

Abstract
Vertebrate polyadenylation sites are identified by the AAUAAA signal and by GU-rich sequences downstream of the cleavage site. These are recognized by a heterotrimeric protein complex (CstF) through its 64 kDa subunit (CstF-64); the strength of this interaction affects the efficiency of poly(A) site utilization. We present the structure of the RNA-binding domain of CstF-64 containing an RNA recognition motif (RRM) augmented by N- and C-terminal helices. The C-terminal helix unfolds upon RNA binding and extends into the hinge domain where interactions with factors responsible for assembly of the polyadenylation complex occur. We propose that this conformational change initiates assembly. Consecutive Us are required for a strong CstF–GU interaction and we show how UU dinucleotides are recognized. Contacts outside the UU pocket fine tune the protein–RNA interaction and provide different affinities for distinct GU-rich elements. The protein–RNA interface remains mobile, most likely a requirement to bind many GU-rich sequences and yet discriminate against other RNAs. The structural distinction between sequences that form stable and unstable complexes provides an operational distinction between weakly and strongly processed poly(A) sites.
Keywords: nuclear magnetic resonance/polyadenylation/RNA-binding proteins/RNA processing/RNA recognition
Introduction
Maturation of mRNA precursors is a constitutive process for most transcripts but is also a major regulatory event in eukaryotic cells, producing multiple isoforms of the same mRNA by alternative processing. Alternative splicing has long been recognized as a major source of diversity in gene expression, while polyadenylation has been considered largely constitutive. However, ∼30% of all human mRNAs contain alternative polyadenylation signals (Beaudoing et al., 2000). Furthermore, an increasing number of developmental and differentiation decisions are now known to be executed by alternative polyadenylation of the same mRNA (Barabino and Keller, 1999; Zhao et al., 1999). Differential usage of alternative poly(A) sites produces mature transcripts with distinctive 3′-UTRs and gene expression profiles and, sometimes, even different coding sequences.
In polyadenylation, as in splicing, the efficiency with which different processing sites are utilized is controlled by regulatory cis-acting RNA elements and their interactions with trans-acting protein factors. In some cases, of which U1A is the best understood example (Boelens et al., 1993), the timing and efficiency of polyadenylation decisions are controlled by gene-specific factors and their interaction with specific RNA elements and with the polyadenylation machinery. More puzzling are situations where poly(A) site selection is controlled instead by altering the intracellular levels of general processing factors such as CstF-64 (Takagaki et al., 1996; Colgan and Manley, 1997; Takagaki and Manley, 1998). Despite its obvious importance, structural and mechanistic insight on how alternative mRNA processing decisions are executed remains sparse.
In higher eukaryotes, the core polyadenylation signal comprises two elements in addition to the site where the mRNA is cleaved [often a CpA dinucleotide (Figure 1)]: the highly conserved AAUAAA hexamer 10–30 nucleotides upstream of the cleavage site, and a more diffuse GU-rich downstream sequence element (DSE) (Wahle and Keller, 1996; Colgan and Manley, 1997). These RNA sequences are specifically recognized by two protein complexes that assemble onto the pre-mRNA to form the cleavage complex (Figure 1, top). The poly(A) signal (AAUAAA) is recognized by the cleavage and polyadenylation specificity factor (CPSF) through its 160 kDa component (CPSF-160). The DSE is recognized by the heterotrimeric cleavage and stimulation factor (CstF) through its 64 kDa subunit CstF-64 (Beyer et al., 1997; Takagaki and Manley, 1997). Both CPSF and CstF bind RNA weakly, but form a strong cooperative complex when bound together to the same pre-mRNA (Bienroth et al., 1993). Enzymatic factors responsible for the phosphodiesterase and polymerase activities (CF Im, CF IIm, PAP etc.) are then recruited to the CstF–CPSF–pre-mRNA complex. Because the AAUAAA sequence is very highly conserved, the relative strength of competing poly(A) sites is defined primarily by the distance between cis-acting sequences (AAUAAA and DSE) and the affinity of the DSE–CstF interaction. However, how the affinity and positioning of the CstF–pre-mRNA interaction regulate assembly of the 3′-end processing machinery remains unclear.

Fig. 1. Composition of the polyadenylation complex in higher eukaryotes. The poly(A) signal is recognized by the heterotetramer CPSF, while downstream regulatory GU-rich sequences are recognized by the heterotrimeric protein complex CstF. CPSF and CstF interact with each other when bound to the pre-mRNA and recruit enzymatic and regulatory components (PAP, CFI, CFII, etc.) to this cleavage complex to form the functional polyadenylation complex (adapted from Shatkin and Manley, 2000). The domain structure of CstF-64 is shown in the lower part.
In addition to recognizing the pre-mRNA and each other, CPSF and CstF also link polyadenylation with other RNA processing events and with transcription. For example, CstF-64 binds PC4, a factor involved in transcriptional termination, and symplekin, a probable architectural component of the polyadenylation apparatus (Takagaki and Manley, 2000; Calvo and Manley, 2001). CstF-77 mediates the interaction between CstF and CPSF and also interacts with CstF-50, which in turn binds to the CTD of RNA polymerase II and to the DNA repair factor BARD1 (Kleiman and Manley, 1999, 2001). At least some of the polyadenylation factors are recruited to the transcription machinery on the promoter (Dantonel et al., 1997) and a functional poly(A) signal is necessary for transcriptional termination (Proudfoot et al., 2002).
At present there is little structural information on any of the components of the polyadenylation apparatus or their RNA and protein interactions, the exceptions being the catalytic domain of poly(A) polymerase (Bard et al., 2000; Martin et al., 2000) and the U1A system (Varani et al., 2000). As a first step towards characterizing the polyadenylation reaction and its regulation at the structural level, we have studied how CstF-64 binds G/U RNA DSEs. CstF-64 is a multidomain protein (Figure 1, bottom) that is both necessary and sufficient for DSE recognition by CstF (Beyer et al., 1997; Takagaki and Manley, 1997). Immediately following the RNA-binding region at the N-terminus of the protein is the hinge domain responsible for the interaction with CstF-77 (Hatton et al., 2000) and with symplekin (Takagaki and Manley, 2000). The C-terminal half of the protein contains two Pro/Gly-rich domains separated by 11 repeats of the MEARA sequence proposed to form a long α-helix (Takagaki et al., 1992). The final 100 amino acids bind the transcriptional coactivator PC4 (Calvo and Manley, 2001). Thus, recognizable protein domains are separated by regions of low sequence complexity; CstF-64 very likely possesses a ‘bead-on-a-string’ structure. Because of this structural characteristic and because different interactions of CstF-64 map to specific regions of the protein, it is possible to study different domains of CstF-64 and their interactions in isolation. Therefore we have characterized the structure and RNA interaction of the RNA-binding domain of CstF-64 using NMR. Because recognition of G/U-rich elements by CstF-64 is an early event during assembly of the cleavage and polyadenylation complex and its regulation, the data presented here also provide insight into how polyadenylation is executed and regulated.
