

Abstract
Although the essential function of checkpoint kinase 1 (Chk1) in DNA damage response has been well established, the role of Chk1 in normal cell cycle progression is unclear. By using RNAi to specifically deplete Chk1, we determined loss-of-function phenotypes in HeLa cells. A vector-based RNAi approach showed that Chk1 is required for normal cell proliferation and survival, inasmuch as a dramatic cell-cycle arrest at G2/M phase and massive apoptosis were observed in Chk1-deficient cells. Coupling of siRNA with cell synchronization further revealed that Chk1 depletion leads to metaphase block, as indicated by various mitotic markers. Neither bipolar spindle formation nor centrosome functions were affected by Chk1 depletion; however, the depleted cells exhibited chromosome misalignment during metaphase, chromosome lagging during anaphase, and kinetochore defects within the regions of misaligned/lagging chromosomes. Moreover, we showed that Chk1 is a negative regulator of polo-like kinase 1 (Plk1), in either the absence or presence of DNA damage. Finally, Chk1 depletion leads to the activation of the spindle checkpoint because codepletion of spindle checkpoint proteins rescues the Chk1 depletion-induced mitotic arrest.
Keywords: cell cycle, mitosis
Upon DNA damage, two phosphor-inositide kinase-related proteins, ataxia telangiectasia mutated (Atm) and Atm-related (Atr), are activated and play a critical role in DNA damage response. Two downstream effector kinases of Atm/r, checkpoint kinase 1 (Chk1) and Chk2, are activated by Atm/r-mediated phosphorylation (1). Activated Chk1/2 have a full spectrum of substrates that are key cell cycle regulators. For example, Chk1/2-associated p53 phosphorylation leads to stabilization of the tumor suppressor and causes subsequent cell cycle arrest and apoptosis (2). In contrast, in response to DNA damage, Cdc25A, a positive regulator of cyclin-dependent kinases, is hyperphosphorylated by Chk1/2 and degraded rapidly through an ubiquitin-mediated pathway to block cell cycle progression (3). In the absence of DNA damage, Chk1 controls the activity of cytoplasmic Cdc25B. Centrosome-associated Chk1 shields centrosomal Cdc2 from unscheduled activation by Cdc25B and subsequently prevents premature mitotic entry (4).
Polo-like kinases (Plks) have emerged as key regulators of cell cycle progression, especially mitosis. Biochemical and genetic data from various organisms have documented that polo kinases are involved in many cell-cycle-related events, such as centrosome maturation, bipolar spindle formation, sister chromatid segregation, and cytokinesis (5). Plk1 is a target of the DNA damage checkpoint, and DNA damage-induced Plk1 inactivation is Atm/r-dependent (6, 7). Considering that Chk1/2 are activated in response to DNA damage, it is reasonable to speculate that Chk1/2 might act as negative regulators of Plk1. In this article, we focus on the potential connection of Chk1/Plk1 during the normal cell cycle in the absence of DNA damage.
Results
Chk1 Is Required for Cell Proliferation and Survival.
To investigate the function of Chk1 during normal cell cycle progression in the absence of DNA damage, we first used vector-based RNAi to specifically deplete Chk1 in HeLa cells. As indicated by Western blotting, Chk1 was depleted efficiently with this approach (Fig. 1A). We next determined whether Chk1 depletion influences the proliferation of HeLa cells. Although transfection with the control vector did not affect the growth rate of the cells, transfection with the plasmid pBS/U6-Chk1 strongly inhibited cell proliferation (Fig. 1B). We also examined the viability of Chk1-depleted cells. Transfection with the control vector showed little effect on cell viability, whereas <10% of Chk1-depleted cells were still attached to the culture dishes at 6 d after transfection (Fig. 1C). To characterize the inhibition of cell growth by Chk1 depletion, cell cycle progression was analyzed by FACS. As shown in Fig. 1D, transfection with the control vector did not affect the cell cycle profile, whereas Chk1 depletion induced an obvious increase in the percentage of cells with 4N DNA content, suggesting that Chk1-depleted cells block at G2/M phase. At 4 d after transfection, Chk1-depleted cells showed a significant sub-G1 DNA population, suggesting that these cells were undergoing apoptosis. To further analyze this phenotype in Chk1-depleted cells, anti-caspase 3 Western blotting was performed (Fig. 7A, which is published as supporting information on the PNAS web site). Caspase 3, the executioner caspase in apoptosis, was clearly activated, as shown by the cleavage of full-length protein. In addition, at 3 d after transfection ≈40% of Chk1-depleted cells showed positive staining with active caspase 3 antibody, compared with <1% of control cells (Fig. 7 B and C). Collectively, these data indicate that Chk1-depleted HeLa cells arrested at G2/M phase, followed by massive apoptosis.
