

Abstract
Insulin stimulates glucose uptake in fat and muscle cells via the translocation of the GLUT4 glucose transporter from intracellular storage vesicles to the cell surface. The signaling pathways linking the insulin receptor to GLUT4 translocation in adipocytes involve activation of the Rho family GTPases TC10α and β. We report here the identification of TCGAP, a potential effector for Rho family GTPases. TCGAP consists of N-terminal PX and SH3 domains, a central Rho GAP domain and multiple proline-rich regions in the C-terminus. TCGAP specifically interacts with cdc42 and TC10β through its GAP domain. Although it has GAP activity in vitro, TCGAP is not active as a GAP in intact cells. TCGAP translocates to the plasma membrane in response to insulin in adipocytes. The N-terminal PX domain interacts specifically with phos phatidylinositol-(4,5)-bisphosphate. Overexpression of the full-length and C-terminal fragments of TCGAP inhibits insulin-stimulated glucose uptake and GLUT4 translocation. Thus, TCGAP may act as a downstream effector of TC10 in the regulation of insulin-stimulated glucose transport.
Keywords: cdc42/GLUT4 translocation/insulin/TC10/TCGAP
Introduction
Insulin stimulates glucose transport into muscle and adipocytes by triggering an increase in the exocytosis of the glucose transporter GLUT4 from intracellular vesicles to the plasma membrane (Pessin et al., 1999; Saltiel and Kahn, 2001). Although the cellular dynamics of GLUT4 vesicle trafficking are well described, the signaling pathways linking the insulin receptor to GLUT4 translocation remain largely unresolved. Activation of type Ia phosphatidylinositol (PtdIns) 3-kinase is required but not sufficient for the action of insulin (Saltiel and Pessin, 2002). Recent studies have revealed that a second pathway occurs as a consequence of c-Cbl tyrosine phosphorylation. Cbl and the adaptor protein CAP (c-Cbl-associated protein; Ribon et al., 1998) are recruited to the insulin receptor by APS [adaptor molecules containing pleckstrin homology (PH) and Src-homology 2 (SH2) domains; Liu et al., 2002]. Once tyrosine phosphorylated by the receptor, Cbl can recruit the adaptor protein CrkII to lipid rafts, along with the guanyl nucleotide exchange factor (GEF) C3G (Chiang et al., 2001). C3G, in turn, can activate the Rho family GTPases TC10α and β (Chiang et al., 2002), which reside in lipid rafts due to tandem acylation (Watson et al., 2001). Activation of both the PtdIns 3-kinase and CAP/Cbl/C3G pathways provides synergistic signals in the regulation of glucose transport (Chiang et al., 2001).
Small GTP-binding proteins of the Rho family, such as RhoA, Rac1, cdc42, TC10α and TC10β, play important roles in membrane trafficking, cytoskeletal remodeling, transcriptional regulation and cell growth (reviewed in Tapon and Hall, 1997; Kaibuchi et al., 1999). These GTPases cycle between active GTP-bound and inactive GDP-bound states, regulated by the opposing effects of GEFs, which catalyze the exchange of bound GDP for GTP, and GTPase-activating proteins (GAPs), which increase hydrolysis of bound GTP.
In addition to the sequences responsible for GAP activity, GAPs may also serve as downstream effectors (Bollag and McCormick, 1991; Hall, 1992). For example, n-chimaerin, a Rho GAP mainly expressed in the central nervous system, mediates lamelipodia and filopodia induction (Kozma et al., 1996). Interestingly, an n-chimaerin mutant lacking GAP activity remains capable of producing cytoskeletal rearrangements (Kozma et al., 1996). In contrast, some Rho GAP domain-containing proteins do not possess GAP activity. These GAP domains mediate specific interactions with the Rho protein. For example, the GAP domain of IQGAP2 is devoid of GAP activity, but interacts with Rac1 and cdc42 in a nucleotide-independent manner through its GAP domain (Brill et al., 1996).
We report here the cloning and characterization of a novel multidomain-containing protein, named TCGAP (TC10/cdc42 GTPase activating protein). TCGAP binds to cdc42 and TC10β specifically through its GAP domain, and may play an important role in the regulation of glucose transport by insulin.
Results
Cloning and characterization of TCGAP
We isolated a novel full-length 4 kb cDNA from mouse 3T3-L1 adipocytes. This open reading frame (ORF) started with an ATG codon in a Kozak consensus sequence and encoded a putative protein of 1305 amino acids with a predicted Mr of 140 kDa (Figure 1A). A stop codon was found upstream of the start codon. Protein structural analysis using the SMART program revealed that this cDNA encoded a novel multidomain protein (Figure 1B). In addition to the three blocks conserved among Rho GAPs, the encoded protein exhibits an N-terminal phox homology (PX) domain and an SH3 domain. The entire coding sequence contains 22 proline-rich sequences (PXXP), most of which were found in the C-terminus. The C-terminal half of the protein (residues 560–1305) comprises a proline-/serine-rich region in which the proline and serine content is ∼35%. Interestingly, five PEST motifs (residues 25–83, 579–590, 683–734, 735–752 and 768–795) commonly found in proteins with a short half-life were also found in TCGAP, suggesting that the protein is of low abundance. Multiple tissue northern blot and RT–PCR showed TCGAP highly expressed in brain and testis, and also expressed in WAT and muscle at a low level (see Supplementary figure 1C and D available at The EMBO Journal Online).

Fig. 1. Amino acid sequence, domain structure and RNA expression of TCGAP. (A) Predicted amino acid sequence of TCGAP. (B) Schematic diagram of domains in TCGAP analyzed by the SMART program.
TCGAP exhibits GAP activity in vitro but not in vivo
To examine the GAP activity of TCGAP on Rho family proteins, Purified GST–GTPases were loaded with [γ-32P]GTP and incubated in the presence or absence of the GST–TCGAP/GAP domain fusion protein. As shown in Supplementary figure 2, the isolated GAP domain stimulated the intrinsic GTPase activity of Cdc42 and Rac1, and was less active on RhoA and TC10.
To investigate the GAP activity of the protein in vivo, plasmids encoding hemagglutinin (HA)-tagged RhoA, Rac1, cdc42, TC10α or TC10β were transiently transfected into Cos-1 cells in combination with either a vector control or Myc-tagged full-length TCGAP. The effect of TCGAP on GTP binding of these Rho family GTPases was evaluated by precipitation with GST–rhotekin for RhoA and GST–PAK1-CRIB for Rac1, cdc42, TC10α and TC10β. Surprisingly, the overexpression of full-length TCGAP did not attenuate the GTP-bound fraction of any GTPases, compared with vector control (Supple mentary figure 3). In contrast, overexpression of p50GAP significantly reduced the GTP loading of cdc42, assayed by its precipitation with GST–PAK1 (data not shown). Thus, although it exhibits activity in vitro, TCGAP does not function as a GAP when overexpressed in intact cells.