Results
Structure of CstF-64 N-terminal RNA recognition motif
The solution structure of the N-terminal RNA-binding domain human CstF-64 was determined by NMR as described in Materials and methods. Experimental and structural statistics are summarized in Table I and a superposition of converged structures is displayed in Figure 2A. The domain adopts the classical RNA recognition motif (RRM) fold (Nagai et al., 1990; Varani and Nagai, 1998) augmented by two additional features. A long C-terminal α-helix (three turns) lies across the β-sheet while a shorter N-terminal helix (two turns) precedes the first β-strand. The first nine and last six residues of the construct are unstructured. The protein is highly stable. For example, the hydroxyl proton of Tyr36 is observable even at pH 6.0 and 300 K, providing a large number of experimental restraints.
Table I. Experimental NMR and structural statistics for CstF-64(1–111).
| Experimental restraints | |
|---|---|
| Distance restraints | |
| Intra | 312 |
| Short | 301 |
| Medium | 275 |
| Long | 597 |
| Total | 1486 |
| Angular constraintsa | |
| φ | 54 |
| ψ | 61 |
| Total | 115 |
| Structure statistics | |
| DYANA target functionb | 0.47–1.21 |
| R.m.s.d. (residues 12–104)c | |
| Backbone | 0.57 ± 0.11 Å |
| Side-chains | 1.39 ± 0.12 Å |
aObtained from Cα, Cβ and C′ chemical shifts using the program TALOS.
bMinimum and maximum values within 20 converged structures.
cPairwise among the 20 converged structures.
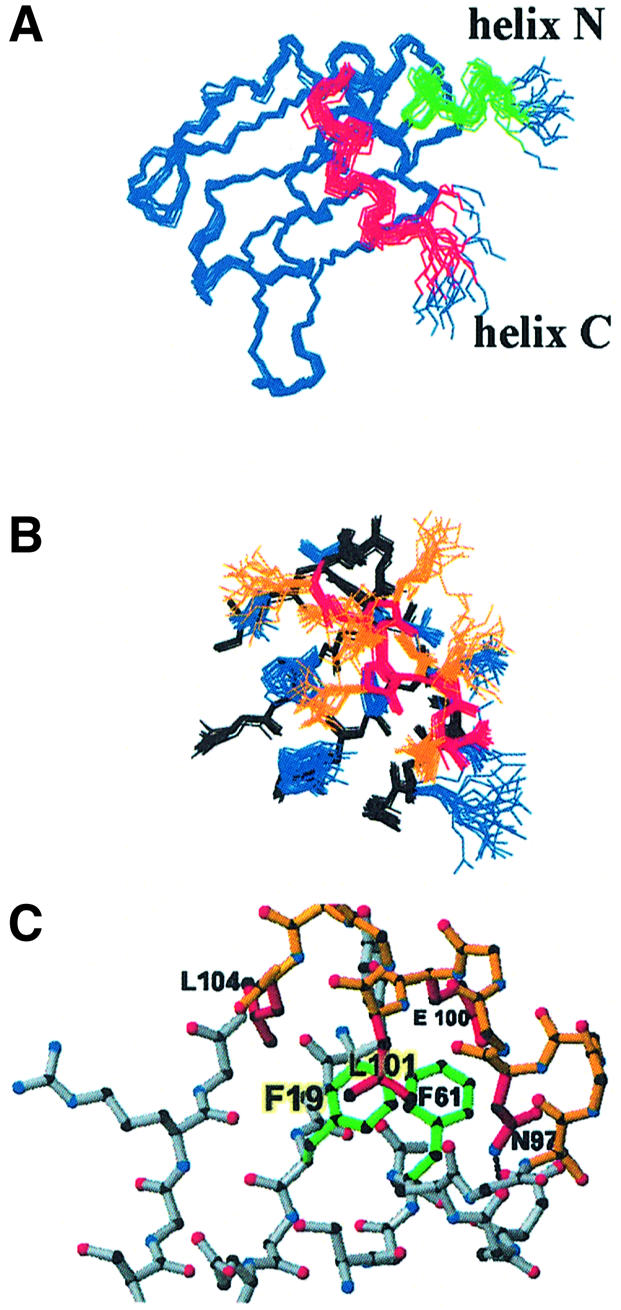
Fig. 2. (A) Structure of the N-terminal domain of human CstF-64 (20 best converged structures). The protein forms an anomalous RRM augmented by two α-helices: helix N (green) and helix C (red). (B) Side-chain packing at the interface between the β-sheet (backbone black; side-chains blue) and helix C (backbone red; side-chains orange). (C) Helix C is stabilized by hydrophobic contacts between the side-chains of Glu100, Leu101 and Leu104 (in helix C) with Phe19 and Phe61 (in RNP2 and RNP1) and by a hydrogen bond between Asn97 (side-chain) and Asn91 (backbone). The figures were generated using MOLMOL (Koradi et al., 1996).
The short α-helix at the N-terminus of the protein is reminiscent of the shorter 310 helix observed at the N-terminus of the first RRM of human hnRNP A1 (Shamoo et al., 1997; Xu et al., 1997). The CstF-64 N-terminal helix has few contacts with the rest of the domain and its formation is probably driven by intrinsic residue propensities. In contrast, helix C (beginning at Ser94 and ending at Gly105) runs nearly perpendicular to the main β-sheet, masking the RNA recognition surface (Figure 2A). Hydrophobic residues within this helix (Leu101 and Leu104) and the side-chain of Glu100 form a small hydrophobic core with the aromatic residues within the RNP1 and RNP2 motifs (Phe19 and Phe61) (Figure 2B and C). Helix C is further stabilized by a hydrogen bond between the side-chain amide of Asn97, in the first turn of the helix, and the backbone carbonyl oxygen of Asn91. Each of these residues is conserved in vertebrates at least and conservatively substituted in Drosophila melanogaster as well (although not in plants and yeast), suggesting very strongly that helix C is a conserved structural feature at least for vertebrate CstF-64 proteins (Figure 3). These positions within CstF-64 have not been probed by site-directed mutagenesis with respect to the RNA binding activity of the protein or its function; our structure identifies these residues as interesting targets for future studies.

Fig. 3. Sequence alignment for the C-terminus of the RNA-binding domain of CstF-64. Helix C is absolutely conserved in all vertebrates and residues positioning the helix with respect to the RRM (Asn91, Asn97, Glu100, Leu101 and Leu104) are also conserved or conservatively substituted in D.melanogaster. Sequence conservation is lost in plants and fungi.