Fig. 1.
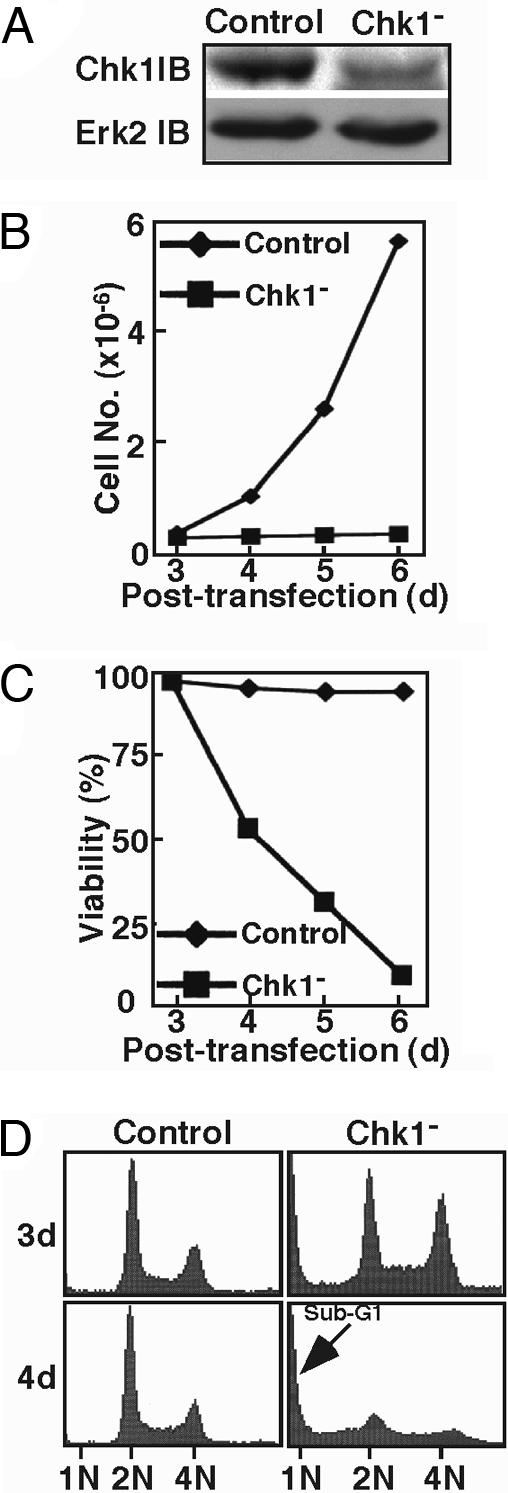
Chk1 is required for cell proliferation and survival. (A) HeLa cells were transfected with pBS/U6-Chk1 (targeting site 351) and pBabe-puro at a ratio of 9:1. At 1 d after transfection, puromycin was added for 2 d to select for transfection-positive cells. After floating cells were washed away, the remaining attached cells were harvested and analyzed by Western blotting. (B and C) Cell proliferation (B) and viability (C) were determined after Chk1 depletion. (D) FACS profiles of Chk1-depleted cells.
Chk1 Depletion Leads to Mitotic Arrest.
To confirm that Chk1 depletion leads to G2/M arrest in HeLa cells, we next analyzed the level of Plk1 in Chk1-depleted cells by using direct transfection of siRNA. Chk1-depleted cells were treated with nocodazole before harvest, and cell lysates were analyzed by anti-Plk1 Western blotting. An obvious increase observed in the level of Plk1 in Chk1-depleted cells suggested premature mitotic entry of these cells (Fig. 2A, compare lane 1 with lane 3 and lane 2 with lane 4). To determine whether Chk1 depletion leads to G2- or M-phase arrest, we measured the level of phospho-H3 (a mitosis marker) in Chk1-depleted cells and found an increased level of phospho-H3 relative to controls (Fig. 2B), suggesting that Chk1 depletion causes M-phase arrest in HeLa cells.
Fig. 2.
