pH 7.5, 150 mM NaCl, 0.05% TWEEN 20), and then blocked for 1 h at room temperature with 3% casein in PBS (pH 7.2). After washing three times with TBST, 100 μl of serial 10-fold dilutions of mouse plasma (102–108) in TBST were added to the wells and incubated at 37°C for 2.5 h. The wells were washed four times with TBST, then 100 μl of TBST containing a 1:500 dilution of peroxidase conjugated goat anti-mouse IgG were added to the wells and incubated at room temperature for 1 h. The wells were washed three times with TBST and twice with TBS (10 mM Tris, pH 7.5, 150 mM NaCl). Peroxidase substrate (ABTS solution, Roche Molecular Biochemicals, Basel, Switzerland) was added (100 μl per well) and plates were incubated at room temperature for 10 min. The reaction was stopped with the addition of 2.5 μl of 20% SDS and the plates read at optical density 400 nm on an automatic ELISA plate reader. To test tolerance to mouse GUS, the same procedure was performed using four MPS VII (Gustm(hE540A·mE536A)Sly) and four MPS VII (gusmps/mps) mice with purified mouse GUS beginning at 2 months of age.
Acknowledgments
M. Rafiqul Islam and Yanhua Bi provided valuable technical assistance. This work was supported by National Institutes of Health Grants GM34182 and DK40163 to W.S.S.
References
- 1.Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta-glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82:249–257. doi: 10.1016/s0022-3476(73)80162-3. [DOI] [PubMed] [Google Scholar]
- 2.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 3. McGraw-Hill, Medical Publishing Division; New York: 2001. pp. 3421–3452. [Google Scholar]
- 3.Paigen K. Mammalian beta-glucuronidase: genetics, molecular biology, and cell biology. Prog Nucl Acid Res Mol Biol. 1989;37:155–205. doi: 10.1016/s0079-6603(08)60698-4. [DOI] [PubMed] [Google Scholar]
- 4.Tomatsu S, Sukegawa K, Ikedo Y, Fukuda S, Yamada Y, Sasaki T, Okamoto H, Kuwabara T, Orii T. Molecular basis of mucopolysaccharidosis type VII: replacement of Ala619 in beta-glucuronidase with Val. Gene. 1990;89:283–287. doi: 10.1016/0378-1119(90)90019-n. [DOI] [PubMed] [Google Scholar]
- 5.Tomatsu S, Fukuda S, Sukegawa K, Ikedo Y, Yamada S, Yamada Y, Sasaki T, Okamoto H, Kuwahara T, Yamaguchi S. Mucopolysaccharidosis type VII: characterization of mutations and molecular heterogeneity. Am J Hum Genet. 1991;48:89–96. [PMC free article] [PubMed] [Google Scholar]
- 6.Shipley JM, Klinkenberg M, Wu BM, Bachinsky DR, Grubb JH, Sly WS. Mutational analysis of a patient with mucopolysaccharidosis type VII, and identification of pseudogenes. Am J Hum Genet. 1993;52:517–526. [PMC free article] [PubMed] [Google Scholar]
- 7.Wu BM, Sly WS. Mutational studies in a patient with the hydrops fetalis form of mucopolysaccharidosis type VII. Hum Mutat. 1993;2:446–457. doi: 10.1002/humu.1380020605. [DOI] [PubMed] [Google Scholar]
- 8.Vervoort R, Lissens W, Liebaers I. Molecular analysis of a patient with hydrops fetalis caused by beta-glucuronidase deficiency, and evidence for additional pseudogenes. Hum Mutat. 1993;2:443–445. doi: 10.1002/humu.1380020604. [DOI] [PubMed] [Google Scholar]
- 9.Wu BM, Tomatsu S, Fukuda S, Sukegawa K, Orii T, Sly WS. Overexpression rescues the mutant phenotype of L176F mutation causing beta-glucuronidase deficiency mucopolysaccharidosis in two Mennonite siblings. J Biol Chem. 1994;269:23681–23688. [PubMed] [Google Scholar]
- 10.Vervoort R, Islam MR, Sly WS, Zabot MT, Kleijer WJ, Chabas A, Fensom A, Young EP, Liebaers I, Lissens W. Molecular analysis of patients with beta-glucuronidase deficiency presenting as hydrops fetalis or as early mucopolysaccharidosis VII. Am J Hum Genet. 1996;58:457–471. [PMC free article] [PubMed] [Google Scholar]