TCGAP specifically interacts with cdc42 and TC10β
Some GAP proteins, such as IQGAP2, act as downstream effectors of Rho family GTPases (Brill et al., 1996). In these cases, the GAP domain serves as a GTPase recognition motif. To examine the binding specificity of TCGAP toward different Rho family members in vivo, HA-tagged GTPase-deficient mutants of RhoA (V14), Rac1 (V12), cdc42 (Q61L), TC10α (Q67L) or TC10β (Q69L) were transiently transfected into Cos-1 cells in combination with a Myc-tagged full-length or N-terminal fragment (amino acids 1–655) of TCGAP. Lysates were immunoprecipitated with either anti-Myc polyclonal antibody or non-specific IgG as control, and immunoblotted with an anti-HA monoclonal antibody to detect bound Rho proteins. Only Cdc42 (Q61L) and TC10β (Q69L) specifically co-immunoprecipitated with both full-length TCGAP (Figure 2A) and the N-terminal fragment, which includes the GAP domain (Figure 2B).

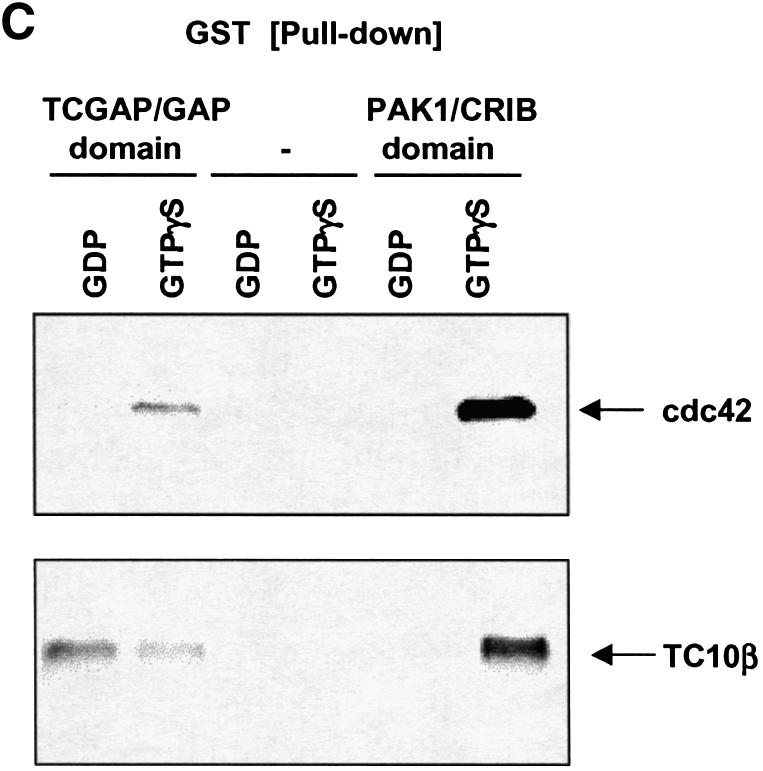
Fig. 2. TCGAP interacts with active Rho GTPases in Cos-1 cells. Cos-1 cells were transiently co-transfected with Myc-TCGAP full-length (A) or Myc-N-terminal TCGAP (B) and HA-tagged constitutively active Rho GTPase constructs as indicated. At 24 h after transfection, cells were collected and lysed in HNTG lysis buffer. Total cell lysates (lower panels) and immunoprecipitates with anti-Myc poly clonal antibody (+) or IgG as control (–) from transfected cells (upper panel) were subjected to immunoblotting with anti-Myc monoclonal antibody (9E10) or anti-HA monoclonal antibody. (C) Cos-1 cells were transiently transfected with HA-tagged wild-type Cdc42 and TC10β. At 24 h after transfection, cells were collected and lysed in NP-40 lysis buffer. Cell lysates were loaded with GDP or GTPγS and incubated with 5 µg of purified GST–GAP/TCGAP, GST–PAK1-CRIB or GST alone beads. Bound proteins were subjected to immunoblotting with anti-HA monoclonal antibody.
These results indicate that TCGAP interacts with cdc42 and TC10β through its N-terminal region, most probably via the GAP domain. We next investigated whether the GAP domain of TCGAP alone is sufficient for Rho family GTPase binding. Lysates derived from serum-deprived cells expressing HA-tagged cdc42 and TC10β were loaded with GDP or GTPγS in vitro and incubated with a purified GST–TCGAP/GAP domain fusion protein, or the GST–PAK1 p21-binding motif (CRIB domain) as positive control. Bound lysates were then incubated with glutathione beads, eluted and subject to immunoblotting (Figure 2C). Both GTPγS-loaded cdc42 and TC10β bound to GST–PAK1/CRIB with high affinity, whereas the GDP-bound forms did not bind, as previously shown (Benard et al., 1999; Vignal et al., 2000). The GAP domain of TCGAP alone bound to cdc42 in a GTP-dependent manner and bound to TC10β in a nucleotide-independent manner.
At this point, we cannot rule out the possibility that TCGAP may interact with cdc42 or TC10β through other cdc42- or TC10β-binding proteins. However, the GAP domain itself is necessary and sufficient for the interaction with Rho family GTPases. Moreover, these studies point out a novel feature of TCGAP, its different nucleotide dependence toward cdc42 and TC10β through the GAP domain. Together, these data suggest that TCGAP may play different roles in cdc42 and TC10β signaling.
TCGAP translocates to the plasma membrane in response to insulin
The interaction of TCGAP with cdc42 and TC10β suggests that TCGAP may localize in close proximity to these Rho family members on the plasma membrane in response to activating extracellular stimuli. Because of the unique role of TC10 in the regulation of glucose transport by insulin, we examined the subcellular localization of TCGAP in 3T3-L1 adipocytes treated with or without the hormone. Myc-tagged full-length TCGAP was expressed in 3T3-L1 adipocytes, followed by treatment with or without insulin. Cells were then immunostained with monoclonal anti-Myc (9E10) and polyclonal anti-caveolin 1 antibodies. As shown in Figure 3A, TCGAP protein was found predominantly in the cytoplasmic/perinuclear region. Interestingly, a significant fraction of TCGAP translocated to the plasma membrane in response to insulin stimulation. Quantitation of the plasma membrane localization of cells expressing TCGAP showed that the protein was translocated in response to insulin in 60% of transfected cells (Figure 3B).

Fig. 3. TCGAP translocates to the plasma membrane in response to insulin. (A) 3T3-L1 adipocytes were electroporated with Myc-TCGAP. After 48 h, cells were starved and treated without (a, b and c) or with 100 nM insulin (d, e and f) for 5 min, followed by co-immunostaining with anti-caveolin polyclonal antibody and anti-Myc monoclonal antibody. Confocal microscopy was used to detect endogenous caveolin 1 (green) (a and d) and transfected Myc-TCGAP (red) (b and e). Overlapping images are shown in (c) and (f). (B) Images from cells expressing Myc-TCGAP treated without (a) or with 100 nM insulin (b) for 5 min. This image is a representative collection of four cell images per field. Quantitation of TCGAP localization. Each value is representative of 100 cells that were counted. The number of cells that show plasma membrane localization out of 100 cells is represented as a percentage of localization. The data are the average of two independent experiments. (C) 3T3-L1 adipocytes were electroporated with Myc-TCGAP N-terminal fragment. After 48 h, cells were starved and treated without (a, b and c) or with 100 nM insulin (d, e and f) for 5 min and followed by co-immunostaining with propidium iodide and anti-Myc monoclonal antibody. Confocal microscopy was used to detect Myc-TCGAP/N (green) (a and d) and propidium iodide (red) (b and e). Overlapping images are shown in (c) and (f). (D) 3T3-L1 adipocytes were electroporated with a Myc-tagged C-terminal fragment of TCGAP. After 48 h, cells were starved for 3 h and treated without (a, b and c) or with 100 nM insulin (d, e and f) for 2 min and followed by co-immunostaining with anti-caveolin polyclonal antibody and anti-Myc monoclonal antibody. Confocal microscopy was used to detect endogenous caveolin 1 (green) (a and d) and transfected Myc-TCGAP (red) (b and e). Overlapping images are shown in (c) and (f).