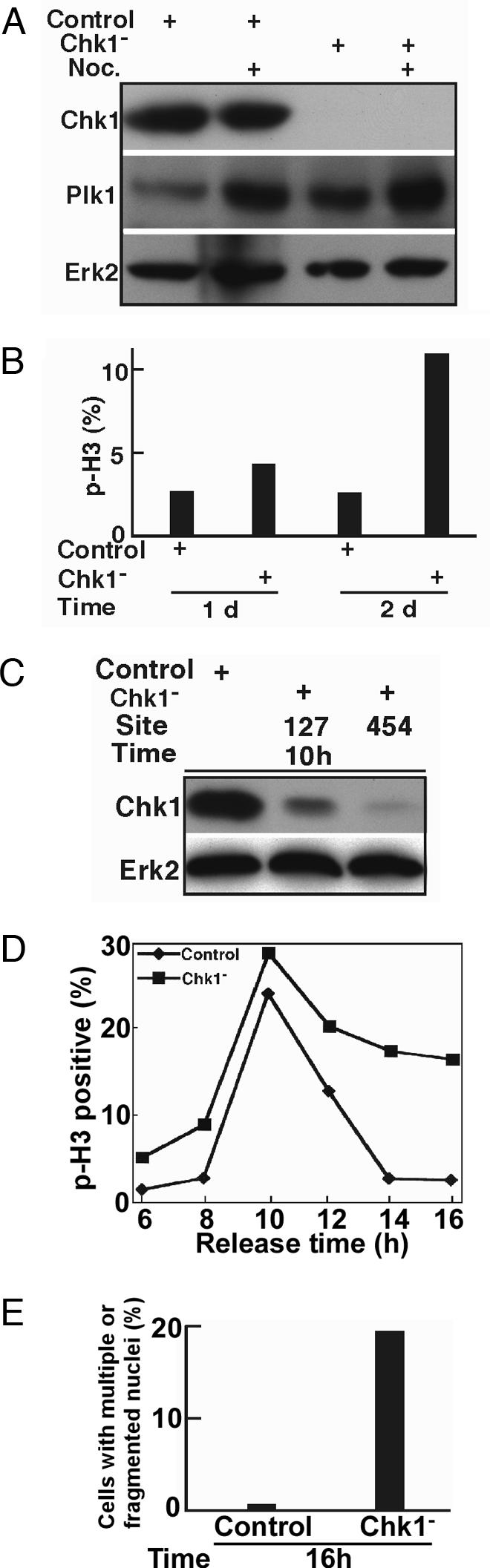
Chk1 depletion causes mitotic arrest. (A) HeLa cells were transfected with siRNA targeting site 454 of Chk1. At 2 d after transfection, cells were treated with 200 ng/ml nocodazole (Noc.) for 10 h and harvested for Western blotting using the antibodies indicated. (B) HeLa cells growing on coverslips were transfected with siRNA targeting site 454 of Chk1, harvested at 1 or 2 d after transfection, and stained with phospho-H3 antibody. (C) Chk1 was depleted with siRNA in well-synchronized HeLa cells by using the double thymidine block as described in the text. Ten hours after release from the block, cells were harvested and analyzed by Western blotting using the antibodies indicated. (D) At different times after release from the double thymidine block, cells were analyzed by anti-phospho-H3 immunofluorescence staining. (E) Histograms quantifying the results from D indicate the formation of cells with multiple or fragmented nuclei after Chk1 depletion.
To more carefully examine the effect of Chk1 depletion, we next used a protocol to couple siRNA transfection with cell synchronization. Briefly, HeLa cells were transfected with siRNA targeting site 127 or site 454 and subjected to a double thymidine block (16 h of thymidine treatment, 8 h of release, followed by a second 16-h incubation with thymidine). After depletion/synchronization, cells were released from the block. Cells harvested at different times were first subjected to Western blotting to examine the depletion efficiency. As shown in Fig. 2C, Chk1 was depleted effectively by this protocol, especially by siRNA targeting site 454. Thus, we chose siRNA targeting site 454 for the remainder of this series of experiments.
Synchronized HeLa cells growing on coverslips were subjected to Chk1 depletion by using the protocol described above. On release from the block into fresh medium, cells were costained with phospho-H3 and tubulin antibodies. Control cells began to enter mitosis at 8 h, reached a peak of phospho-H3 staining at 10 h, and had exited mitosis at 14 h after release. In contrast, Chk1-depleted cells began to enter mitosis at 6 h after release, indicating that Chk1 depletion led to premature mitotic entry, in agreement with the data obtained by using Chk1 inhibitors (4). A more striking difference was observed after a longer time following release; at 14 h after release, ≈18% of Chk1-depleted cells remained phospho-H3 positive, whereas <2% of control cells were still in mitosis (Fig. 2D). Careful inspection of DNA and tubulin staining patterns revealed the formation of cells with multiple or fragmented nuclei (Fig. 2E), consistent with the cell division defects and apoptotic phenotype described above.
