

Abstract
We report direct evidence for the existence of an iron(IV)–hydroxide. Resonance Raman measurements on chloroperoxidase compound II (CPO-II) reveal an isotope (18O and 2H)-sensitive band at νFe–O = 565 cm−1. Preparation of CPO-II in H2O using H218O2 results in a red-shift of 22 cm−1, while preparation of CPO-II in 2H2O using H2O2 results in a red-shift of 13 cm−1. These values are in good agreement with the isotopic shifts predicted (23 and 12 cm−1, respectively) for an Fe–OH harmonic oscillator. The measured Fe–O stretching frequency is also in good agreement with the 1.82-Å Fe–O bond reported for CPO-II. A Badger’s rule analysis of this distance provides an Fe–O stretching frequency of νBadger = 563 cm−1. We also present X-band electron nuclear double resonance (ENDOR) data for cryoreduced CPO-II. Cryogenic reduction (77 K) of the EPR-silent Fe(IV)OH center in CPO-II results in an EPR-active Fe(III)OH species with a strongly coupled (13.4 MHz) exchangeable proton. Based on comparisons with alkaline myoglobin, we assign this resonance to the hydroxide proton of cryoreduced CPO-II.
Keywords: cytochrome P450, ferryl, protonated-ferryl
Iron(IV)–hydroxides have been implicated in the catalytic cycles of a number of heme and nonheme enzymes. Bridging or terminal iron(IV)–hydroxides have been proposed for the di-iron centers in the mixed-valent form of ribonucleotide reductase (intermediate X) (1, 2) and the bound radical state of methane monooxygenase (intermediate R) (3). In heme enzymes, the putative rebound intermediates of cytochrome P450 (4) and nitric oxide synthase (5) are both thought to be iron(IV)–hydroxides, and the ferryl forms of chloroperoxidase (CPO) (6), horseradish peroxidase (HRP) (7), cytochrome c peroxidase (8), myoglobin (9), and catalase (10) all have recently been reported to be protonated. To date, however, there has been no direct evidence to support the existence of an Fe(IV)OH species in these or any other chemical system.
Reports of iron(IV)–hydroxides are notable. No synthetic or mineral iron–hydroxide above the ferric state has been structurally characterized, and no signal directly associated with the proton of an iron(IV)–hydroxide has ever been reported. Claims of Fe(IV)OH species in heme enzymes have been based on Fe–O bond distances obtained from x-ray structural techniques. Crystal structures of the ferryl forms of HRP, cytochrome c peroxidase, myoglobin, and catalase have yielded Fe–O bond lengths ranging from 1.84 to 1.92 Å (7–10), whereas x-ray absorption measurements on the ferryl form of CPO have provided an Fe–O distance of 1.82 Å (6). X-ray crystallography and x-ray absorption spectroscopy do not allow for the direct observation of protons. In each case, the assignment of an Fe(IV)OH species was based on the observed Fe–O bond length (1.84–1.92 Å), which was deemed too long to be indicative of an authentic Fe(IV)O species (rFe–O ≈ 1.65 Å).
The existence of iron(IV)–hydroxides in heme systems has been controversial. The long Fe–O bond lengths reported in crystal structures of the ferryl forms of HRP, cytochrome c peroxidase, and myoglobin are at odds with the much shorter (1.64–1.70 Å) oxo-like bond distances obtained from x-ray absorption spectroscopy (11–13). In hopes of clarifying the issue, we recently examined the applicability of Badger’s rule to heme iron–oxygen bonds (14). Badger’s rule is an empirical formula relating bond distance and vibrational frequency (15). A theoretical parameterization of Badger’s rule was obtained by examining >30 complexes. Calculations were performed on compounds with different axial-ligands, iron-oxidation, and oxygen-protonation states. The results were impressive. Given the calculated iron–oxygen bond length, Badger’s rule predicted the calculated vibrational frequency with an average error of 9 cm−1 for a 17-molecule training set and an average error of 19 cm−1 for a previously unexamined collection of heme and nonheme systems (14 molecules). Our study suggests that Badger’s rule is applicable to the iron-oxygen bonds of mononuclear oxo and hydroxo complexes.