The presence of a C-terminal helix in an RRM was first reported for U1A (Avis et al., 1996) and the C-terminus of several RRM proteins folds into a helical conformation upon RNA/DNA binding: Pab (Deo et al., 1999), Sxl (Handa et al., 1999), HuD (Wang and Tanaka-Hall, 2001), nucleolin (Allain et al., 2000) and hnRNP A1 (Ding et al., 1999). CstF-64 resembles U1A and not these other proteins, but the interaction between the C-terminal helices and the β-sheet differs between CstF-64 and U1A (Figure 4). In U1A, helix C mainly contacts the edges of the β-sheet; in CstF-64, it is perpendicular to the RRM. Most importantly, aromatic residues of RNP1 and RNP2 are considerably more buried in CstF-64 than in U1A. The implications of this result for RNA binding are discussed later.
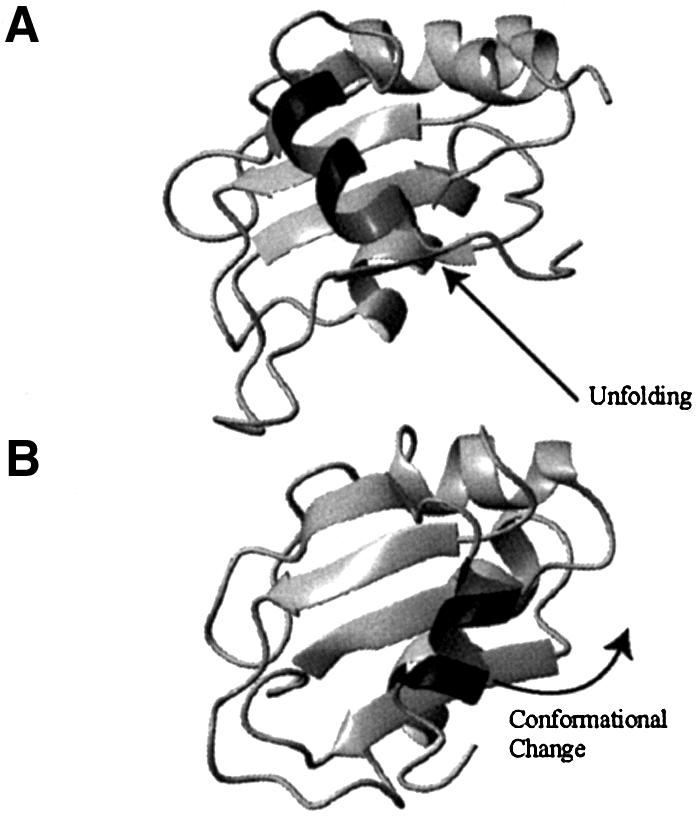
Fig. 4. The orientation of the C-terminal helices with respect to the β-sheet differ between CstF-64 (top) and U1A (bottom) proteins, and between their free and RNA-bound conformations. (A) The CstF-64 helix C (dark grey) lies almost perpendicular to the β-sheet strands and unfolds upon RNA binding. (B) In contrast, the U1A helix C is positioned at the edge of the β-sheet and changes its position without unfolding upon RNA binding.
RNA recognition
CstF-64 plays a key role in polyadenylation by recognizing the DSEs. However, these sequences lack strong sequence conservation, being simply characterized by an abundance of Gs and Us. Therefore CstF-64 must somehow bind a class of sequences without strong preference for any particular sequence, and yet must also discriminate effectively against other RNAs. Thus, CstF-64 appears to provide an example of RNA recognition distinct from that of other well studied members of the superfamily (U1A, Pabp or Sxl) that bind a distinctive sequence with high selectivity. Consistent with the functional requirements, in vitro selection experiments have demonstrated that CstF-64 binds a wide range of GU-rich sequences with comparable affinity, but not G/A or C repeats for example, yet no unique highly specific target sequence could be identified (Beyer et al., 1997; Takagaki and Manley, 1997). A similar study of the complete CstF complex also identified U-rich stretches ubiquitously among sequences selected to bind with high affinity, and GU dinucleotides as lower-efficiency ligands (Beyer et al., 1997; Takagaki and Manley, 1997). How does CstF-64 recognize sequences that are rich in Us and Gs without requiring a strict consensus, and yet discriminate so effectively against other RNAs? To address this question, we elected to compare a number of GU-rich sequences based on the results of the selection experiments. A compilation of all sequences studied is provided in Table II.
Table II. G/U-rich RNA sequences studied in the present work.
| No. | Sequence | Size | Affinity |
|---|---|---|---|
| 1 | GUGUGUG | 7 | Weak |
| 2 | GUGUGUGU | 8 | Weak |
| 3 | UGUGUGUU | 8 | Strong |
| 4 | GUGUGUGUUG | 10 | Strong |
| 5 | GUGUGUGUUUG | 11 | Strong |
| 6 | GUGUGUGUUUUG | 12 | Strong |
| 7 | GUUGUGUGUGUUG | 13 | Strong |
The annotation on binding affinity is based on the NMR observations reported here.
The NMR experiments confirmed that the RNA recognition surface of CstF-64 is the β-sheet (Figure 5) and allowed us to divide the sequences studied into two groups. The first group includes sequences containing exclusively GU repeats which tend to bind CstF-64 with lower affinity (µM) (Takagaki and Manley, 1997), i.e. (GU)6G and (GU)7. The second group contains at least a pair of consecutive Us within the GU-rich repeat and bind RNA tighter. The presence or absence of two or more consecutive uracils has a remarkable effect on CstF-64 binding. For example, GUGUGUGU belongs to the ‘weak’ class and titrates in the NMR fast-exchange regime, resulting in dissociation that is faster than ∼10–3 s. In contrast, UGUGUGUU (very similar to the previous sequence except for the presence of two consecutive Us at the 3′ end) forms a much more stable complex with off rates slow enough to allow the observation of two sets of signals for free and bound protein during the titration (Figure 5). The NMR spectra of the first class of complexes display minor changes scattered across the entire RNA-binding surface, suggesting diffuse and perhaps non-specific binding. Members of the second class of sequences induce much larger changes in the position of the NMR signals as well as in their linewidth upon binding (Figure 6).

Fig. 5. [1H–15N]HSQC spectra of free CstF-64 (black) superimposed on spectra of 1:1 complexes of the same domain bound to (A) (GU)4 (red) and (B) (GU)4UG (red). Chemical shift changes upon complex formation are shown for the two complexes in (C) and (D), respectively. (E) Low-affinity sequences form complexes with fast off rates on the NMR chemical shift timescale; only one set of signals migrating from free to complex chemical shift is observed during titrations (highlighted in green). (F) High-affinity complexes with much slower off rates display instead two sets of signals at substoichiometric protein-to-RNA ratios.