Chk1 Depletion Causes Chromosome Misalignment and Lagging During Mitosis.
Chk1-depleted cells showed obvious defects in chromosome behavior during mitosis. For control cells, highly condensed chromosomes aligned along the equatorial planes during metaphase, and sister chromatids segregated at the onset of anaphase. In Chk1-depleted cells, neither chromosome condensation during prophase nor bipolar spindle formation during metaphase were affected. However, the condensed chromosomes failed to align properly along the equatorial planes during metaphase but instead spread throughout the cell, as clearly detected by both DAPI and phospho-H3 staining. Cells attempting to enter anaphase showed obvious chromosome lagging (Fig. 3A). Chromosome misalignment during metaphase was further confirmed by the H2B pattern in Chk1-depleted cells stably expressing GFP-H2B (Fig. 8A, which is published as supporting information on the PNAS web site). On the basis of chromosome characteristics and tubulin staining patterns, the phospho-H3-positive cells were subgrouped into three categories: prophase, metaphase, and anaphase. Analysis of the data indicated that Chk1-depleted cells were blocked mainly at metaphase 16 h after release, with the formation of normal bipolar spindles but misaligned chromosomes (Fig. 3B). At the onset of anaphase, cyclin B begins to be degraded via the ubiquination pathway to inactivate Cdc2 kinase activity and drive mitotic exit. This phenomenon was demonstrated by a very low percentage of cyclin B-positive staining in control cells 14 h after release, whereas the percentage of cells staining cyclin B-positive was clearly increased in Chk1-depleted cells (Fig. 8B). Upon exit from mitosis, Cdc2 is also inactivated by phosphorylation at tyrosine 15 by the inhibitory kinase Wee1 (8). Consistent with this fact, immunofluorescence analysis showed that phospho-Tyr-15-Cdc2-positive cells were clearly detected in control cells, but not in Chk1-depleted cells, at 14 h after release (Fig. 8C). Finally, cells were stained with phospho-H2AX antibody, a marker for DNA damage. As expected, phospho-H2AX signals were detected only in Chk1-depleted cells and not in control cells (Fig. 8D). In agreement with this observation, it was recently reported (9) that Chk1 inhibition by siRNA and by two drugs, 7-hydroxystaurosporine (UCN-01) and CEP-3891, leads to phosphorylation of Atr targets such as H2AX and p53, accompanied by induction of DNA strand breaks.
Fig. 3.

Chk1 is involved in proper chromosome alignment during metaphase. Synchronized HeLa cells were depleted of Chk1 by using the protocol described in Fig. 2C. At different times after release from the block, cells were stained with the antibodies indicated and analyzed by immunofluorescence microscopy. (A) Representative images of cells during mitosis. (Scale bar, 5 μm.) (B) Relative percentages of different subphases of mitosis of Chk1-depleted cells.
Kinetochore Defects in Chk1-Depleted Cells.
To further examine the chromosome alignment defect induced by Chk1 depletion, we analyzed kinetochore–microtubule attachments in these cells. For that purpose, HeLa cells stably expressing GFP-tubulin were Chk1-depleted and stained with Hec1 antibody, a kinetochore marker. A series of Z-stack images was taken from typical metaphase cells, and projections of the total Z-stack are shown in Fig. 4A. In control cells, the well-aligned chromosomes were clearly attached to kinetochore–microtubule bundles. In Chk1-depleted cells, Hec1 staining was not observed in misaligned chromosomes, and kinetochore fibers were detected only in the region with well-aligned chromosomes. Similarly, positive immunofluorescent antikinetochore (CREST) staining was observed only in the kinetochores with normal tubulin attachment but not in the regions without tubulin attachment in Chk1-depleted cells (Fig. 4B). Because Plk1 also localizes to the kinetochores during mitosis, we examined the subcellular localization of Plk1 in Chk1-depleted cells (Fig. 4C). As with Hec1 staining, Plk1 was detected only in the kinetochores of well-aligned chromosomes but not in the kinetochores of misaligned chromosomes of Chk1-depleted cells.
Fig. 4.