Translocation of TCGAP to the plasma membrane might result from the insulin-dependent release of a cytoplasmic retention signal, and/or the exposure of a plasma membrane targeting signal, potentially through protein–lipid or protein–protein interactions. To delineate the regions responsible for the translocation of TCGAP, Myc-tagged N- and C-terminal fragments were expressed in 3T3-L1 adipocytes and immunostained with anti-Myc antibody to visualize the localization of the fragments. Unlike the full-length protein, the N-terminal fragment of TCGAP, which contains the PX, SH3 and Rho GAP domains, was found predominantly in a nuclear compartment and at the plasma membrane. The localization of this N-terminal fragment was unaffected by exposure of the cells to insulin. Co-staining with propidium iodide, which stains DNA, confirmed that N-terminal TCGAP localized in the nucleus and at the plasma membrane (Figure 3C).
A Myc-tagged C-terminal fragment of TCGAP, which contains the multiple PXXP motifs, was also expressed in 3T3-L1 adipocytes and immunostained with anti-Myc antibodies. Interestingly, this C-terminal fragment was found exclusively at the plasma membrane (co-stained with caveolin), even in the absence of insulin (Figure 3D). The differences in the subcellular localization of full-length TCGAP and its truncation mutants, as well as their differential insulin responsiveness, suggest that insulin might produce a conformational change in the protein, in turn exposing plasma membrane targeting signals at both termini, and/or masking the nuclear targeting signal at the N-terminus.
The PX domain of TCGAP preferentially binds to PtdIns(4,5)P2
The N-terminal PX domain of TCGAP contains the consensus motif, RR(F/Y)S(D/E)F, previously shown to be critical for phospholipid binding (Supplementary figure 1A). Recent reports (reviewed in Ago et al., 2001; Sato et al., 2001; Xu et al., 2001; Ellson et al., 2002; Itoh and Takenawa, 2002) have suggested that PX domains are responsible for the interactions of proteins with specific phospholipids, and thus direct subcellular localization. PX domains identified so far have shown distinct phospholipid-binding specificity. For example, the PX domains of p40phox and Vam7p interact with PtdIns-3-phosphate (PtdIns3P), the PX domain of p47phox binds to PtdIns-(3,4)-bisphosphate [PtdIns(3,4)P2], while the PX domain of CPK PtdIns-3 kinase selectively binds to PtdIns(4,5)P2.
Vam7p, which is involved in vacuolar morphogenesis, requires a PX domain for its function (Sato et al., 1998). A tyrosine to alanine mutation in Vam7p abolished PtdIns3P binding and vacuolar trafficking in yeast (Cheever et al., 2001). In order to explore the importance of this domain in TCGAP, the consensus tyrosine was mutated to alanine in the Myc-tagged N-terminal fragment of TCGAP (Y124A). We then performed a protein–lipid overlay assay (Cheever et al., 2001) to study the phospholipid-binding properties of the mutant and wild-type proteins. In vitro transcription/translation reactions were performed to generate a Myc-tagged vector alone, N-terminal wild type, and Y124A mutants of TCGAP. Nitrocellulose membranes spotted with various phospholipids were incubated with identical amounts of in vitro translation lysates from the Myc-tagged N-terminal fragments or vector as control (Figure 4A). The interaction of the PX domain with phospholipids was detected by immunoblotting with an anti-Myc 9E10 antibody. As shown in Figure 4B, the N-terminal TCGAP strongly interacted with PtdIns(3)P, PtdIns(4)P, PtdIns (3,4)P2 and PtdIns (4,5)P2, whereas the Y124A mutant showed a significant decrease in PtdIns(3)P and PtdIns (4,5)P2 binding compared with the wild type. The control lysate did not interact with any of the phospholipids, even after longer exposure of the autoradiogram (data not shown).



Fig. 4. The PX domain of TCGAP preferentially binds to PtdIns(4,5)P2. Equal amounts of Myc fusion proteins (A) or Myc alone (negative control not shown) were used to perform protein–phospholipid binding assays (B and C). (B) PIP Strips™ are spotted with 100 pmol of each phospholipid. (C) PIP Arrays™ are spotted with serial dilutions of phospholipids. (D) Densitometry was performed on the arrays shown in (C) that are representative of three experiments. (E) 3T3-L1 adipocytes were electroporated with Myc-TCGAP/FL wild type (a and b) or Y124A (c and d). After 48 h, cells were starved and treated without (a and c) or with 100 nM insulin for 5 min (b and d), followed by immunostaining with anti-Myc monoclonal antibody. Confocal microscopy was used to detect transfected Myc-TCGAP. This image is a representative collection of four cell images per field. Quantitation of TCGAP wild type and Y124A mutant localization in the absence or presence of insulin. Each condition depicts a representative of 100 cells that were counted. The number of cells that showed plasma membrane localization out of 100 cells is represented as a percenatge of localization. The data are the average of two independent experiments.
To analyze the affinity of phosphoinositide binding, PIP arrays with various concentrations of phospholipids were incubated with the in vitro translated products of the N-terminal fragments, and binding was detected by blotting with an anti-Myc antibody. As shown in Figure 4C, the binding of the wild-type protein to PtdIns(4,5)P2 was observed at low concentrations. Interestingly, binding affinity was significantly decreased in the Y124A mutant. Quantitation of these data revealed an ∼5-fold difference in PtdIns(4,5)P2 binding affinity between the wild-type and mutant proteins (Figure 4D).
The PX domain of sorting nexin 1 (snx1) specifically binds to PtdIns(3)P and targets the protein to early endosomes where PtdIns(3)P is highly enriched (Zhong et al., 2002), providing evidence of a role for pleckstrin homology (PH) domains in protein targeting. PtdIns(4,5)P2 plays a key role in the plasma membrane targeting of proteins (Sechi and Wehland, 2000; Martin, 2001; Payrastre et al., 2001). The PH domain of phospho lipase Cδ (PLCδ) binds specifically to PtdIns(4,5)P2 and localizes at the plasma membrane (Garcia et al., 1995; Lemmon et al., 1995; Varnai et al., 2002). To evaluate the distribution of PtdIns(4,5)P2 in 3T3-L1 adipocytes, a cDNA encoding the green fluorescent protein (GFP)– PLCδPH domain was transfected into cells, which were subsequently immunostained with the plasma membrane marker caveolin 1. As shown in Supplementary figure 4, the PLCδPH domain exclusively localized at the plasma membrane, indicating the membrane localization of PtdIns(4,5)P2 in adipocytes. Insulin did not increase the cell surface labeling of this marker, consistent with the notion that insulin does not modulate PtdIns(4,5)P2 synthesis or degradation.