- 11.Birkenmeier EH, Davisson MT, Beamer WG, Ganschow RE, Vogler CA, Gwynn B, Lyford KA, Maltais LM, Wawrzyniak CJ. Murine mucopolysaccharidosis type VII. Characterization of a mouse with beta-glucuronidase deficiency. J Clin Invest. 1989;83:1258–1266. doi: 10.1172/JCI114010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogler C, Birkenmeier EH, Sly WS, Levy B, Pegors C, Kyle JW, Beamer WG. A murine model of mucopolysaccharidosis VII. Gross and microscopic findings in beta-glucuronidase-deficient mice. Am J Pathol. 1990;136:207–217. [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfe JH, Sands MS. Protocols for gene delivery using cells. In: Lowenstein PR, Enquist LW, editors. Protocols for Gene Transfer in Neuroscience: Towards Gene Therapy of Neurological Disorders. Wiley; Chichester: 1996. pp. 2–45. [Google Scholar]
- 14.Birkenmeier EH, Barker JE, Vogler CA, Kyle JW, Sly WS, Gwynn B, Levy B, Pegors C. Increased life span and correction of metabolic defects in murine mucopolysaccharidosis type VII after syngeneic bone marrow transplantation. Blood. 1991;78:3081–3092. [PubMed] [Google Scholar]
- 15.Sands MS, Barker JE, Vogler C, Levy B, Gwynn B, Galvin N, Sly WS, Birkenmeier E. Treatment of murine mucopolysaccharidosis type VII by syngeneic bone marrow transplantation in neonates. Lab Invest. 1993;68:676–686. [PubMed] [Google Scholar]
- 16.Bastedo L, Sands M, Lambert D, Pisa M, Birkenmeier E, Chang P. Behavioral consequences of bone marrow transplantation in the treatment of murine mucopolysaccharidosis type VII. J Clin Invest. 1994;94:1180–1186. doi: 10.1172/JCI117434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poorthuis BJ, Romme AE, Willemsen R, Wagemaker G. Bone marrow transplantation has a significant effect on enzyme levels and storage of glycosaminoglycans in tissues and in isolated hepatocytes of mucopolysaccharidosis type VII mice. Pediatr Res. 1994;36:187–193. doi: 10.1203/00006450-199408000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Sands MS, Erway LC, Vogler C, Sly WS, Birkenmeier EH. Syngeneic bone marrow transplantation reduces the hearing loss associated with murine mucopolysaccharidosis type VII. Blood. 1995;86:2033–2040. [PubMed] [Google Scholar]
- 19.Vogler C, Sands M, Higgins A, Levy B, Grubb J, Birkenmeier EH, Sly WS. Enzyme replacement with recombinant beta-glucuronidase in the newborn mucopolysaccharidosis type VII mouse. Pediatr Res. 1993;34:837–840. doi: 10.1203/00006450-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 20.Sands MS, Vogler C, Kyle JW, Grubb JH, Levy B, Galvin N, Sly WS, Birkenmeier EH. Enzyme replacement therapy for murine mucopolysaccharidosis type VII. J Clin Invest. 1994;93:2324–2331. doi: 10.1172/JCI117237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogler C, Sands MS, Levy B, Galvin N, Birkenmeier EH, Sly WS. Enzyme replacement with recombinant beta-glucuronidase in murine mucopolysaccharidosis type VII: impact of therapy during the first six weeks of life on subsequent lysosomal storage, growth, and survival. Pediatr Res. 1996;39:1050–1054. doi: 10.1203/00006450-199606000-00019. [DOI] [PubMed] [Google Scholar]