Fig. 6. Binding of GU-rich RNAs containing at least two consecutive uracils produces large changes in the CstF-64 NMR spectra. (A) Chemical shift changes mapped onto the CstF-64 structure: amino acids showing the largest changes (>200 Hz) are in red, while less strongly perturbed sites (>100 Hz) are in green. (B) RNA binding leads to changes in the linewidth of certain regions of the CstF-64 NMR spectrum: amides whose signals become sharper are coloured green, while amino acids whose signals broaden upon RNA binding are identified in red (larger effects) and orange.
When RNA sequences from the second class bind CstF-64, the entire helix C (residues 94 onwards) displays sharper linewidths as it unfolds (see below); at the same time most of the β-sheet signals exhibit broad lines in the complex (Figure 6B). Broadening is particularly severe for clusters of residues in strands β2 and β4 (e.g. 45–48, 54, 57–59) and in the loop between β4 and the C-terminal helix (88, 91, 93 and 94). It undoubtedly reflects local motion in the microsecond to millisecond timescale that is only present in the complex and not in the free protein. Neither line broadening nor helix C unfolding was observed in the complexes with the first two sequences.
The dynamic behaviour of CstF-64 is unusual. For example, in U1A quenching of motion occurred upon binding (Mittermaier et al., 1999). However, it may be a functionally important characteristic; it may be critical for the ability of CstF-64 to recognize a wide range of sequences rich in Us and Gs without specifically binding a single well defined sequence or consensus. Slow binding kinetics indicative of tight and specific binding are also uncommon in single-RRM proteins, except for U1A (Allain et al., 1996). The RNA complexes of RRMs derived from hnRNP D0 (Nagata et al., 1999), U2AF65 (Ito et al., 1999) and HuC (Inoue et al., 2000) all show fast exchange behaviour, consistent with complexes with short lifetimes. Stable complexes with longer lifetimes are achieved in many cases by cooperative binding of two RRMs to single-stranded RNA–DNA (e.g Sxl, Pabp, HuD and HnRNP A1). The single RRM from CstF-64 binds G/U-rich sequences tightly enough to display slow kinetics, unlike all other known examples of single RRMs binding to single-stranded RNA.
Taken together, our results strongly suggest that CstF-64 has a high-affinity binding pocket that specifically recognizes a pair of consecutive uracils. Nucleotides flanking the UU anchor point contact other regions of the protein surface while retaining significant mobility on the microsecond–millisecond timescale. Those other contacts fine tune the protein–RNA interaction and provide different affinities for distinct GU-rich elements. These observations have several implications for the analysis of the intrinsic strength of different poly(A) sites (see below).
Recognition of UU dinucleotides by the CstF-64 RRM
The NMR and binding data discussed above highlight the importance of UU recognition by CstF-64. Therefore dissecting how the dinucleotide is recognized by CstF-64 is important to understanding how the protein binds RNA. However, the broadened NMR spectra, a reflection of the diffuse specificity of this protein, have so far made it impossible to observe many intermolecular nuclear Overhauser effects (NOEs) at the protein–RNA interface and thereby to determine the structure of the complex, despite the exploration of a wide range of conditions and temperatures. Fortunately, we were able to use the remarkable sequence and structural homology between CstF-64 and HuD to define the UU binding pocket. HuDs are neuronal expressed proteins that bind AU-rich elements within the 3′-UTR of certain mRNAs using the first two of three highly conserved RRMs, and affect the stability and half-lives of such mRNAs (Chen and Shyu, 1995).
The superposition of CstF-64 (1–111) on RRM2 of HuD (Wang and Tanaka-Hall, 2001) reveals how similar these proteins are (Figure 7A), even in the orientation of the variable loops, with one significant exception. The C-terminal helix of CstF-64 (green) is located exactly where the uracil bases are placed in the HuD–c-fos complex. In the HuD complex, uracil bases make several base-selective intermolecular hydrogen bonds with the side-chains of Asn126, Arg155 and Lys201 and the backbone of Ala203 (Figure 7B). Arg46 in CstF-64 is equivalent to Arg155 in HuD and is likely to play a similar role in the recognition of the 3′-uracil, while Ser17 is analogous to Asn126 in HuD. The aromatic side-chains from RNP-1 and RNP-2 that position helix C in CstF-64 (Phe19 and Phe61) have the same conformation as Tyr128 and Phe170 in the HuD complex (Figure 7B). Since these two amino acids in HuD form intermolecular stacking interactions with the two uracils, Phe19 and Phe61 appear to be prepositioned in CstF-64 for RNA binding by the long-range interactions with helix C. Asn91 in CstF-64 makes a very important hydrogen bond with the Asn97 side-chain within the helix. This interaction is lost upon RNA binding because of the unfolding of the helix, allowing base-specific contacts analogous to those observed in the HuD complex to be formed instead.

Fig. 7. (A) Superposition of the CstF-64 structure (red) with the HuD–c-fos complex (Wang and Tanaka-Hall, 2001) (blue). Helix C of CstF-64 (green) covers the site occupied by Ura3 and Ura4 (yellow) in the HuD complex. (B) A detailed view of the RNA binding pocket showing the high degree of similarity, even in side-chain orientation, of residues in HuD (blue) responsible for RNA (yellow) recognition and equivalent residues in CstF-64 (red).
Having identified the UU binding pocket, we used the homology between HuD and CstF-64 to build a model of the complex between CstF-64 and a UU dinucleotide. The structure of the model was calculated (see Materials and methods) by combining our NMR data on the CstF-64 complex with putative intermolecular interactions identified from the sequence and structural homology between HuD and CstF-64. A superposition of 10 calculated model structures is shown in Figure 8. According to our model, the 5′-uracil is recognized specifically by base-selective hydrogen bonds between its O4 carbonyl and the Ser17 side-chain and between its O2 carbonyl and the Arg46 side-chain. Together, these two interactions discriminate against cytosine; G and A are also discriminated against on the basis of their different size in addition to those hydrogen bonds. In the HuD–c-fos structure, the NH of the equivalent uracil base forms a water-mediated hydrogen bond; the model would accommodate this feature as well. Concerning the second position, the equivalent uracil in HuD–c-fos is specifically recognized by hydrogen bonds between the Lys201 side-chain and the O4 carbonyl group and by a main-chain Ala203 carbonyl to base NH contact. In our model, Asn91 in CstF-64 (once its side-chain is released following unfolding of helix C) plays the same role as Lys201 in HuD by recognizing the O4 carbonyl, while an equivalent main-chain contact is provided to the uracil NH. Cytosine and adenine bases would not be able to form equivalent hydrogen bonds; guanine could form the same hydrogen bonds but would be discriminated on the basis of its different size. A reorganization of the protein–RNA interface and a subsequent loss in affinity would be required to accommodate G instead of U at this position.