To investigate further whether PtdIns(4,5)P2 is a target for the PX domain of TCGAP in a physiological setting, the Myc-TCGAP/N-terminal fragment (wild type and Y124A mutant) was expressed in 3T3-L1 adipocytes and visualized by immunostaining with anti-Myc antibody (Supplementary figure 5). The wild-type N-terminal fragment, which contains SH3, PX and Rho GAP domains, was found in the nucleus and at the plasma membrane, co-staining with caveolin 1, and did not change in response to insulin. Thus, it is likely that this protein contains a nuclear targeting signal other than the PX domain in the N-terminal fragment (Supplementary figure 6). Interest ingly, the Y124A mutant showed significantly less plasma membrane staining than did the wild-type protein, suggesting that the highly conserved residue Tyr124 in the PX domain is critical for plasma membrane localization. PtdIns(3)P and PtdIns(3,5)P2 are enriched in endosomes and are thought to be involved in vacuole trafficking (Wurmser and Emr, 1998; Ellson et al., 2002). Full-length Myc-TCGAP or its N-terminal fragment did not costain with EEA1-positive early endosomes or Golgi/endoplasmic reticulum (data not shown), suggesting that PtdIns(3)P and PtdIns(3,5)P2 do not interact with TCGAP in vivo, even though modest binding to both phospholipids was detected in the overlay assay.
As was shown in Figure 3A, TCGAP translocates from cytoplasmic/perinuclear regions to the plasma membrane in response to insulin. This finding, along with the specificity of PX domain–phospholipid interactions, suggests that the generation of PtdIns(4,5)P2 at the plasma membrane may recruit TCGAP to the plasma membrane via interactions with the PX domain. To investigate this hypothesis, Myc-tagged wild-type TCGAP and its PX domain mutant (Y124A) were expressed in 3T3-L1 adipocytes, followed by immunostaining with anti-Myc and anti-caveolin 1 antibodies. Compared with wild type, the TCGAP Y124A mutant did not translocate to the plasma membrane in response to insulin. Quantitation of these data revealed that translocation of the PX domain mutant was decreased by 60% compared with the wild-type control (Figure 4E). Thus, although insulin does not produce an increase in PtdIns(4,5)P2 levels in the plasma membrane (Supplementary figure 4), these data suggest that the interactions of the PX domain of TCGAP with plasma membrane phosphoinositides are necessary but not sufficient for the translocation of the protein to the plasma membrane.
The translocation of TCGAP is mediated by a CrkII/CAP-dependent pathway
In the C-terminal region of TCGAP (amino acids 560–1300), the proline and serine content is ∼35%. One notable candidate for such an interaction is the adaptor protein CrkII, which translocates to the plasma membrane in response to insulin as a consequence of binding to tyrosine-phosphorylated Cbl (Baumann et al., 2000). A CrkII SH3 domain consensus binding sequence (PXLPXK/R) (Knudsen et al., 1995) was found at residues 1223–1228 (PRLPQK). To test the interaction between TCGAP and CrkII, Myc-tagged TCGAP (full-length, N- and C-terminal) constructs were expressed in 293T cells. Lysates were incubated with a purified GST–CrkII full-length fusion protein conjugated to glutathione beads. As shown in Figure 5A, both the full-length and C-terminal fragment of TCGAP interacted with CrkII, whereas the N-terminal fragment did not. This result suggests that a proline-rich sequence in the C-terminal region of TCGAP interacts with CrkII through an SH3 domain of the latter protein. We next incubated the lysates from cells overexpressing TCGAP or its two fragments with the purified GST–CrkII SH3(N) domain only, which binds to proteins with the consensus sequence PXLPXK/R. As shown in Figure 5B, the interaction between CrkII and TCGAP is mediated by the N-terminal SH3 domain of CrkII and the C-terminal sequences of TCGAP.


Fig. 5. TCGAP interacts with the N-terminal SH3 domain of c-Crk II through a C-terminal proline-rich sequence. 293T cells were transiently transfected with Myc-TCGAP full-length, N- or C-terminal constructs. At 28 h after transfection, cells were collected and lysed in NP-40 lysis buffer. Total cell lysates (left) were incubated with an equal amount of purified GST–CrkII full-length or GST alone (A) or SH3 N (B). Bound proteins were subjected to immunoblotting with an anti-Myc monoclonal antibody (9E10). (C) Myc-TCGAP was co-transfected with HA-CrkII into CHO-IR cells. After 36 h, cells were starved overnight followed by treatment with insulin for the indicated times. Lysates were immunoprecipitated with anti-Myc antibody and blotted with anti-HA antibody. (D) Wild-type Myc-TCGAP and a CrkII binding mutant (K1228A) were co-transfected with HA-CrkII into Cos-1 cells. At 24 h after transfection, cells were lysed in HNTG lysis buffer. Equal amounts of lysates were immunoprecipitated with anti-HA antibody and blotted with anti-HA or anti-Myc antibody. Quantitation of TCGAP:crkII K1228A mutant localization in the absence or presence of insulin in 3T3-L1 adipocytes. Each picture is a representative of 100 cells that were counted. The number of cells that show plasma membrane localization out of 100 cells is represented as a percentage of localization. The data are the average of two independent experiments.
To evaluate the effect of insulin on the interaction of CrkII with TCGAP, Myc-tagged TCGAP was co-expressed with HA-CrkII in Chinese hamster ovary cells expressing the human insulin receptor (CHO/IR). Cells were treated with insulin, and lysates were immunoprecipitated with an anti-Myc antibody. As shown in Figure 5C, insulin increased the interaction of TCGAP and CrkII. These data suggest that after insulin stimulation, a conformational change in TCGAP may expose the proline-rich domain in the C-terminus, leading to increased binding to CrkII via its N-terminal SH3 domain.
Since the studies outlined above suggested that the CrkII-binding sequence may be responsible for plasma membrane targeting, a CrkII-binding mutant (K1228A) of TCGAP was generated. HA-tagged CrkII was transfected into Cos-1 cells in combination with Myc-tagged full-length TCGAP or its K1228A mutant. Lysates were immunoprecipitated with an anti-HA polyclonal antibody and immunoblotted with anti-Myc antibody to detect the binding to TCGAP. As shown in Figure 5D, introduction of the K1228A substitution in TCGAP resulted in a marked decrease in binding to CrkII. This tagged TCGAP mutant (K1228A) was then expressed in 3T3-L1 adipocytes to evaluate the effect of CrkII binding on the translocation of the protein to the plasma membrane. While this mutant exhibited a reduction in plasma membrane binding in both basal and insulin-stimulated cells, insulin still produced an ∼2-fold increase in plasma membrane localization, similar to that observed for the wild-type protein. Thus, the precise role of the CrkII–TCGAP interaction in mediating the translocation of TCGAP to the plasma membrane remains uncertain. Since there are 19 potential SH3-binding sites in TCGAP, it is possible that other residues may be able to substitute for the sequences around K1228 for this or another SH3-mediated interaction.
Previous studies have suggested that CrkII is recruited to the plasma membrane in response to insulin by interacting with tyrosine-phosphorylated Cbl (Chiang et al., 2001). Deletions of the SH3 domains or the sorbin homology domain of CAP (CAPΔSH3 and CAPΔSoHo) behave as dominant-negative mutants, blocking Cbl, C3G and CrkII translocation to lipid rafts, GLUT4 translocation to the plasma membrane and glucose uptake in response to insulin (Baumann et al., 2000; Kimura et al., 2001). To investigate the importance of the Cbl/CAP pathway in the stimulation of TCGAP translocation, TCGAP was co-transfected into 3T3-L1 adipocytes with wild-type CAP, CAPΔSH3 or CAPΔSoHo. Cells were treated with or without insulin, and the localization of TCGAP was visualized by immunostaining with anti-Myc antibody. Co-expression of 3T3-L1 adipocytes with either CAPΔSH3 or CAPΔSoHo blocked the translocation of TCGAP to the plasma membrane in response to insulin (Figure 6). These data indicate that the translocation of TCGAP occurs downstream of a CAP/Cbl-dependent pathway, consistent with an important role for CrkII in this process.