- 22.O’Connor LH, Erway LC, Vogler CA, Sly WS, Nicholes A, Grubb J, Holmberg SW, Levy B, Sands MS. Enzyme replacement therapy for murine mucopolysaccharidosis type VII leads to improvements in behavior and auditory function. J Clin Invest. 1998;101:1394–1400. doi: 10.1172/JCI1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogler C, Levy B, Galvin NJ, Thorpe C, Sands MS, Barker JE, Baty J, Birkenmeier EH, Sly WS. Enzyme replacement in murine mucopolysaccharidosis type VII: neuronal and glial response to beta-glucuronidase requires early initiation of enzyme replacement therapy. Pediatr Res. 1999;45:838–844. doi: 10.1203/00006450-199906000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Soper BW, Pung AW, Vogler CA, Grubb JH, Sly WS, Barker JE. Enzyme replacement therapy improves reproductive performance in mucopolysaccharidosis type VII mice but does not prevent postnatal losses. Pediatr Res. 1999;45:180–186. doi: 10.1203/00006450-199902000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Daly TM, Lorenz RG, Sands MS. Abnormal immune function in vivo in a murine model of lysosomal storage disease. Pediatr Res. 2000;47:757–762. doi: 10.1203/00006450-200006000-00012. [DOI] [PubMed] [Google Scholar]
- 26.Wolfe JH, Sands MS, Barker JE, Gwynn B, Rowe LB, Vogler CA, Birkenmeier EH. Reversal of pathology in murine mucopolysaccharidosis type VII by somatic cell gene transfer. Nature. 1992;360:749–753. doi: 10.1038/360749a0. [DOI] [PubMed] [Google Scholar]
- 27.Moullier P, Bohl D, Heard JM, Danos O. Correction of lysosomal storage in the liver and spleen of MPS VII mice by implantation of genetically modified skin fibroblasts. Nat Genet. 1993;4:154–159. doi: 10.1038/ng0693-154. [DOI] [PubMed] [Google Scholar]
- 28.Marechal V, Naffakh N, Danos O, Heard JM. Disappearance of lysosomal storage in spleen and liver of mucopolysaccharidosis VII mice after transplantation of genetically modified bone marrow cells. Blood. 1993;82:1358–1365. [PubMed] [Google Scholar]
- 29.Taylor RM, Wolfe JH. Decreased lysosomal storage in the adult MPS VII mouse brain in the vicinity of grafts of retroviral vector-corrected fibroblasts secreting high levels of beta-glucuronidase. Nat Med. 1997;3:771–774. doi: 10.1038/nm0797-771. [DOI] [PubMed] [Google Scholar]
- 30.Gao C, Sands MS, Haskins ME, Ponder KP. Delivery of a retroviral vector expressing human beta-glucuronidase to the liver and spleen decreases lysosomal storage in mucopolysaccharidosis VII mice. Mol Ther. 2000;2:233–244. doi: 10.1006/mthe.2000.0121. [DOI] [PubMed] [Google Scholar]
- 31.Ohashi T, Yokoo T, Iizuka S, Kobayashi H, Sly WS, Eto Y. Reduction of lysosomal storage in murine mucopolysaccharidosis type VII by transplantation of normal and genetically modified macrophages. Blood. 2000;95:3631–3633. [PubMed] [Google Scholar]
- 32.Brooks AI, Stein CS, Hughes SM, Heth J, McCray PM, Jr, Sauter SL, Johnston JC, Cory-Slechta DA, Federoff HJ, Davidson BL. Functional correction of established central nervous system deficits in an animal model of lysosomal storage disease with feline immunodeficiency virus-based vectors. Proc Natl Acad Sci USA. 2002;99:6216–6221. doi: 10.1073/pnas.082011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L, Mango RL, Sands MS, Haskins ME, Ellinwood NM, Ponder KP. Evaluation of pathological manifestations of disease in mucopolysaccharidosis VII mice after neonatal hepatic gene therapy. Mol Ther. 2002;6:745–758. doi: 10.1006/mthe.2002.0809. [DOI] [PubMed] [Google Scholar]