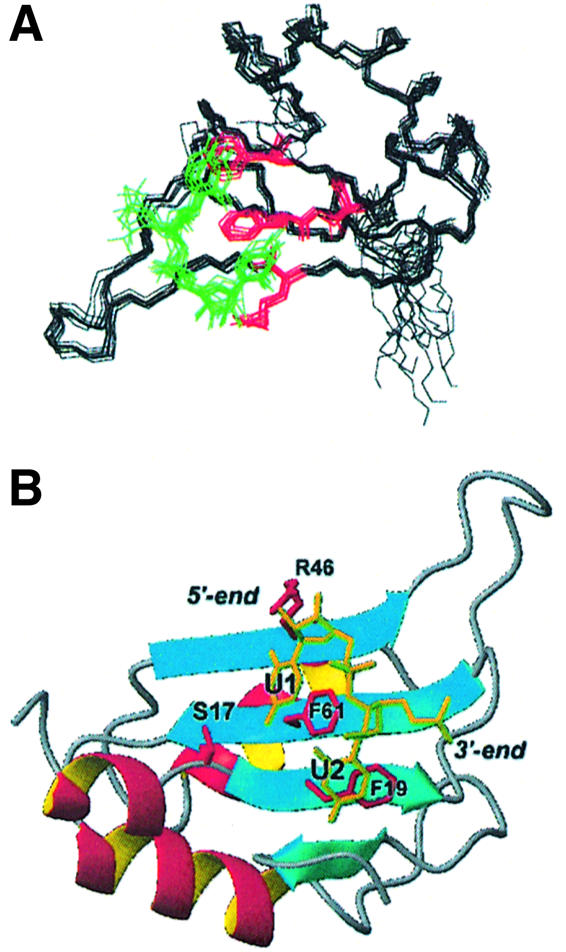
Fig. 8. Model of the CstF-64 RNA complex based on the present NMR analysis of the CstF-64 complexes and the homology with HuD. (A) Superposition of backbone traces for 10 calculated structures: the protein backbone is black, the side-chains at the protein–RNA interface are red and the UU dinucleotide is yellow. (B) Ribbon representation of one of the converged structures, showing explicitly the side-chains of residues involved in base-specific contacts.
Discussion
We have determined the structure of the CstF-64 RNA-binding domain and studied its interaction with GU-rich sequences mimicking downstream polyadenylation regulatory elements. Binding of CstF-64 to these GU-rich elements defines a distinct example of recognition from all other well studied cases where RRM proteins bind a well defined sequence. However, it is also distinct from non-specific binding, since the protein discriminates effectively between GU-rich and random sequence RNA. The observation of a conformational change at the C-terminus of the domain and the different binding and kinetic characteristics of complexes involving different GU-rich sequences are most informative of the mechanism by which polyadenylation is regulated.
CstF-64 binding to GU-rich sequences represents a new example of RNA recognition
The function of CstF-64 requires it to bind sequences characterized only by a preponderance of Gs and Us, and yet to discriminate effectively against all other RNAs. By comparing a number of RNA sequences characterized by different binding affinity, we have shown that the protein achieves this distinct specificity profile through a binding pocket for UU dinucleotides highly homologous to the HuD–UU interface (Wang and Tanaka-Hall, 2001). Contacts outside this pocket fine tune the protein–RNA interaction and provide different affinities for distinct GU-rich elements, and therefore are likely to be critical for the regulatory activity of this protein. However, unlike HuD, CstF-64 lacks a second RRM that helps define a tight and specific complex. Unlike all other single RRMs studied so far (of which U1A is an exception), CstF-64 binding to G/U-rich sequences leads to the formation of stable complexes with slow off rates. Remarkably, the protein retains extensive mobility on the microsecond–millisecond timescale at its RNA interface, though not in its RNA-free state. This is the opposite of what we observed for U1A where slow motional processes at the RNA interface disappeared upon RNA binding, leading to a rigid (and highly specific) interface (Mittermaier et al., 1999). Motion at the interface is very likely to be functionally important by providing CstF-64 with the ability to recognize a wide range of sequences rich in Us and Gs without specifically binding a single well defined sequence or consensus. Therefore it is intrinsic to its functional requirement to bind all DSEs and yet to discriminate against non-GU-rich sequences.
Does the conformational change at the C-terminus of CstF-64 provide a signal for assembly of the polyadenylation apparatus?
The location of helix C within CstF-64 suggests that unfolding on G/U-rich RNA binding may play a role in controlling formation of the precleavage complex. Both CPSF and CstF are preassembled and recruited to the CTD early during transcription (Dantonel et al., 1997). These two protein complexes must somehow be ‘activated’ to a conformation capable of interacting with each other and then to recruit cleavage factors and poly(A) polymerase once the poly(A) site is transcribed. The hinge domain of CstF-64 immediately follows the N-terminal RRM and includes helix C; it interacts with symplekin, an architectural component of the polyadenylation machinery, and with CstF-77 (Hatton et al., 2000). This last protein interacts with CPSF (Takagaki and Manley, 2000). If helix C is also present within the assembled CstF complex, as is very likely given the high stability of the structure presented here, then it must unfold for G/U binding to occur and before the precleavage complex is assembled. The conformational change could affect the CstF-64 interaction with CstF-77 and symplekin, and potentially the CstF–CPSF interaction as well. In this model, the C-terminal helix would act as a molecular switch in gating assembly of the polyadenylation complex by communicating the signal that RNA is bound to CPSF and to the transcription machinery. Whether the signal is transmitted by a rigid-body-like motion or the maturation of the cleavage complex is promoted by an early destabilization event, helix C unfolding upon RNA binding is a good candidate to trigger assembly.
Consistent with the essential functional role suggested by our model, helix C is absolutely conserved in vertebrates, and residues positioning helix C with respect to the RRM (Asn91, Asn97, Glu100, Leu101 and Leu104) are also conserved or conservatively substituted in D.melanogaster (Figure 3). Although sequence conservation does not extend to plants and fungi, it is worth remarking that the mechanism of polyadenylation differs somewhat between yeast and vertebrates and involves differences in both cis-acting sequences and trans-acting factors (Wahle and Keller, 1996; Zhao et al., 1999).