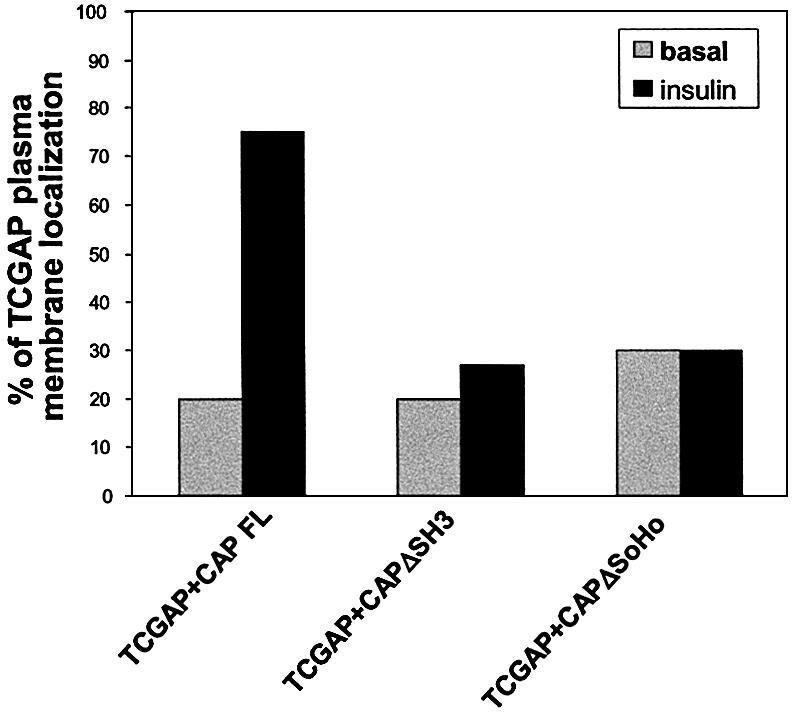
Fig. 6. Effect of CAP mutants on TCGAP translocation. 3T3-L1 adipocytes were co-electroporated with Myc-TCGAP FL and FLAG tagged-CAP FL, ΔSH3 or ΔSoHo. After 48 h, cells were starved for 3 h and treated with or without 100 nM insulin for 5 min, and co-immunostained with anti-FLAG monoclonal antibody and anti-Myc polyclonal antibody. Confocal microscopy was used to detect transfected FLAG-CAP proteins and transfected Myc-TCGAP. Quantitation of TCGAP localization in the absence or presence of insulin. Each picture is a representative of 50 cells that were counted. The number of cells that show plasma membrane localization out of 50 cells is represented as a percentage of localization. The data are representative of three independent experiments.
TCGAP plays a role in insulin-stimulated glucose transport and GLUT4 translocation
The interaction of TCGAP with Rho family GTPases suggests that it might play a role in insulin signaling. To examine the effect of TCGAP expression on insulin-stimulated glucose transport, differentiated 3T3-L1 adipocytes were electroporated with a LacZ control vector or full-length, N- or C-terminal constructs of TCGAP. Cells were transfected with ∼40–50% efficiency. In cells electroporated with the LacZ control, insulin produced an ∼10- to 12-fold stimulation of 2-deoxyglucose uptake. Interestingly, overexpression of either full-length or the C-terminal fragment of TCGAP significantly attenuated the insulin-stimulated uptake of 2-deoxyglucose, without any effect on basal uptake (Figure 7A). Taking into account the transfection efficiency, the reduction in glucose transport suggested that overexpression of TCGAP significantly blocked this action of insulin. In contrast, overexpression of the N-terminal fragment of TCGAP, which targets primarily to the nucleus and plasma membrane, did not have any effect on insulin-stimulated glucose uptake.


Fig. 7. Overexpression of TCGAP mutants blocks insulin-stimulated glucose uptake and GLUT4 translocation. (A) 3T3-L1 adipocytes were electro porated with Myc-LacZ, TCGAP full-length (FL), and N- and C-terminal cDNA constructs. After 48 h of recovery, cells were starved, followed by treatment with or without 100 nM insulin for 5 min. [14C]2-deoxyglucose uptake was measured in triplicate and counted. Glucose uptake is normalized to LacZ basal state and is represented as fold simulation. This data are representative of four independent experiments. (B) 3T3-L1 adipocytes were co-electroporated with GLUT4–EGFP plus Myc-LacZ, CAPΔSH3, TCGAP FL, N- and C-terminal cDNA constructs. After 48 h of recovery, cells were starved, followed by treatment with or without 100 nM insulin for 30 min. Cells were washed in PBS, fixed with paraformaldehyde, and mounted on microscope slides for analysis. Fluorescence was examined under a confocal microscope. Each sample shows two cells that are representative of 50 cells that were counted. The number of cells that show plasma membrane staining out of 50 cells is represented as a percentage of PM translocation. The data are representative of three independent experiments. (C) 3T3-L1 adipocytes were co-electroporated with GLUT4–EGFP plus Myc-vector control, TCGAP FL wild type, PX mutant (Y124A) or CrkII binding mutant (K1228A) cDNA constructs. After 48 h of recovery, cells were starved, followed by treatment with or without 100 nM insulin for 30 min. The number of cells that show plasma membrane staining out of 50 cells is represented as a percentage of PM translocation. The data are representative of three independent experiments.
To confirm that the inhibitory effects of TCGAP on glucose transport were due to the inhibition of GLUT4 translocation, we co-expressed the LacZ control vector or TCGAP constructs with a GLUT4–EGFP fusion construct in 3T3-L1 adipocytes. In cells co-transfected with the control vector, insulin stimulation resulted in the appearance of strong plasma membrane GLUT4–EGFP rim fluorescence, indicative of translocation (Figure 7B). Co-expression of full-length TCGAP or its C-terminal fragment with GLUT4–EGFP markedly decreased the number of cells responding to insulin. In contrast, co-expression of N-terminal TCGAP was without effect on insulin-stimulated GLUT4–EGFP translocation. Quantit ation by counting the number of cells displaying a con tinuous cell surface GLUT4–EGFP fluorescence showed a 66% decrease in insulin-stimulated GLUT4 translocation after expression of the full-length TCGAP or its C-terminal fragment. This result indicates that the effect of TCGAP on glucose uptake is due to the blockade of GLUT4 translocation to the plasma membrane.
Since mutations in the PX domain and CrkII-binding sequences decreased the insulin-stimulated translocation of the protein to the plasma membrane, we sought to determine what role these domains might play in the putative effector function of TCGAP. GLUT4–EGFP was expressed in differentiated 3T3-L1 adipocytes in combination with the PX domain mutant (Y124A) or the CrkII-binding mutant (K1228A) of TCGAP. The impact of these TCGAP mutants on GLUT4 translocation was quantitated by counting the cells displaying GLUT4–EGFP rim fluorescence. As shown in Figure 7C, the TCGAP Y124A mutant produced a 56% inhibition of GLUT4 translocation, ∼80% of the effect seen with the wild-type protein. The K1228A mutant blocked GLUT4 translocation to an extent comparable with that seen with the wild-type TCGAP. These data suggest that the plasma membrane localization of TCGAP contributes to, but is not absolutely required for its effect on GLUT4 translocation. However, we cannot rule out the possibility that the decreased affinity for binding to phospholipids or CrkII alone may not be sufficient to abolish this inhibitory effect.