- 34.Li T, Davidson BL. Phenotype correction in retinal pigment epithelium in murine mucopolysaccharidosis VII by adenovirus-mediated gene transfer. Proc Natl Acad Sci USA. 1995;92:7700–7704. doi: 10.1073/pnas.92.17.7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohashi T, Watabe K, Uehara K, Sly WS, Vogler C, Eto Y. Adenovirus-mediated gene transfer and expression of human beta-glucuronidase gene in the liver, spleen, and central nervous system in mucopolysaccharidosis type VII mice. Proc Natl Acad Sci USA. 1997;94:1287–1292. doi: 10.1073/pnas.94.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghodsi A, Stein C, Derksen T, Yang G, Anderson RD, Davidson BL. Extensive beta-glucuronidase activity in murine central nervous system after adenovirus-mediated gene transfer to brain. Hum Gene Ther. 1998;9:2331–2340. doi: 10.1089/hum.1998.9.16-2331. [DOI] [PubMed] [Google Scholar]
- 37.Ghodsi A, Stein C, Derksen T, Martins I, Anderson RD, Davidson BL. Systemic hyperosmolality improves beta-glucuronidase distribution and pathology in murine MPS VII brain following intraventricular gene transfer. Exp Neurol. 1999;160:109–116. doi: 10.1006/exnr.1999.7205. [DOI] [PubMed] [Google Scholar]
- 38.Kosuga M, Takahashi S, Sasaki K, Li XK, Fujino M, Hamada H, Suzuki S, Yamada M, Matsuo N, Okuyama T. Adenovirus-mediated gene therapy for mucopolysaccharidosis VII: involvement of cross-correction in wide-spread distribution of the gene products and long-term effects of CTLA-4Ig coexpression. Mol Ther. 2000;1:406–413. doi: 10.1006/mthe.2000.0067. [DOI] [PubMed] [Google Scholar]
- 39.Watson GL, Sayles JN, Chen C, Elliger SS, Elliger CA, Raju NR, Kurtzman GJ, Podsakoff GM. Treatment of lysosomal storage disease in MPS VII mice using a recombinant adeno-associated virus. Gene Ther. 1998;5:1642–1649. doi: 10.1038/sj.gt.3300775. [DOI] [PubMed] [Google Scholar]
- 40.Daly TM, Vogler C, Levy B, Haskins ME, Sands MS. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc Natl Acad Sci USA. 1999;96:2296–2300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daly TM, Okuyama T, Vogler C, Haskins ME, Muzyczka N, Sands MS. Neonatal intramuscular injection with recombinant adeno-associated virus results in prolonged beta-glucuronidase expression in situ and correction of liver pathology in mucopolysaccharidosis type VII mice. Hum Gene Ther. 1999;10:85–94. doi: 10.1089/10430349950019219. [DOI] [PubMed] [Google Scholar]
- 42.Skorupa AF, Fisher KJ, Wilson JM, Parente MK, Wolfe JH. Sustained production of beta-glucuronidase from localized sites after AAV vector gene transfer results in widespread distribution of enzyme and reversal of lysosomal storage lesions in a large volume of brain in mucopolysaccharidosis VII mice. Exp Neurol. 1999;160:17–27. doi: 10.1006/exnr.1999.7176. [DOI] [PubMed] [Google Scholar]
- 43.Sferra TJ, Qu G, McNeely D, Rennard R, Clark KR, Lo WD, Johnson PR. Recombinant adeno-associated virus-mediated correction of lysosomal storage within the central nervous system of the adult mucopolysaccharidosis type VII mouse. Hum Gene Ther. 2000;11:507–519. doi: 10.1089/10430340050015707. [DOI] [PubMed] [Google Scholar]
- 44.Bosch A, Perret E, Desmaris N, Heard JM. Long-term and significant correction of brain lesions in adult mucopolysaccharidosis type VII mice using recombinant AAV vectors. Mol Ther. 2000;1:63–70. doi: 10.1006/mthe.1999.0005. [DOI] [PubMed] [Google Scholar]
- 45.Kakkis ED, McEntee MF, Schmidtchen A, Neufeld EF, Ward DA, Gompf RE, Kania S, Bedolla C, Chien SL, Shull RM. Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem. Mol Med. 1996;58:156–167. doi: 10.1006/bmme.1996.0044. [DOI] [PubMed] [Google Scholar]
- 46.Shull RM, Lu X, McEntee MF, Bright RM, Pepper KA, Kohn DB. Myoblast gene therapy in canine mucopolysaccharidosis. I: abrogation by an immune response to alpha-L-iduronidase. Hum Gene Ther. 1996;7:1595–1603. doi: 10.1089/hum.1996.7.13-1595. [DOI] [PubMed] [Google Scholar]