The presence of a helical appendix to the RRM was first considered an anomaly (Howe et al., 1994; Avis et al., 1996), but every RRM studied so far is augmented by an α-helix at its C-terminus, usually in the presence of RNA. However, the helices play different structural and functional roles in different proteins. The C-terminal helix of CstF-64 does not participate directly in RNA recognition but rather prevents it by occluding the RRM surface. In contrast, the C-terminal α-helix of U1A does not unfold and is repositioned upon RNA binding through a small number of changes in the protein backbone to form protein–RNA and protein–protein contacts (Allain et al., 1996; Varani et al., 2000). The behaviour of helix C is also completely different from what is observed in Pab, Sxl, HuD and hnRNP A1, where folding of unstructured linkers between neighbouring RRMs to form α-helices, rather than unfolding, is observed upon RNA binding. In these proteins, the α-helical linker regions participate in base-specific contacts and therefore contribute to specific binding both directly and by controlling the topological arrangement of the two RRMs (Sachs and Varani, 2000). In CstF-64, helix C is stably associated with the domain but unfolds upon RNA binding and therefore does not contribute directly to RNA recognition.
CstF-64–DSE interaction and alternative polyadenylation
A substantial number of RNA transcripts in ver tebrates (30%) present alternative polyadenylation sites (Beaudoing et al., 2000). In the IgM-G heavy chain, perhaps the best understood example of regulated alternative polyadenylation (reviewed in Zhao et al., 1999), and in several other well characterized examples, poly(A) site usage is correlated with intracellular levels of CstF-64. The ‘strength’ of a poly(A) site appears to be determined at least in part by the affinity of the CstF64–DSE interaction. Our data show that sequences containing at least two consecutive Us form more stable complexes with longer lifetimes and presented a model of how recognition of UU dinucleotides occur. Analysis of sequences selected to bind CstF-64 confirms that sequences containing multiple successive Us bind more tightly to both CstF and to isolated CstF-64, and tend to be processed more efficiently (Beyer et al., 1997; Takagaki and Manley, 1997; Chao et al., 1999).
In several well studied examples of regulated polyadenylation, weak sites are utilized only at high cellular levels of CstF-64 (Takagaki and Manley, 1998; Chuvpilo et al., 1999). Normally, the majority of CstF-64 is associated into the heterotrimeric CstF complex and recruited to the CTD during transcription. When cellular levels of CstF-64 are increased, free CstF-64 (or free CstF, i.e. the fraction not associated with the elongating polymerase) may saturate even weak GU-rich sequences found in upstream polyadenylation sites, perhaps increasing the chance of the formation of a complex committed to cleavage and transcription termination.
Kinetic competition with transcription may affect polyadenylation site definition
The affinity of the CstF–DSE interaction may control not just the efficiency of poly(A) site usage but also the kinetics of cleavage complex assembly. Surprisingly, 3′-end processing requires 10 s or more (Chao et al., 1999) and is faster for ‘stronger’ than for ‘weaker’ poly(A) signals. According to this result, selection of the weak sites may be inefficient because of the slow assembly rate of the cleavage complex compared with transcriptional elongation rates (Chao et al., 1999). We notice that the organization of transcripts undergoing alternative polyadenylation follows a common pattern: proximal polyadenylation sites are utilized with low efficiency while more distal sites are utilized by default. If assembly is fast enough, the transcriptional machinery might be directed to termination, while assembly at inefficient sites may occur so slowly that transcription has progressed far enough to allow skipping of the polyadenylation site. According to this ‘kinetic model’, weak sites are skipped because the reorganization of the 3′-end processing machinery into a complex ‘committed’ to cleavage is slow compared with transcriptional elongation; in other words, weak sites are kinetically silenced. We suggested that the unfolding transition in helix C might be an early step in the assembly of the cleavage complex, introducing a further kinetic element to regulation. Perhaps the structural change in CstF-64 observed with the high-affinity sequences might speed up the assembly of a functional cleavage complex. Other mechanisms may influence processing efficiency: for example, the presence of transcription pause sites, competition between CstF-64 and other factors binding the pre-mRNA (e.g. hnRNPs) and competition between splicing and polyadenylation. However, kinetic competition between RNA processing and transcription is an attractive model that may be relevant not just to polyadenylation but also to other situations, such as pre-mRNA splicing, where general processing factors necessary for basal processing reactions also play distinct regulatory roles on different sets of mRNAs.
Materials and methods
Protein expression and purification
Full-length human CstF-64 was amplified from a vector kindly provided by Professor J.Manley (Columbia University) and subcloned into the pET-28a vector (Novagen) between the NdeI and NotI restriction sites. The construct corresponding to the N-terminal RRM (amino acids 1–111) was generated by introducing a stop codon in the pET-28a–CstF64 full-length construct using site-directed mutagenesis. Cell cultures [Bl21(DE3) Escherichia coli] were grown in M9 minimal media supplemented with appropriately isotope-labelled NH4Cl and glucose. Cultures were induced with isopropyl-β-d-thiogalactopyranoside (IPTG) during mid-logarithmic phase and harvested 4 h post-induction. Protein purification was by metal chelate affinity chromatography (Amersham-Pharmacia Hi-trap), followed by thrombin (Sigma) cleavage to remove the His tag. The sample was reloaded onto the Ni2+ affinity column following thrombin digestion and the flowthrough was passed through a benzamidine minicolumn (Amersham-Pharmacia Hi-trap) for thrombin removal. The protein was then further purified by anion exchange (Q-Sepharose) and size exclusion chromatography (Amersham-Pharmacia Superdex™ 75). No impurities were detected by SDS gel electrophoresis or mass spectroscopy.
RNA synthesis
All RNA sequences studied (Table II) were prepared by in vitro run-off transcription using T7 RNA polymerase and subsequently purified essentially as described (Price et al., 1998), with the exception of sequence number 3, which was chemically synthesized (Dharmacom). After de-protection following the manufacturer’s instructions, the RNA was purified by gel electrophoresis as for the RNAs prepared in vitro.
NMR spectroscopy
NMR experiments where conducted on Bruker AMX500, DMX600 and AV800 MHz spectrometers equipped with triple resonance probes and gradient units. All the experiments were performed under the same buffer conditions (10 mM phosphate pH 6.0) and at a temperature of 300 K. Backbone Cα, Cβ, C′ and N assignments of CstF-64 (1–111) free and in complex with the RNA (GU)3GUUG sequence were obtained using triple resonance HNCA, HNCO, HN(CO)CA and CBCA(CO)HN experiments (Sattler et al., 1999). Side-chain assignments were completed with an HCCH-COSY experiment. Titration experiments were performed by adding increasing amounts of unlabelled oligonucleotides to 15N-labelled protein and by following the changes in [15N]HSQC protein spectra. Two-dimensional NOESY experiments (in H2O and D2O) and heteronuclear edited three-dimensional (3D) 15N- and 13C-HSQC-NOESYs recorded at 600 MHz at mixing times of 80 ms were used to obtain distance constraints for free CstF-64 (1–111) protein. Two-dimensional NOESY spectra were recorded at 800 MHz for the RNA complex.