Discussion
We have identified a cDNA encoding a novel multidomain-containing Rho GAP protein, TCGAP. TCGAP binds to cdc42 and TC10β specifically through its GAP domain. TCGAP translocates to the plasma membrane in response to insulin in adipocytes, and may play an important role in the stimulation of glucose transport by insulin. Like other genes crucial to insulin signaling (Tontonoz et al., 1994; Ribon et al., 1998), TCGAP mRNA expression is dramatically induced during adipocyte differentiation. Preliminary results suggested that TCGAP possesses intrinsic GAP activity, as determined by an in vitro GAP assay. However, overexpression of full-length TCGAP had no significant effect on the GTP-binding status of the Rho family GTPases in vivo. Thus, although it remains possible that the GAP activity might be masked (Dumenil et al., 2000; Jenna et al., 2002; Miura et al., 2002), TCGAP appears to behave as an effector rather than as a signal terminator, similar to what has been observed for IQGAP. TCGAP could also act as a scaffolding protein by binding to Cdc42 and TC10β through its GAP domain, thus bringing other proteins to the membrane.
Cdc42, TC10α and TC10β share a high degree of homology compared with other members of the Rho GTPase family. Although Cdc42 is not sensitive to insulin, TC10α and β are both activated by the hormone in 3T3-L1 adipocytes, and may play an important part in the stimulation of GLUT4 translocation and glucose transport stimulation by insulin (Chiang et al., 2001; Watson et al., 2001). Interestingly, the α form of TC10, which is 83% identical to TC10β, shares identical sequences in the five canonical Ras boxes, and diverges mainly in the N- and C-terminal residues (Chiang et al., 2002). Thus, the selective interaction between TCGAP and the β but not the α form of TC10 suggests differences in the responsible binding motif and the ensuing downstream signaling pathways.
Most proteins in the Rho GAP family contain additional interaction domains, suggesting that they are multifunctional. In addition to its centrally located GAP domain, TCGAP contains an SH3 domain, an N-terminally located PX domain, numerous PXXP motifs and a serine-/proline-rich region in the C-terminal half of the protein. The presence of a serine-/proline-rich region predicts that TCGAP may be the target of proline-directed serine/threonine kinases, perhaps regulating the conformation, stability or activity of the protein. We previously observed that CrkII translocates to a lipid raft subdomain of the plasma membrane in response to insulin, due to its specific interaction with tyrosine-phosphorylated Cbl, via the SH2 domain of CrkII (Chiang et al., 2001). Thus, it is likely that the interaction of TCGAP with Crk may recruit TCGAP to a specific compartment in the plasma membrane. In this regard, overexpression of mutant forms of CAP lacking the SH3 or SoHo domain, which block the translocation of Crk to the plasma membrane, also attenuates the translocation of TCGAP. These data support a central role for CrkII in mediating the translocation of TCGAP.
One unusual feature of TCGAP is the presence at the N-terminus of a PX domain. In addition to binding to phosphoinositides, PX domains can in some cases bind to SH3 domains, since they often contain a polyproline motif (XPpXP) in the middle of the sequence (Hiroaki et al., 2001; Wientjes and Segal, 2003). Such a sequence is also found in the PX domain of TCGAP, which represents the first identified Rho GAP protein with both PX and SH3 domains. One possibility is that these domains work in concert to regulate the localization and function of the protein. For example, the intracellular retention of TCGAP may result from an intramolecular interaction between the PX and SH3 domains. After exposure of cells to insulin, the interaction with certain protein(s) may induce a conformational change that reduces the PX–SH3 interaction, thus permitting the PX domain to bind PtdIns(4,5)P2 at the plasma membrane, and stabilizing the membrane translocation produced by insulin (Karathanassis et al., 2002; Endo et al., 2003), and thus explaining why the PX domain may be necessary but not sufficient for translocation. Binding of the protein to TC10β or cdc42 might also stabilize this plasma membrane interaction further. Alternatively, the N-terminal SH3 domain may interact with a C-terminal proline-rich sequence (Gmeiner et al., 2001) and prevent its interaction with CrkII at the basal state. As a consequence, insulin stimulation might induce a conformational change that exposes the C-terminal proline-rich sequence to CrkII, recruiting TCGAP to the plasma membrane.
It is thought that in order for adaptor proteins to facilitate signaling, the relative concentrations of the components must be in balance (Ferrell, 2000). For example, overexpression of APS, a member of the Lnk family of adaptor proteins, binds to the insulin receptor and enhances insulin-stimulated tyrosine phosphorylation of Cbl. However, when overexpressed, APS inhibited the phosphorylation of endogenous Cbl in response to insulin, presumably due to the relative excess of APS over endogenous Cbl and insulin receptor (Liu et al., 2002). In adipocytes, insulin stimulates the translocation of TCGAP from the cytoplasm to the plasma membrane, where TC10β resides. TCGAP is expressed at relatively low levels in adipocytes, presumably due to the presence of PEST motifs that are commonly found in proteins with a short half-life. Thus, the blockade of insulin-stimulated glucose uptake and GLUT4 translocation observed after overexpression of TCGAP probably reflects a disruption of the balance between TC10β and its upstream or downstream regulators. Taken together, the multidomain structure of TCGAP implicates interactions with other proteins that might play an important role in glucose transport activation by insulin. Current efforts are focusing on identifying such proteins.
Materials and methods
Reagents
Anti-HA and anti-Myc monoclonal (9E10) and polyclonal antibodies were obtained from Santa Cruz Biotech. Anti-caveolin 1 antibody was purchased from Transduction Laboratories. Enhanced chemiluminesence (ECL) reagents were purchased from NEN, Inc. GST–PAK1-CRIB agarose beads were purchased from Cytoskeleton, Inc. GST–rhotekin agarose beads were purchased from Upstate Biotechnology. QuikChange™ XL site-directed mutagenesis kit was purchased from Stratagene.
Cloning of full-length TCGAP
The partial 3′ cDNA fragment of TCGAP was cloned originally from a yeast two-hybrid screening using full-length Synip (Min et al., 1999) as bait. 5′ RACE–PCRs were employed to obtain the entire ORF using the Marathon cDNA Amplification kit and Advantage cDNA polymerase (Clontech, Inc.) following the manufacturer’s instructions. After obtaining the full-length sequence of TCGAP by RACE–PCR, another PCR was performed to amplify the full-length cDNA using the d17 embryonic cDNA RACE library. The specific 5′ transcript of TCGAP expressing in 3T3-L1 adipocytes was confirmed by RT–PCR. The Blast program from NCBI and the SMART program from EMBL were used for sequence analysis. The TCGAP cDNA sequence has been submitted to the GenBank database under accession No. AY217764.
Cell culture
Cos-1 and human embryonic kidney (HEK) 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C with 5% CO2. CHO/IR cells were a generous gift from Dr Jeffrey Pessin (Waters et al., 1995). These cells were maintained in minimal Eagle’s medium (MEM) containing nucleotides plus 10% FBS at 37°C with 5% CO2. 3T3-L1 pre-adipocytes were cultured and differentiated as previously described (Olson et al., 1997).