A Badger’s rule analysis of the available resonance Raman, x-ray absorption, and crystallographic data suggests that the ferryl forms of HRP (pKa ≤ 4), cytochrome c peroxidase (pKa ≤ 4), and myoglobin (pKa ≤ 4) are not iron(IV)–hydroxides (14). The extended x-ray absorption fine structure and resonance Raman data reported for these systems are in good agreement with Badger’s rule (6, 11–13, 16–19), whereas the crystallographically determined bond distances deviate from it substantially (7–9). The long Fe–O bonds that have been reported in the crystal structures of these enzymes appear to be the hallmarks of ferric and ferrous hydroxides (14).
One complex is a clear outlier in the Fe(IV)OH debate: the ferryl form of the thiolate-ligated heme-enzyme CPO. It is the only ferryl system for which an Fe–O stretching frequency has been unobservable (20–22). Several workers have unsuccessfully attempted to locate an 18O-sensitive stretch in the region where authentic iron(IV)oxo stretching occurs, 655–875 cm−1 (20, 21). The assignment of CPO compound II (CPO-II) as an Fe(IV)OH is consistent with their results. X-ray absorption measurements put the Fe–O bond in CPO-II at 1.82 Å, in good agreement with calculations on a thiolate-ligated Fe(IV)OH porphyrin (rFe–O = 1.81 Å) (6). Based on the reported Fe–O bond length, we predicted that the Fe–OH stretch in CPO-II would be visible near νBadger = 563 cm−1 (14).
Here we report direct evidence for the existence of an Fe(IV)OH species in CPO-II. Resonance Raman measurements reveal an isotope (18O and 2H)-sensitive band at νFe–O = 565 cm−1. Preparation of CPO-II in H2O with H218O2 results in a red-shift of 22 cm−1, while preparation of CPO-II in 2H2O with H2O2 results in a red-shift of 13 cm−1. These shifts are in good agreement with the values expected for an Fe–OH harmonic oscillator (23 and 12 cm−1, respectively). We also present X-band electron nuclear double resonance (ENDOR) data for cryoreduced CPO-II. Reduction of the EPR-silent Fe(IV)OH center in CPO-II at 77 K results in an EPR-active ferric species with a strongly coupled (13.4 MHz) exchangeable proton. Based on comparisons with alkaline myoglobin, we assign this resonance to the hydroxide proton of cryoreduced CPO-II.
Results and Discussion
Resonance Raman Spectroscopy.
Low-frequency resonance Raman data (pH 6.5) for ferric CPO and the H216O2/H216O, H216O2/D216O, and H218O2/H216O preparations of CPO-II are shown in Fig. 1. Also shown in Fig. 1 are the oxidation-state marker band (ν4) and EPR spectrum of each sample (23). The EPR spectra and the position of ν4 indicate that our CPO-II samples are ≈95% pure: The EPR measurements reveal that the samples contain <5% ferric enzyme, while ν4 at 1,377 cm−1 (2 and 28 cm−1 higher than in ferric and ferrous CPO, respectively) is indicative of CPO-II. The radical signal near g = 2 is attributed to oxidized ascorbate (24).
Fig. 1.

Low-frequency resonance Raman data (454.5-nm excitation), oxidation-state marker band (ν4), and EPR spectra of ferric CPO and CPO-II samples (pH 6.5). EPR spectra indicate that CPO-II samples contained <5% ferric enzyme. The radical signal near g = 2 is attributed to oxidized ascorbate (24). Arrows highlight movement of the Fe(IV)–OH stretch with isotopic substitution. ∗, labels the 466-nm line of the argon-ion laser. No movement of the oxidation-state marker band (ν4) was observed during data collection. EPR measurements taken after sample irradiation revealed no detectable change in sample composition.
The frequency range associated with Fe(IV)–OH stretching in CPO-II is highlighted in Fig. 2. There, an overlay of the CPO-II spectra as well as their differences can be seen. The resonance Raman spectrum of the H216O2/H216O preparation of CPO-II (blue line; Fig. 2 Upper) reveals a peak at 565 cm−1. This stretch, which is not present in the ferric spectrum, shifts to 552 cm−1 in D2O (black line; Upper), and to 543 cm−1 when CPO-II is prepared with H218O2 (red line; Upper). The difference spectra (Fig. 2 Lower) indicate isotopic shifts of 13 and 22 cm−1, which are in good agreement with the values predicted for an Fe–OH harmonic oscillator (12 and 23 cm−1). The data presented in Figs. 1 and 2 provide direct evidence for the existence of an Fe(IV)OH species in CPO-II.