Structure calculation
The 3D structure of CstF-64 (1–111) protein was calculated using the program DYANA (Güntert et al., 1997). A total of 1486 relevant NOE-derived distance restraints (upper limits only), corresponding to an average of 14 constraints per residue, were used in the calculation. A statistical analysis of the Cα, Cβ and C′ chemical shifts with the program TALOS (Cornilescu et al., 1999) provided 123 φ and ψ angle restraints that were used in the later stages of the structural refinement. Calculations were performed starting from 40 randomly generated conformers, which were then subjected to a simulated annealing protocol of 5000 torsion angle dynamics (TAD) cycles. Hydrogen atoms were included later, followed by a second low-temperature simulated annealing step of 800 TAD cycles. The protocol showed very good convergence; typically, only two or three of 40 starting structures failed to converge and showed large values of the target function representing deviations from experimental constraints. The 20 structures with the lowest target function were selected to represent the ensemble of converged CstF-64 (1–111) structures. Experimental data and structural statistics are summarized in Table I. The quality of the structure was analysed with the suite of programs PROCHECK-NMR (Laskowski et al., 1996): 83.1% of residues have backbone angles within the most populated regions of the Ramachandran plot, 14.8% are in additional allowed regions, 1.9% are in generously allowed regions and only 0.2% are in disallowed regions.
CstF–UU complex
The >30% identity between CstF-64 and HuD was used in combination with the experimental data to model the CstF–UU interaction based on the HuD–RNA complex structure (Wang and Tanaka-Hall, 2001). Modelling was initiated from the constraint set obtained for RNA-free CstF-64, since the data recorded on protein–RNA complexes demonstrated conclusively that there are no structural changes except at the C-terminal helix. Since the NMR data clearly show that the helix unfolds upon RNA binding, all distances involving the C-terminal helix were removed from the distance constraint set. A set of intermolecular short distances (all 153 restraints corresponding to distances <6 Å) involving protons from U3 and U4 in the HuD complex was generated from the HuD coordinate set. These non-experimental constraints were arbitrarily given more generous upper bounds (30% larger) to account for possible structural differences between HuD and CstF-64. We then recalculated the structure of CstF-64(1–111) bound to a UU dinucleotide following the same simulated annealing protocol used for free CstF-64. No intramolecular distance restraints were used to define the RNA conformation.
Accession codes
Coordinates, experimental restraints and other NMR parameters have been deposited under PDB code 1P1T and BioMagRes accession number 5774.
Acknowledgments
Acknowledgements
We are grateful to Professor James Manley for plasmids and for several discussions on the project. This work was supported by NIH (RO1 GM64440-01A1), the Medical Research Council, the Human Frontier of Science and EMBO (ALTF 42-2000) and by a Marie Curie Fellowship of the European Community human potential programme (HPRMF-CT-2000-00722) to J.M.P.C.
References
- Allain F.H.-T., Gubser,C.C., Howe,P.W.A., Nagai,K., Neuhaus,D. and Varani,G. (1996) Specificity of ribonucleoprotein interaction determined by RNA folding during complex formation. Nature, 380, 646–650. [DOI] [PubMed] [Google Scholar]
- Allain F.H.-T., Gilbert,D.E., Bouvet,P. and Feigon,J. (2000) Solution structure of the two N-terminal RNA-binding domains of nucleolin and NMR study of the interaction with its RNA target. J. Mol. Biol., 303, 227–241. [DOI] [PubMed] [Google Scholar]
- Avis J., Allain,F.H.-T., Howe,P.W.A., Varani,G., Neuhaus,D. and Nagai,K. (1996) Solution structure of the N-terminal RNP domain of U1A protein: the role of C-terminal residues in structure stability and RNA binding. J. Mol. Biol., 257, 398–411. [DOI] [PubMed] [Google Scholar]
- Barabino S.M.L. and Keller,W. (1999) Last but not least: regulated poly(A) tail formation. Cell, 99, 9–11. [DOI] [PubMed] [Google Scholar]
- Bard J., Zhelkovsky,A.M., Helmling,S., Earnest,T.N., Moore,C.L. and Bohm,A. (2000) Structure of yeast poly(A) polymerase alone and in complex with 3′-dATP. Science, 289, 1346–1349. [DOI] [PubMed] [Google Scholar]
- Beaudoing E., Freier,S., Wyatt,J.R., Claverie,J.M. and Gautheret,D. (2000) Patterns of variant polyadenylation signal usage in human genes. Genome Res., 10, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer K., Dandekar,T. and Keller,W. (1997) RNA ligands selected by cleavage stimulation factor contain distinct sequence motifs that function as downstream elements in 3′-end processing of pre-mRNA. J. Biol. Chem., 272, 26769–26779. [DOI] [PubMed] [Google Scholar]
- Bienroth S., Keller,W. and Wahle,E. (1993) Assembly of a processive polyadenylation complex. EMBO J., 12, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens W.C., Jansen,E.J.R., van Venrooij,W.J., Stripecke,R., Mattaj,I.W. and Gunderson,S.I. (1993) The human U1 snRNP-specific U1A protein inhibits polyadenylation of its own pre-mRNA. Cell, 72, 881–892. [DOI] [PubMed] [Google Scholar]
- Calvo O. and Manley,J.L. (2001) Evolutionarily conserved interaction between CtsF-64 and PC4 links transcription, polyadenylation and termination. Mol. Cell, 7, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Chao L., Jamil,A., Kim,S., Huang,L. and Martinson,H.G. (1999) Assembly of the cleavage and polyadenylation apparatus requires about 10 seconds in vivo and is faster for strong than for weak poly(A) sites. Mol. Cell. Biol., 19, 5588–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-Y.A. and Shyu,A.-B. (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- Chuvpilo S. et al. (1999) Alternative polyadenylation events contribute to the induction of NF-ATc in effector T cells. Immunity, 10, 261–269. [DOI] [PubMed] [Google Scholar]
- Colgan D.F. and Manley,J.L. (1997) Mechanism and regulation of mRNA polyadenylation. Genes Dev., 11, 2755–2766. [DOI] [PubMed] [Google Scholar]
- Cornilescu G., Delaglio,F. and Bax,A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR, 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Dantonel J.-C., Murthy,K.G.K., Manley,J.L. and Tora,L. (1997) Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature, 389, 399–402. [DOI] [PubMed] [Google Scholar]