Transfection of Cos-1, HEK293T, CHO/IR cells and 3T3-L1 adipocytes
Full-length TCGAP was subcloned into another mammalian expression vector (pCS2-MT) downstream of six Myc epitopes under a cytomegalovirus (CMV) promoter using an EcoRI site. The N-terminal TCGAP construct (amino acids 1–655) was generated by subcloning the EcoRI–SalI fragment from the original N-terminal construct into the EcoRI–StuI sites of the pCS2 vector. The C-terminal TCGAP construct (amino acids 656–1305) was generated by subcloning the BglII–EcoRV fragment from the original C-terminal construct into the XbaI site of the pCS2 vector.
HEK293T cells were transfected with a mammalian CaPO4 transfection kit as described by the manufacturer (Promega). Cos-1 cells were transfected with the Fugene 6 method (Life Technologies). CHO/IR cells and 3T3-L1 adipocytes were transfected by electroporation as previously described (Min et al., 1999).
Immunoprecipitation and immunoblotting
Whole-cell detergent extracts were prepared by detergent solubilization in HNTG buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol) and 1 mM EDTA, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM sodium vanadate and complete protease inhibitor (Roche) for 15 min at 4°C. Immunoprecipitations were performed by using the amount of lysates indicated pre-cleared with protein A beads for 30 min. Supernatants were incubated with 5 µg of polyclonal antibody for 2 h and then incubated with protein A beads for another 1 h at 4°C. Beads were washed three times with HNTG buffer. The immunoprecipitates were subjected to SDS–PAGE, and western blotting was performed using the indicated antibodies.
Protein–phospholipid overlay assay
Myc fusion proteins were expressed in vitro using the TNT™ Coupled Reticulocyte Lysate System (Promega) following the manufacturer’s instructions. Phospholipid-spotted membranes (PIP Strips™ and PIP Arrays™, Echelon) were blocked with 3% fatty acid-free bovine serum albumin (BSA) in Tris-buffered saline–Tween (TBST). The strips were incubated with Myc fusion proteins resulting from the in vitro transcription/translation assay. Membranes were washed three times with TBST, then incubated with anti-Myc monoclonal antibody. After washing, blots were incubated with horseradish peroxidase (HRP)-coupled anti-mouse IgG, washed, developed by chemiluminescence and then visualized by autoradiography. Protein–phospholipid interaction was quantified by densitometry.
Assay of glucose transport
Determination of 2-deoxyglucose uptake was performed as previously described (Baumann et al., 2000). Construction of all EGFP fusion proteins and translocation assays were carried out using the method of Thurmond et al. (2000). Briefly, 3T3-L1 adipocytes were electroporated with 80 µg of EGFP-tagged plasmid and 250 µg of either empty vector or TCGAP constructs. Adipocytes were replated, allowed to recover overnight and stimulated with or without 100 nM insulin for 30 min. The cells were then fixed and EGFP fluorescence was analyzed by confocal immunofluorescence microscopy.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Dr Bruce Mayer (University of Connecticut) for providing GST–Crk II (SH3), Dr Jun Liu for GST–CrkII full-length, Dr Ian Macara for GST–RhoA, Rac1 and cdc42 constructs, Dr R.W.Holz for the GFP–PLCδ/PH domain construct, Dr Jeffrey Pessin for the GFP–Grp1/PH domain and GLUT4–EGFP fusion constructs, and Drs Cheryl Neudauer and Jeffrey Pessin for valuable discussions and comments.
References
- Ago T., Takeya,R., Hiroaki,H., Kuribayashi,F., Ito,T., Kohda,D. and Sumimoto,H. (2001) The PX domain as a novel phosphoinositide-binding module. Biochem. Biophys. Res. Commun., 287, 733–738. [DOI] [PubMed] [Google Scholar]
- Baumann C.A., Ribon,V., Kanzaki,M., Thurmond,D.C., Mora,S., Shigematsu,S., Bickel,P.E., Pessin,J.E. and Saltiel,A.R. (2000) CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature, 407, 202–207. [DOI] [PubMed] [Google Scholar]
- Benard V., Bohl,B.P. and Bokoch,G.M. (1999) Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem., 274, 13198–13204. [DOI] [PubMed] [Google Scholar]
- Bollag G. and McCormick,F. (1991) Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature, 351, 576–579. [DOI] [PubMed] [Google Scholar]
- Brill S., Li,S., Lyman,C.W., Church,D.M., Wasmuth,J.J., Weissbach,L., Bernards,A. and Snijders,A.J. (1996) The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol. Cell. Biol., 16, 4869–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever M.L., Sato,T.K., de Beer,T., Kutateladze,T.G., Emr,S.D. and Overduin,M. (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol., 3, 613–618. [DOI] [PubMed] [Google Scholar]
- Chiang S.H., Baumann,C.A., Kanzaki,M., Thurmond,D.C., Watson,R.T., Neudauer,C.L., Macara,I.G., Pessin,J.E. and Saltiel,A.R. (2001) Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature, 410, 944–948. [DOI] [PubMed] [Google Scholar]
- Chiang S.H., Hou,J.C., Hwang,J., Pessin,J.E. and Saltiel,A.R. (2002) Cloning and functional characterization of related TC10 isoforms, a subfamily of Rho proteins involved in insulin-stimulated glucose transport. J. Biol. Chem., 277, 13067–13073. [DOI] [PubMed] [Google Scholar]
- Dumenil G., Sansonetti,P. and Tran Van Nhieu,G. (2000) Src tyrosine kinase activity down-regulates Rho-dependent responses during Shigella entry into epithelial cells and stress fibre formation. J. Cell Sci., 113, 71–80. [DOI] [PubMed] [Google Scholar]
- Ellson C.D., Andrews,S., Stephens,L.R. and Hawkins,P.T. (2002) The PX domain: a new phosphoinositide-binding module. J. Cell Sci., 115, 1099–1105. [DOI] [PubMed] [Google Scholar]
- Endo M., Shirouzu,M. and Yokoyama,S. (2003) The Cdc42 binding and scaffolding activities of the fission yeast adaptor protein Scd2. J. Biol. Chem., 278, 843–852. [DOI] [PubMed] [Google Scholar]
- Ferrell J.E. Jr (2000) What do scaffold proteins really do? Sci. STKE, 2000, PE1. [DOI] [PubMed] [Google Scholar]
- Garcia P., Gupta,R., Shah,S., Morris,A.J., Rudge,S.A., Scarlata,S., Petrova,V., McLaughlin,S. and Rebecchi,M.J. (1995) The pleckstrin homology domain of phospholipase C-δ 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry, 34, 16228–16234. [DOI] [PubMed] [Google Scholar]
- Gmeiner W.H., Xu,I., Horita,D.A., Smithgall,T.E., Engen,J.R., Smith,D.L. and Byrd,R.A. (2001) Intramolecular binding of a proximal PPII helix to an SH3 domain in the fusion protein SH3Hck:PPIIhGAP. Cell Biochem. Biophys., 35, 115–126. [DOI] [PubMed] [Google Scholar]
- Hall A. (1992) Ras-related GTPases and the cytoskeleton. Mol. Biol. Cell, 3, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroaki H., Ago,T., Ito,T., Sumimoto,H. and Kohda,D. (2001) Solution structure of the PX domain, a target of the SH3 domain. Nat. Struct. Biol., 8, 526–530. [DOI] [PubMed] [Google Scholar]