Kinetochore defects in Chk1-depleted cells. HeLa cells stably expressing GFP-tubulin were Chk1-depleted by using the protocol described in Fig. 2C, released for 14 h, and stained with antibodies as indicated. DNA was stained with DAPI. (A) Representative images of Hec1 (red) antibody staining of control and Chk1-depleted HeLa cells. A series of Z-stack images was taken from mitotic cells; projections of the total Z-stack are shown. (B and C) Representative images of anti-CREST (B) or anti-Plk1 (C) staining patterns of control and Chk1-depleted cells during metaphase. (Scale bar, 5 μm.)
Chk1 Is a Negative Regulator of Plk1 Activation.
Because Chk1 depletion leads to various mitotic defects, we next sought to determine whether its protein level or kinase activity is cell-cycle-regulated in the absence of DNA damage. For that purpose, HeLa cells were synchronized by using the double thymidine block, released at different times, and harvested. The level of Plk1 peaked at 10 h after release, then decreased gradually 12 h after release. In contrast, the level of Chk1 remained constant along the different phases of the cell cycle (Fig. 5A). Both the Plk1 and Chk1 kinase activities of cells harvested at different phases of the cell cycle were determined by using immunoprecipitation (IP) kinase assays. Consistent with its protein level, Plk1 kinase activity peaked at 10 h after release and decreased quickly at 12 h after release. The autophosphorylation signal of Chk1 was detected at 12 h after release, 2 h behind the mitotic peak, supporting the potential role of Chk1 during mitotic exit (Fig. 5B). Next, we examined the influence of Chk1 depletion on Cdc2 and Plk1 activation in well-synchronized cultures by using the protocol described in Fig. 2C. Upon release from the block at different times, the cells were harvested, lysates were immunoprecipitated with Cdc2 or Plk1 antibody, and the immunocomplexes were incubated with histone H1 or GST-translationally controlled tumor protein (TCTP) in the presence of [γ-32P]ATP (Fig. 5 C and D). For control cells, the protein kinase activities of both Cdc2 and Plk1 peaked at 10 h after release and then decreased at 14 h after release, indicating the mitotic exit. For Chk1-depleted cells, both Cdc2 and Plk1 activities remained higher than those of control cells, especially at 14 h after release, consistent with the M-phase block described above.
Fig. 5.

Chk1 is a negative regulator of Plk1 and Cdc2. (A) HeLa cells were synchronized by using the double thymidine block, released at different times, and analyzed with the antibodies indicated. (B) Cell lysates prepared as in A were subjected to anti-Plk1 IP kinase assays using TCTP as a substrate (Upper), or the autophosphorylation of Chk1 was examined by anti-Chk1 IP kinase assay (Lower). (C and D) Plk1 fails to be inactivated in Chk1-depleted cells during mitotic exit. HeLa cells were Chk1-depleted by using the protocol described in Fig. 2C, released at different times, and subjected to Western blotting using the antibodies indicated (C) and to Cdc2 or Plk1 IP kinase assay using H1 (Upper) or GST-TCTP (Lower), respectively, as a substrate (D). (E) Random-growing HeLa cells were Chk1-depleted by direct transfection of dsRNA targeting site 454. Two days after transfection, cells were UV-irradiated with 100 J/m2 and then treated with 100 ng/ml nocodazole for 10 h before harvest. Cell lysates were subjected to anti-Cdc2 (Top) or anti-Plk1 (Middle) IP kinase assay, or to anti-Plk1 immunoblotting (Bottom). (F) HeLa cells were Chk1-depleted by using the protocol described in Fig. 2C. After release into medium containing 100 ng/ml nocodazole for 10 h, cells were UV-irradiated with 100 J/m2 and incubated for a further 2 h in the presence of nocodazole before harvest. Cell lysates were subjected to anti-Plk1 IP kinase assay with GST-TCTP as a substrate. Lane 1, cells harvested after the double thymidine block as a G1 control; lanes 2–4, cells harvested 12 h after release.
Plk1 has been reported to be a target of the DNA damage checkpoint (6). In response to DNA damage, inhibition of Plk1 occurs in an Atm/r-dependent manner (7) and Chk1 is activated by Atm/r-associated phosphorylation. To investigate a possible connection between Chk1 and Plk1, Chk1-depleted cells were UV-irradiated with 100 J/m2, incubated with nocodazole for 10 h, and harvested. The cell lysates were subjected to anti-Cdc2 and anti-Plk1 IP kinase assays (Fig. 5E). In agreement with the previous data, UV irradiation caused significant inhibition of Plk1 activity. Chk1 depletion partially rescued the activity of Plk1, indicating that Chk1 may be a negative regulator of Plk1 in response to DNA damage. To rule out the possibility that the UV-induced Plk1 inactivation was due to a G2 arrest, we depleted Chk1 in synchronized cells as described in Fig. 2C. After release into medium containing nocodazole for 10 h to block cells at mitosis, cells were UV-irradiated and incubated for a further 2 h. Chk1 depletion also reversed UV irradiation-induced Plk1 inactivation under this condition, further suggesting that Chk1 negatively regulates Plk1 function during mitosis (Fig. 5F).