Fig. 2.

Low-frequency-resonance Raman data (454.5-nm excitation) of CPO-II. (Upper) Overlay of CPO-II spectra: 16OH (blue), 16OD (black), and 18OH (red). (Lower) Difference spectra reveal changes in νFe(IV)–OH upon deuterium (black) and 18O (red) substitutions.
The location of the iron–oxygen stretch in CPO-II and its 13-cm−1 downshift in D2O are unprecedented in an Fe(IV) complex. They are indicative of hydroxide ligation. Previous examinations of enzymatic ferryl species have reported iron–oxygen stretching frequencies ranging from 745 to 821 cm−1 (25). In some cases, small (≈3 cm−1 on average) upshifts in these frequencies have been reported in D2O (25, 26). These upshifts have been attributed to decreased hydrogen-bonding between the ferryl oxygen and a protonated distal residue. In contrast, the 13-cm−1 downshift in CPO-II results from a change from −OH to −OD ligation. This shift is similar to the change observed in myoglobin hydroxide. The alkaline form of myoglobin has an iron–oxygen stretch at νFe–OH = 550 cm−1, which shifts 12 cm−1 to νFe–OH = 538 cm−1 in D2O (27).
The νFe–OH = 565 cm−1 (rBadger = 1.82 Å) measured in CPO-II is in good agreement with the Fe–O bond distances obtained from density functional calculations (1.81 Å) and x-ray absorption measurements (1.82 Å) on CPO-II (6). A Badger’s rule analysis of these bond distance predicts νFe–OH ranging from 563 to 573 cm−1. The application of Badger’s rule to the νFe(III)–OH = 550 cm−1 stretch in myoglobin hydroxide provides an Fe–O bond distance of 1.83 Å, which is in good agreement with calculations on an imidazolate-ligated Fe(III)OH porphyrin (rFe–OH = 1.85 Å, νBadger = 533 cm−1) and an x-ray absorption near-edge spectroscopy investigation of alkaline myoglobin (rFe–OH = 1.84 Å, νBadger = 543 cm−1) (14, 28). It is interesting that the Fe(IV)–OH bond in CPO-II is only slightly (≈0.02 Å) shorter than the Fe(III)–OH bond in alkaline myoglobin. This result, which is consistent with theoretical calculations (14), reflects the strong electron-donating ability of the axial-thiolate. It is this donating ability that stabilizes the unusual iron(IV)–hydroxide state in CPO-II.
ENDOR Spectroscopy.
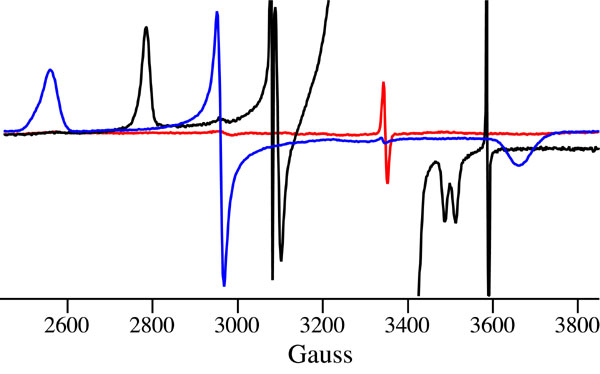
To further support our assignment of the Fe(IV)OH center in CPO-II, we performed X-band continuous-wave ENDOR measurements on the cryogenically reduced form of CPO-II (CR-CPO-II). The S=1 Fe(IV)OH center in CPO-II is EPR silent (see EPR spectra in Fig. 1). Radiolytic reduction of CPO-II at 77 K results in the formation of an EPR-active ferric hydroxide species (Fig. 3). The ENDOR spectrum of CR-CPO-II reveals a strongly coupled exchangeable proton resonance (13.4 MHz, 3,100 G) that is similar to the hydroxide resonance (11.4 MHz, 3,100 G) found in alkaline myoglobin. The ENDOR spectra of both species are shown in Fig. 4. By comparison, we assign the 13.4-MHz resonance of CR-CPO-II to a hydroxide proton.