- Itoh T. and Takenawa,T. (2002) Phosphoinositide-binding domains: functional units for temporal and spatial regulation of intracellular signalling. Cell Signal., 14, 733–743. [DOI] [PubMed] [Google Scholar]
- Jenna S., Hussain,N.K., Danek,E.I., Triki,I., Wasiak,S., McPherson,P.S. and Lamarche-Vane,N. (2002) The activity of the GTPase-activating protein CdGAP is regulated by the endocytic protein intersectin. J. Biol. Chem., 277, 6366–6373. [DOI] [PubMed] [Google Scholar]
- Kaibuchi K., Kuroda,S. and Amano,M. (1999) Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem., 68, 459–486. [DOI] [PubMed] [Google Scholar]
- Karathanassis D., Stahelin,R.V., Bravo,J., Perisic,O., Pacold,C.M., Cho,W. and Williams,R.L. (2002) Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J., 21, 5057–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A., Baumann,C.A., Chiang,S.H. and Saltiel,A.R. (2001) The sorbin homology domain: a motif for the targeting of proteins to lipid rafts. Proc. Natl Acad. Sci. USA, 98, 9098–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen B.S., Zheng,J., Feller,S.M., Mayer,J.P., Burrell,S.K., Cowburn,D. and Hanafusa,H. (1995) Affinity and specificity requirements for the first Src homology 3 domain of the Crk proteins. EMBO J., 14, 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R., Ahmed,S., Best,A. and Lim,L. (1996) The GTPase-activating protein n-chimaerin cooperates with Rac1 and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol. Cell. Biol., 16, 5069–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M.A., Ferguson,K.M., O’Brien,R., Sigler,P.B. and Schlessinger,J. (1995) Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl Acad. Sci. USA, 92, 10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Kimura,A., Baumann,C.A. and Saltiel,A.R. (2002) APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol. Cell. Biol., 22, 3599–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T.F. (2001) PI(4,5)P(2) regulation of surface membrane traffic. Curr. Opin. Cell Biol., 13, 493–499. [DOI] [PubMed] [Google Scholar]
- Min J. et al. (1999) Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol. Cell, 3, 751–760. [DOI] [PubMed] [Google Scholar]
- Miura K., Jacques,K.M., Stauffer,S., Kubosaki,A., Zhu,K., Hirsch,D.S., Resau,J., Zheng,Y. and Randazzo,P.A. (2002) ARAP1: a point of convergence for Arf and Rho signaling. Mol. Cell, 9, 109–119. [DOI] [PubMed] [Google Scholar]
- Olson A.L., Knight,J.B. and Pessin,J.E. (1997) Syntaxin 4, VAMP2 and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol., 17, 2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payrastre B., Missy,K., Giuriato,S., Bodin,S., Plantavid,M. and Gratacap,M. (2001) Phosphoinositides: key players in cell signalling, in time and space. Cell Signal., 13, 377–387. [DOI] [PubMed] [Google Scholar]
- Pessin J.E., Thurmond,D.C., Elmendorf,J.S., Coker,K.J. and Okada,S. (1999) Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J. Biol. Chem., 274, 2593–2596. [DOI] [PubMed] [Google Scholar]
- Ribon V., Printen,J.A., Hoffman,N.G., Kay,B.K. and Saltiel,A.R. (1998) A novel, multifuntional c-Cbl binding protein in insulin receptor signaling in 3T3-L1 adipocytes. Mol. Cell. Biol., 18, 872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel A.R. and Kahn,C.R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 414, 799–806. [DOI] [PubMed] [Google Scholar]
- Saltiel A.R. and Pessin,J.E. (2002) Insulin signaling pathways in time and space. Trends Cell Biol., 12, 65–71. [DOI] [PubMed] [Google Scholar]
- Sato T.K., Darsow,T. and Emr,S.D. (1998) Vam7p, a SNAP-25-like molecule and Vam3p, a syntaxin homolog, function together in yeast vacuolar protein trafficking. Mol. Cell. Biol., 18, 5308–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.K., Overduin,M. and Emr,S.D. (2001) Location, location, location: membrane targeting directed by PX domains. Science, 294, 1881–1885. [DOI] [PubMed] [Google Scholar]
- Sechi A.S. and Wehland,J. (2000) The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J. Cell Sci., 113, 3685–3695. [DOI] [PubMed] [Google Scholar]
- Tapon N. and Hall,A. (1997) Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol., 9, 86–92. [DOI] [PubMed] [Google Scholar]
- Thurmond D.C., Kanzaki,M., Khan,A.H. and Pessin,J.E. (2000) Munc18c function is required for insulin-stimulated plasma membrane fusion of GLUT4 and insulin-responsive amino peptidase storage vesicles. Mol. Cell. Biol., 20, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P., Hu,E., Graves,R.A., Budavari,A.I. and Spiegelman,B.M. (1994) mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev., 8, 1224–1234. [DOI] [PubMed] [Google Scholar]
- Varnai P., Lin,X., Lee,S.B., Tuymetova,G., Bondeva,T., Spat,A., Rhee,S.G., Hajnoczky,G. and Balla,T. (2002) Inositol lipid binding and membrane localization of isolated pleckstrin homology (PH) domains. Studies on the PH domains of phospholipase Cδ1 and p130. J. Biol. Chem., 277, 27412–27422. [DOI] [PubMed] [Google Scholar]
- Vignal E., De Toledo,M., Comunale,F., Ladopoulou,A., Gauthier-Rouviere,C., Blangy,A. and Fort,P. (2000) Characterization of TCL, a new GTPase of the rho family related to TC10 andCcdc42. J. Biol. Chem., 275, 36457–36464. [DOI] [PubMed] [Google Scholar]
- Waters S.B., Holt,K.H., Ross,S.E., Syu,L.J., Guan,K.L., Saltiel,A.R., Koretzky,G.A. and Pessin,J.E. (1995) Desensitization of Ras activation by a feedback disassociation of the SOS–Grb2 complex. J. Biol. Chem., 270, 20883–20886. [DOI] [PubMed] [Google Scholar]
- Watson R.T., Shigematsu,S., Chiang,S.H., Mora,S., Kanzaki,M., Macara,I.G., Saltiel,A.R. and Pessin,J.E. (2001) Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. J. Cell Biol., 154, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wientjes F.B. and Segal,A.W. (2003) PX domain takes shape. Curr. Opin. Hematol., 10, 2–7. [DOI] [PubMed] [Google Scholar]
- Wurmser A.E. and Emr,S.D. (1998) Phosphoinositide signaling and turnover: PtdIns(3)P, a regulator of membrane traffic, is transported to the vacuole and degraded by a process that requires lumenal vacuolar hydrolase activities. EMBO J., 17, 4930–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Seet,L.F., Hanson,B. and Hong,W. (2001) The Phox homology (PX) domain, a new player in phosphoinositide signalling. Biochem. J., 360, 513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q., Lazar,C.S., Tronchere,H., Sato,T., Meerloo,T., Yeo,M., Songyang,Z., Emr,S.D. and Gill,G.N. (2002) Endosomal localization and function of sorting nexin 1. Proc. Natl Acad. Sci. USA, 99, 6767–6772. [DOI] [PMC free article] [PubMed] [Google Scholar]