Release of Chk1 Depletion-Induced Mitotic Block by Inactivation of the Spindle Assembly Checkpoint.
Chromosome misalignment caused by Chk1 depletion likely will activate the spindle assembly checkpoint machinery. As shown in Fig. 9 (which is published as supporting information on the PNAS web site), the spindle checkpoint protein BubR1 was clearly detected as a punctate pattern during prophase and prometaphase of control cells and diffused quickly once the bipolar spindle was formed. However, the punctate pattern of BubR1 staining persisted much longer in Chk1-depleted cells during mitosis, suggesting activation of the spindle assembly checkpoint. Next, we determined whether codepletion of the spindle assembly checkpoint proteins would reverse Chk1 depletion-induced metaphase arrest. Toward that end, HeLa cells transfected with siRNA targeting one of two spindle checkpoint proteins, BubR1 or Mad2, were subjected to the double thymidine block, and Chk1 was depleted during the 8-h interval. Fourteen hours after release from the block, cells were harvested and analyzed with either Western blotting or immunofluorescence staining. This protocol efficiently codepleted BubR1 or Mad2 with Chk1, as indicated by Western blotting (Fig. 6A). Staining with phospho-H3 antibody revealed that Chk1 depletion increased the phospho-H3-positive cells from 2.6% to 20%, whereas codepletion of BubR1 or Mad2 decreased the phospho-H3-positive cells back to 2.0% and 7.6%, respectively (Fig. 6B), in agreement with the data presented in Fig. 9.
Fig. 6.
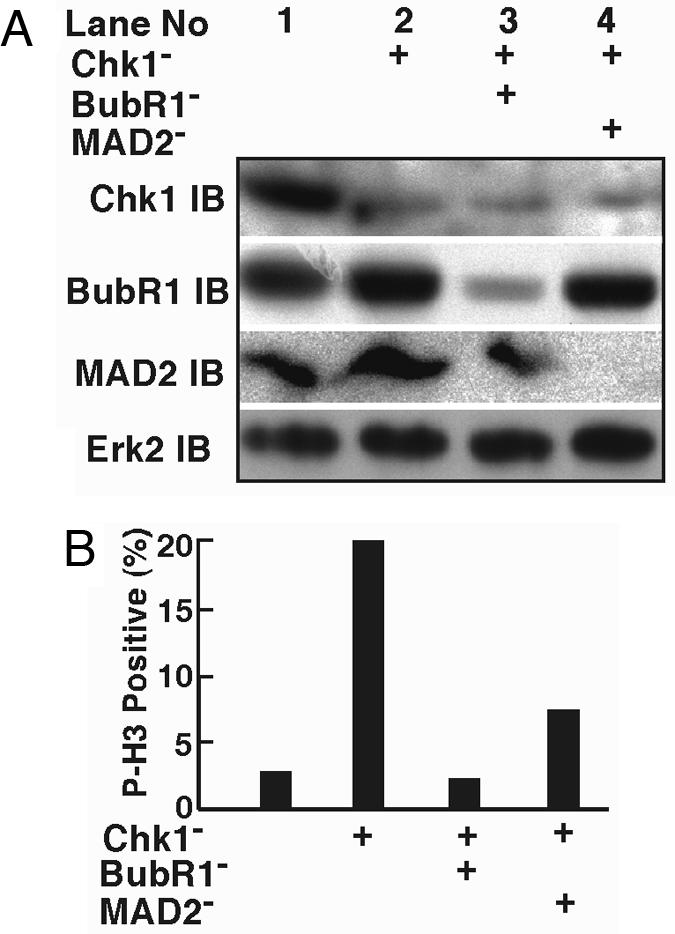
Codepletion of the spindle assembly checkpoint proteins relieves Chk1 depletion-induced mitotic arrest. (A) BubR1 or Mad2 was codepleted with Chk1 in synchronized HeLa cells, as described in the text. Fourteen hours after release from the double thymidine block, cells were harvested, lysed, and analyzed with Western blotting using the antibodies indicated. (B) Fourteen hours after release from the block, Chk1/BubR1- or Chk1/Mad2-codepleted cells were stained with phospho-H3 antibody.