- Deo R.C., Bonanno,J.B., Sonenberg,N. and Burley,S.K. (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell, 98, 835–845. [DOI] [PubMed] [Google Scholar]
- Ding J., Hayashi,M.K., Zhang,Y., Manche,L., Krainer,A.R. and Xu,R.-M. (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev., 13, 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güntert P., Mumenthaler,C. and Wüthrich,K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol., 273, 283–298. [DOI] [PubMed] [Google Scholar]
- Handa N., Nureki,O., Kurimoto,K., Kim,I., Sakamoto,H., Shimura,Y., Muto,Y. and Yokohama,S. (1999) Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature, 398, 579–585. [DOI] [PubMed] [Google Scholar]
- Hatton L.S., Eloranta,J.J., Figureueiredo,L.M., Takagaki,Y., Manley,J.L. and O’Hare,K. (2000) The Drosophila homologue of the 64 kDa subunit of cleavage and stimulation factor interacts with the 77 kDa subunit encoded by the Suppressor of Forked gene. Nucleic Acids Res., 28, 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe P.W.A., Nagai,K., Neuhaus,D. and Varani,G. (1994) NMR studies of U1 snRNA recognition by the N-terminal RNP domain of the human U1A protein. EMBO J., 13, 3873–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M., Muto,Y., Sakamoto,H. and Yokohama,S. (2000) NMR studies on functional structures of the AU-rich element-binding domains of Hu Antigen C. Nucleic Acids Res., 28, 1743–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Muto,Y., Green,M.R. and Yokoyama,S. (1999) Solution structures of the first and second RNA-binding domain of human U2 small nuclear ribonucleoprotein particle auxiliary factor (U2AF65). EMBO J., 18, 4523–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman F.E. and Manley,J.L. (1999) Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science, 285, 1576–1579. [DOI] [PubMed] [Google Scholar]
- Kleiman F.E. and Manley,J.L. (2001) The BARD1–CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell, 104, 743–753. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter,M. and Wuthrich,K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph., 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmann,J.A.C., MacArthur,M.W., Kaptein,R. and Thornton,J.M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR, 8, 477–486. [DOI] [PubMed] [Google Scholar]
- Martin G., Keller,W. and Doublié,S. (2000) Crystal structure of mammalian poly(A) polymerase in complex with an analog of ATP. EMBO J., 19, 4193–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermaier A., Varani,L., Muhandiram,D.R., Kay,L.E. and Varani,G. (1999) Changes in sidechain and backbone dynamics identify determinants of specificity in RNA recognition by human U1A protein. J. Mol. Biol., 294, 967–979. [DOI] [PubMed] [Google Scholar]
- Nagai K., Oubridge,C., Jessen,T.H., Li,J. and Evans,P.R. (1990) Structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature, 348, 515–520. [DOI] [PubMed] [Google Scholar]
- Nagata T., Kurihara,Y., Matsuda,G., Saeki,J.-I., Kohno,T., Yanagida,Y., Ishikawa,F., Uesugi,S. and Katahira,M. (1999) Structure and interactions with RNA of the N-terminal UUAG-specific RNA-binding domain of hnRNP D0. J. Mol. Biol., 287, 221–237. [DOI] [PubMed] [Google Scholar]
- Price S.R., Oubridge,C., Varani,G. and Nagai,K. (1998) Preparation of RNA–protein complexes for X-ray crystallography and NMR. In Smith,C. (ed.), RNA–Protein Interaction: A Practical Approach. Oxford University Press, Oxford, UK, pp. 37–74.
- Proudfoot N.J., Furger,A. and Dye,M.J. (2002) Integrating mRNA processing with transcription. Cell, 108, 501–512. [DOI] [PubMed] [Google Scholar]
- Sachs A. and Varani,G. (2000) Eukaryotic translation initiation: there are (at least) two sides to every story. Nat. Struct. Biol., 7, 356–360. [DOI] [PubMed] [Google Scholar]
- Sattler M., Schleucher,J. and Griesinger,C. (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. NMR Spectrosc., 34, 93–158. [Google Scholar]
- Shamoo Y., Krueger,U., Rice,L.M., Williams,K.R. and Steitz,T.A. (1997) Crystal structure of the two RNA binding domains of human hnRNP A1 at 1.75 Å resolution. Nat. Struct. Biol., 4, 215–222. [DOI] [PubMed] [Google Scholar]
- Shatkin A.J. and Manley,J.L. (2000) The ends of the affair: capping and polyadenylation. Nat. Struct. Biol., 7, 838–842. [DOI] [PubMed] [Google Scholar]
- Takagaki Y. and Manley,J.L. (1997) RNA recognition by the human polyadenylation factor CstF. Mol. Cell. Biol., 17, 3907–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y. and Manley,J.L. (1998) Levels of polyadenylation factor CstF-64 control IgM heavy chain mRNA accumulation and other events associated with B cell differentiation. Mol. Cell, 2, 761–771. [DOI] [PubMed] [Google Scholar]
- Takagaki Y. and Manley,J.L. (2000) Complex protein interactions within the human polyadenylation machinery identify a novel component. Mol. Cell. Biol., 20, 1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y., MacDonald,C.C., Shenk,T. and Manley,J.L. (1992) The human 64-kDa polyadenylation factor contains a ribonucleoprotein-type RNA binding domain and unusual auxiliary motifs. Proc. Natl Acad. Sci. USA, 89, 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y., Seipelt,R.L., Peterson,M.L. and Manley,J.L. (1996) The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell, 87, 941–952. [DOI] [PubMed] [Google Scholar]
- Varani G. and Nagai,K. (1998) RNA recognition by RNP proteins during RNA processing and maturation. Annu. Rev. Biophys. Biomol. Struct., 27, 407–445. [DOI] [PubMed] [Google Scholar]
- Varani L., Gunderson,S., Kay,L.E., Neuhaus,D., Mattaj,I. and Varani,G. (2000) The NMR structure of the 38 kDa RNA–protein complex reveals the basis for cooperativity in inhibition of polyadenylation by human U1A protein. Nat. Struct. Biol., 7, 329–335. [DOI] [PubMed] [Google Scholar]
- Wahle E. and Keller,W. (1996) The biochemistry of polyadenylation. Trends Biochem. Sci., 21, 247–250. [PubMed] [Google Scholar]
- Wang X. and Tanaka-Hall,T. (2001) Structural basis for recognition of AU-rich element RNA by Hu proteins. Nat. Struct. Biol., 8, 141–146. [DOI] [PubMed] [Google Scholar]
- Xu R.-M., Jokhan,L., Cheng,X., Mayeda,A. and Krainer,A.R. (1997) Crystal structure of human UP1, the domain of hnRNP A1 that contains two RNA-recognition motifs. Structure, 5, 559–570. [DOI] [PubMed] [Google Scholar]
- Zhao J., Hyman,L. and Moore,C. (1999) Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation and interrelationship with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev., 63, 405–445. [DOI] [PMC free article] [PubMed] [Google Scholar]