Fig. 3.

Cryoreduction of CPO-II. A sample of CPO-II containing 20% glycerol was γ-irradiated at 77 K, resulting in reduction of the EPR-silent Fe(IV)OH center. The Fe(III)OH species generated by this process is EPR active, and ENDOR measurements revealed a very strongly coupled (13.4 MHz) exchangeable proton.
Fig. 4.

X-band continuous-wave ENDOR spectra of alkaline myoglobin hydroxide (Left) and cryoreduced CPO-II (Right) in H2O (red) and D2O (blue) centered at the proton Larmor frequency. ∗, indicates hydroxide resonances in H2O samples.
Conclusions
We have located the Fe(IV)–OH stretching mode in CPO-II. Resonance Raman measurements provide an iron–oxygen stretching frequency of 565 cm−1. This vibration downshifts 13 cm−1 in D2O, in good agreement with the isotopic shift expected (12 cm−1) for an Fe(IV)–OH harmonic oscillator. Both the location of the band and its shift in D2O are unprecedented in an Fe(IV) complex. The νFe–OH = 565 cm−1 measured in CPO-II is in excellent agreement with the 1.82-Å Fe–O bond reported for CPO-II. A Badger’s rule analysis of this distance provides an Fe–O stretching frequency of νBadger = 563 cm−1. X-band continuous-wave ENDOR measurements on the cryoreduced form of CPO-II further support our assignment of an Fe(IV)–OH center in CPO-II. The ENDOR spectrum of CR-CPO-II shows a strongly coupled exchangeable proton (13.4 MHz), which based on comparisons with myoglobin hydroxide, we assign to the hydroxide proton of CR-CPO-II.
Materials and Methods
CPO was obtained from Caldariomyces fumago and purified according to known procedures (29). CPO-II samples were prepared for resonance Raman spectroscopy by reacting a solution of 450 mM ascorbate and 1.5 mM 57Fe-enriched ferric CPO with 30 mM H2O2 (or H218O2) in a 2:1 mixture.† The reaction was quenched in liquid ethane (−180°C) 7 ms after mixing. Reagents were in 100 mM KPhos buffer (pH 6.5). Ethane was decanted and evaporated. Samples were packed into EPR tubes in liquid N2. Deuterated CPO-II was prepared as described above, but CPO was exchanged four times with D2O buffer, and all reagents were in D2O buffer. EPR measurements at 77 K revealed that the CPO-II samples contained <5% ferric enzyme. Resonance Raman spectra were acquired with a TriVista 555 triple monochromator (900/900/2400 gr/mm; Acton Research, Acton, MA) equipped with a CCD camera (1,340 × 100 pixels; Princeton Instruments, Princeton, NJ). The 454.5-nm line of an argon-ion laser (I-308; Coherent, Santa Clara, CA) was used for excitation. Power was <25 mW at the sample. 900/900/2,400 gr/mm gratings provided an instrumental resolution of 1.5 cm−1 at 454.5 nm (0.51 cm−1 per CCD pixel). Samples were held in an EPR finger-dewar (77 K) in an ≈135° back-scattering arrangement. The 2,326.5-cm−1 vibration of liquid N2 was used for calibration. A photoreduction/degradation study of CPO-II was performed in which the same sample-spot was irradiated continuously for >4 h (see Fig. 5, which is published as supporting information on the PNAS web site). No significant change in the sample’s spectrum was observed. The CPO-II oxidation-state marker band at 1,377 cm−1 did not change position over the course of 4 h (23), and no peaks associated with ferric CPO grew in. The CPO-II data presented in Fig. 2 had <3 h of exposure time. EPR measurements (77 K) taken after laser irradiation showed no detectable change in sample composition. All Raman spectra contained a smoothly varying background, which was removed, but the data were not smoothed or manipulated in any other way. Locations of the difference spectra extrema were obtained by performing a 21-point Savitzky–Golay smoothing. Application of different background subtractions and smoothing procedures resulted in minimal changes (<2 cm−1) in Fe(IV)–OH stretching frequencies.