Discussion
Although the function of Chk1 in response to DNA damage has been well documented, its role in normal cell cycle progression is controversial. In an early study (10), it was shown that knocking out the Chk1 gene in mice is embryonically lethal, suggesting that Chk1 is essential for early embryonic development. To support this notion, a conditional knockout of Chk1 induced apoptosis in embryonic stem cells in one cycle (11) and in proliferating mammary cells of adult mice (12). Very recently, a study using conditional Chk1 knockout mouse embryonic stem cells (13) further revealed that Chk1 depletion leads to premature activation of Cdc2/cyclin B and mitotic catastrophe. In striking contrast, by using gene targeting to eliminate Chk1 function in avian B-lymphoma cell line DT40, Zachos et al. (14) showed that Chk1-deficient tumor cells are viable but exhibit multiple checkpoint and survival defects. Moreover, it was reported that siRNA effectively depleting Chk1 did not alter the cell cycle profile or induce apoptosis in various human cancer cell lines under normal conditions (15).
In this study, we used various RNAi approaches to analyze Chk1 loss-of-function phenotypes in HeLa cells in the absence of DNA damage. We showed that Chk1 is essential for normal mitotic progression of HeLa cells, likely because Chk1 is involved in the proper alignment of chromosomes to the equatorial planes during metaphase. Chk1 depletion causes apparent defects in kinetochore functions, activates the spindle assembly checkpoint, and eventually leads to mitotic catastrophe. Thus, Chk1 is essential for HeLa cell proliferation under normal growth conditions. The apparent discrepancy between our Chk1 siRNA data and the data published in ref. 15 may be explained as follows. First, the vector-based RNAi approach we used allowed us to select transfection-positive cells with puromycin, so that relatively long-term effects after Chk1 depletion could be examined and defects in the cell cycle profile and proliferation could be observed. Second, coupling of siRNA treatment with cell synchronization allowed us to follow events during mitosis, especially potential defects associated with chromosome structure. Third, because several different sites were chosen to target Chk1, an off-target effect was unlikely. An independent investigation using siRNA to deplete Chk1 in human cells (16) also demonstrated that Chk1 is involved in normal cell cycle progression. The researchers proposed that Cdc25A is continually being phosphorylated by Chk1 on Ser-123, a site critical to controlling the stability of Cdc25A, in the absence of checkpoint activation, and that this phosphorylation contributes to its instability during an unperturbed cell cycle. DNA damage activates a pathway that targets phosphorylated Cdc25A for complete destruction (16).
Earlier studies (6, 7) showed that Plk1is a DNA damage target and that DNA damage-induced Plk1 inhibition is Atm/r-dependent. In the present study, we provide further evidence that DNA damage-induced Plk1 inhibition is also Chk1-dependent, suggesting that Chk1 is a negative regulator of Plk1 in response to DNA damage. Moreover, the fact that Plk1 activity increased significantly in Chk1-depleted, well-synchronized HeLa cells indicates that Chk1 acts as a negative regulator of Plk1 even during the normal cell cycle, and especially during late mitosis.
The spindle assembly checkpoint machinery monitors defects during prometaphase, such as kinetochores unattached by microtubules and lack of tension generated on kinetochores by microtubules, to ensure the fidelity of chromosome segregation (17). Of all of the checkpoint proteins identified, BubR1 and Mad2 are the two most extensively studied. Mad2 has been proposed to be a specific marker for unattached kinetochores, whereas BubR1 is involved in checkpoint activation due to either unattached kinetochores or lack of tension (17). In this study, we found that Chk1 depletion led to the formation of kinetochores unattached by microtubules in the chromosome regions that failed to align properly to the equatorial planes during metaphase. Immunofluorescence stainings with several kinetochore markers, such as Hec1, CREST, and Plk1, all indicated defects in kinetochore functions in Chk1-depleted cells, in agreement with the positive BubR1 staining of these cells. Results from reversal of the Chk1 depletion-induced metaphase block by codepletion of BubR1 or Mad2 further strengthen the statement that Chk1 is a negative regulator of the spindle checkpoint during the metaphase–anaphase transition.