- 47.Wong AW, He S, Grubb JH, Sly WS, Withers SG. Identification of Glu-540 as the catalytic nucleophile of human beta-glucuronidase using electrospray mass spectrometry. J Biol Chem. 1998;273:34057–34062. doi: 10.1074/jbc.273.51.34057. [DOI] [PubMed] [Google Scholar]
- 48.Islam MR, Tomatsu S, Shah GN, Grubb JH, Jain S, Sly WS. Active site residues of human beta-glucuronidase. Evidence for Glu(540) as the nucleophile and Glu(451) as the acid-base residue. J Biol Chem. 1999;274:23451–23455. doi: 10.1074/jbc.274.33.23451. [DOI] [PubMed] [Google Scholar]
- 49.Sly WS, Vogler C, Grubb JH, Zhou M, Jiang J, Zhou XY, Tomatsu S, Bi Y, Snella EM. Active site mutant transgene confers tolerance to human beta-glucuronidase without affecting the phenotype of MPS VII mice. Proc Natl Acad Sci USA. 2001;98:2205–2210. doi: 10.1073/pnas.051623698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sands MS, Vogler C, Torrey A, Levy B, Gwynn B, Grubb J, Sly WS, Birkenmeier EH. Murine mucopolysaccharidosis type VII: long term therapeutic effects of enzyme replacement and enzyme replacement followed by bone marrow transplantation. J Clin Invest. 1997;99:1596–1605. doi: 10.1172/JCI119322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crawley AC, Niedzielski KH, Isaac EL, Davey RC, Byers S, Hopwood JJ. Enzyme replacement therapy from birth in a feline model of mucopolysaccharidosis type VI. J Clin Invest. 1997;99:651–662. doi: 10.1172/JCI119208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kakkis ED, Schuchman E, He X, Wan Q, Kania S, Wiemelt S, Hasson CW, O’Malley T, Weil MA, Aguirre GA, Brown DE, Haskins ME. Enzyme replacement therapy in feline mucopolysaccharidosis I. Mol. Genet Metab. 2001;72:199–208. doi: 10.1006/mgme.2000.3140. [DOI] [PubMed] [Google Scholar]
- 53.Tripathy SK, Black HB, Goldwasser E, Leiden JM. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med. 1996;2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 54.Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, Fanselow MS, Suzuki K, Vanier MT, Neufeld EF. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc Natl Acad Sci USA. 1999;96:14505–14510. doi: 10.1073/pnas.96.25.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jain S, Drendel WB, Chen ZW, Mathews FS, Sly WS, Grubb JH. Structure of human beta-glucuronidase reveals candidate lysosomal targeting and active-site motifs. Nat Struct Biol. 1996;3:375–381. doi: 10.1038/nsb0496-375. [DOI] [PubMed] [Google Scholar]
- 56.Wagner TE, Hoppe PC, Jollick JD, Scholl DR, Hodinka RL, Gault JB. Microinjection of a rabbit beta-globin gene into zygotes and its subsequent expression in adult mice and their offspring. Proc Natl Acad Sci USA. 1981;78:6376–6380. doi: 10.1073/pnas.78.10.6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glaser JH, Sly WS. Beta-glucuronidase deficiency mucopolysaccharidosis: methods for enzymatic diagnosis. J Lab Clin Med. 1973;82:969–977. [PubMed] [Google Scholar]
- 58.Peterson GL. Review of the Folin phenol protein quantitation method of Lowry, Rosebrough, Farr and Randall. Anal Biochem. 1979;100:201–220. doi: 10.1016/0003-2697(79)90222-7. [DOI] [PubMed] [Google Scholar]
- 59.Bjornsson S. Simultaneous preparation and quantitation of proteoglycans by precipitation with alcian blue. Anal Biochem. 1993;210:282–291. doi: 10.1006/abio.1993.1197. [DOI] [PubMed] [Google Scholar]
- 60.Whitley CB, Ridnour MD, Draper KA, Dutton CM, Neglia JP. Diagnostic test for mucopolysaccharidosis. I Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin Chem. 1989;35:374–379. [PubMed] [Google Scholar]