Samples for cryoreduction and ENDOR spectroscopy were prepared by reacting a solution of 37.5 mM ascorbate and 4 mM ferric CPO with 75 mM peracetic acid in a 2:1 mixture. Reactions were quenched into liquid ethane 28 ms after mixing and packed into EPR tubes. Reagents were in 100 mM KPhos buffer (pH 6.5) and 20% (vol/vol) glycerol. Samples were examined by EPR and resonance Raman spectroscopy before cryoreduction. The EPR measurements revealed that the samples contained <5% ferric enzyme, whereas resonance Raman measurements indicated they were CPO-II. Samples were γ-irradiated (60Co; total dose of 4.5 Mrad) at the γ-irradiation facility of the Breazeale nuclear reactor at Pennsylvania State University. During irradiation, samples were maintained at 77 K by immersion in liquid N2. Alkaline myoglobin was prepared by dissolving horse heart myoglobin (Sigma, St. Louis, MO) in 50 mM borate buffer (pH/pD 10.4/10.9, where pD = −log[D3O+]). Continuous-wave X-band EPR and ENDOR measurements were performed on an Elexsys E-560 (Bruker, Billerica, MA) equipped with a liquid helium cryostat (Oxford Instruments, Oxon, U.K.) and a 150-W radiofrequency (rf) amplifier (see Fig. 4 and also Fig. 6, which is published as supporting information on the PNAS web site). Typical experimental conditions were as follows: sample temperature, 10 K; microwave frequency, 9.43 GHz; microwave power, 40 mW; rf modulation, 20 kHz; rf modulation depth of the rf field, 100 kHz; rf attenuation, 0 dB.
Supplementary Material
Acknowledgments
We thank Thomas Brunold, Troy Stich, Brian Bennett, Kurt Warncke, and David Britt for helpful discussions and Candace Davison and Paul Rankin for assistance with cryoreduction experiments at the Breazeale nuclear reactor facility. K.L.S. and R.K.B. received training in EPR and ENDOR techniques as well as travel support under the National Biomedical EPR Center Training Program (Candice Klug, Director; National Institutes of Health Grant EB001980). This work was supported by the National Science Foundation, the Petroleum Research Fund, and the Arnold and Mabel Beckman Foundation (Beckman Young Investigators Program). R.K.B. is supported by a grant from the Herman Frasch Foundation. M.T.G. is an Alfred P. Sloan Fellow.
Glossary
Abbreviations
- CPO
chloroperoxidase
- CPO-II
CPO compound II
- CR-CPO-II
cryogenically reduced form of CPO-II
- ENDOR
electron nuclear double resonance.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
†Protein obtained from the growth of C. fumago on minimal medium (57Fe-enriched protein) appeared to have less background fluorescence.