In summary, the data presented here support the following model. During metaphase, Chk1 is required for proper chromosome alignment on the equatorial planes, and the spindle checkpoint is activated to ensure kinetochore attachments by microtubules. At the onset of anaphase, Chk1 negatively regulates Plk1 to inactivate the spindle assembly checkpoint machinery and cause subsequent mitotic exit. However, either the absence of Chk1 or an excess of Plk1 during late mitosis will result in failure to inactivate the spindle checkpoint and lead to mitotic arrest.
Materials and Methods
RNAi.
Two approaches were used to deplete Chk1 in HeLa cells. For direct transfection of siRNA, the targeting sequences are AAGCGTGCCGTAGACTGTCCA and GCAACAGTATTTCGGTATAAT, corresponding to the coding regions of positions 127–147 and 454–474 relative to the first nucleotide of the start codon. For vector-based RNAi, plasmid pBS/U6-Chk1 was constructed to target sequence GGTGGTTTATCTGCATG GTAT, corresponding to the coding region of positions 351–371 relative to the first nucleotide of the start codon. To specifically deplete BubR1 and Mad2, targeting sequences were TTATAGTGATCCTCGATTTCT and GGAAGAGCGCGCTCATAAA GG, corresponding to the coding regions of positions 345–365 and 609–629, respectively, relative to the first nucleotide of the start codon.
Supporting Information.
Cell culture, transfections, and immunoblotting are described in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank Eleanor Erikson for critical reading of the manuscript. This work was supported by National Institutes of Health Grant GM59172. R.L.E. is the John F. Drum American Cancer Society Research Professor. X.L. is a recipient of Howard Temin Award K01 CA114401 from the National Cancer Institute.
Abbreviations
- Chk1
checkpoint kinase 1
- Plk1
polo-like kinase 1
- IP
immunoprecipitation
- TCTP
translationally controlled tumor protein.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Chen Y., Sanchez Y. DNA Repair. 2004;3:1025–1032. doi: 10.1016/j.dnarep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Shieh S. Y., Ahn J., Tamai K., Taya Y., Prives C. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 3.Chen M. S., Ryan C. E., Piwnica-Worms H. Mol. Cell. Biol. 2003;23:7488–7497. doi: 10.1128/MCB.23.21.7488-7497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kramer A., Mailand N., Lukas C., Syljuasen R. G., Wilkinson C. J., Nigg E. A., Bartek J., Lukas J. Nat. Cell Biol. 2004;6:884–891. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- 5.Strebhardt K., Ullrich A. Nat. Rev. Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 6.Smits V. A., Klompmaker R., Arnaud L., Rijksen G., Nigg E. A., Medema R. H. Nat. Cell Biol. 2000;2:672–676. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- 7.van Vugt M. A., Smits V. A., Klompmaker R., Medema R. H. J. Biol. Chem. 2001;276:41656–41660. doi: 10.1074/jbc.M101831200. [DOI] [PubMed] [Google Scholar]
- 8.Kellogg D. R. J. Cell Sci. 2003;116:4883–4890. doi: 10.1242/jcs.00908. [DOI] [PubMed] [Google Scholar]
- 9.Syljuasen R. G., Sorensen C. S., Hansen L. T., Fugger K., Lundin C., Johansson F., Helleday T., Sehested M., Lukas J., Bartek J. Mol. Cell. Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takai H., Tominaga K., Motoyama N., Minamishima Y. A., Nagahama H., Tsukiyama T., Ikeda K., Nakayama K., Nakanishi M., Nakayama K. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q., Guntuku S., Cui X. S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., et al. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 12.Lam M. H., Liu Q., Elledge S. J., Rosen J. M. Cancer Cells. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 13.Niida H., Tsuge S., Katsuno Y., Konishi A., Takeda N., Nakanishi M. J. Biol. Chem. 2005;280:9246–9252. doi: 10.1074/jbc.M505009200. [DOI] [PubMed] [Google Scholar]
- 14.Zachos G., Rainey M. D., Gillespie D. A. EMBO J. 2003;22:713–723. doi: 10.1093/emboj/cdg060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z., Xiao Z., Chen J., Ng S. C., Sowin T., Sham H., Rosenberg S., Fesik S., Zhang H. Mol. Cancer Ther. 2003;2:543–548. [PubMed] [Google Scholar]
- 16.Zhao H., Watkins J. L., Piwnica-Worms H. Proc. Natl. Acad. Sci. USA. 2002;99:14795–14800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinsky B. A., Biggins S. Trends Cell Biol. 2005;15:486–493. doi: 10.1016/j.tcb.2005.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.