References
- 1.Que L., Jr, Dong Y. Acc. Chem. Res. 1996;29:190–196. [Google Scholar]
- 2.Burdi D., Sturgeon B. E., Tong W. H., Stubbe J., Hoffman B. M. J. Am. Chem. Soc. 1996;118:281–282. [Google Scholar]
- 3.Baik M.-H., Gherman B. F., Friesner R. A., Lippard S. J. J. Am. Chem. Soc. 2002;124:14608–14615. doi: 10.1021/ja026794u. [DOI] [PubMed] [Google Scholar]
- 4.Groves J. T. Proc. Natl. Acad. Sci. USA. 2003;100:3569–3574. doi: 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alderton W. K., Cooper C. E., Knowles R. G. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green M. T., Dawson J. H., Gray H. B. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 7.Berglund G. I., Carlsson G. H., Smith A. T., Szöke H., Henrikson A., Hajdu J. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 8.Bonagura C. A., Bhaskar B., Shimizu H., Li H., Sundaramoorthy M., McRee D. E., Goodin D. B., Poulos T. L. Biochemistry. 2003;42:5600–5608. doi: 10.1021/bi034058c. [DOI] [PubMed] [Google Scholar]
- 9.Hersleth H. P., Dalhus B., Gørbitz C. H., Andersson K. K. J. Biol. Inorg. Chem. 2002;7:299–304. doi: 10.1007/s007750100296. [DOI] [PubMed] [Google Scholar]
- 10.Gouet P., Jouve H.-M., Williams P. A., Andersson I., Andreoletti P., Nussaume L., Hajdu J. Nat. Struct. Biol. 1996;3:951–956. doi: 10.1038/nsb1196-951. [DOI] [PubMed] [Google Scholar]
- 11.Penner-Hahn J. E., Eble K. S., McMurry T. J., Renner M., Balch A. J., Groves J. T., Dawson J. H., Hodgson K. O. J. Am. Chem. Soc. 1986;108:7819–7825. doi: 10.1021/ja00284a054. [DOI] [PubMed] [Google Scholar]
- 12.Chance M., Powers L., Poulos T., Chance B. Biochemistry. 1986;25:1266–1270. doi: 10.1021/bi00354a011. [DOI] [PubMed] [Google Scholar]
- 13.Chance M., Powers L., Kumar C., Chance B. Biochemistry. 1986;25:1259–1265. doi: 10.1021/bi00354a010. [DOI] [PubMed] [Google Scholar]
- 14.Green M. T. J. Am. Chem. Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 15.Badger R. M. J. Chem. Phys. 1935;3:710–714. [Google Scholar]
- 16.Kincaid J. R., Zheng Y., Al-Mustafa J., Czarnecki K. J. Biol. Chem. 1996;271:28805–28811. doi: 10.1074/jbc.271.46.28805. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto S., Tatsuno Y., Kitagawa T. Proc. Natl. Acad. Sci. USA. 1986;83:2417–2421. doi: 10.1073/pnas.83.8.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto S., Teraoka J., Inubushi T., Yonetani T., Kitagawa T. J. Biol. Chem. 1986;261:11110–11118. [PubMed] [Google Scholar]
- 19.Sitter A. J., Reczek C. M., Terner J. Biochim. Biophys. Acta. 1985;264:14209–14215. doi: 10.1016/0167-4838(85)90301-2. [DOI] [PubMed] [Google Scholar]
- 20.Egawa T., Proshlyakov D. A., Miki H., Makino R., Ogura T., Kitagawa T., Ishimura Y. J. Biol. Inorg. Chem. 2001;6:46–54. doi: 10.1007/s007750000181. [DOI] [PubMed] [Google Scholar]
- 21.Egawa T., Miki H., Ogura T., Makino R., Ishimura Y., Kitagawa T. FEBS Lett. 1992;305:206–208. doi: 10.1016/0014-5793(92)80668-7. [DOI] [PubMed] [Google Scholar]
- 22.Hosten C. M., Sullivan A. M., Palaniappan V., Fitzgerald M. M., Terner J. J. Biol. Chem. 1994;269:13966–13978. [PubMed] [Google Scholar]
- 23.Sprio T. G. In: Iron Porphyrins. Lever A. P. B., Gray H. B., editors. Vol. II. New York: VCH; 1982. pp. 89–159. [Google Scholar]
- 24.Yamazaki I., Mason H. S., Piette L. J. Biol. Chem. 1960;235:2444–2449. [PubMed] [Google Scholar]
- 25.Terner J., Palaniappan V., Gold A., Weiss R., Fitzgerald M. M., Sullivan A. M., Hosten C. M. J. Inorg. Biochem. 2006;100:480–501. doi: 10.1016/j.jinorgbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Behan R. K., Green M. T. J. Biol. Inorg. Chem. 2006;100:448–459. doi: 10.1016/j.jinorgbio.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 27.Feis A., Marzocchi M. P., Paoli M., Smulevich G. Biochemistry. 1994;33:4577–4583. doi: 10.1021/bi00181a019. [DOI] [PubMed] [Google Scholar]
- 28.Longa S. D., Pin S., Cortès R., Soldatov A. V., Alpert B. Biophys. J. 1998;75:3154–3162. doi: 10.1016/S0006-3495(98)77757-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hashimoto A., Pickard M. A. J. Gen. Microbiol. 1984;130:2051–2058. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}