

Abstract
The Na/K pump establishes essential ion concentration gradients across animal cell membranes. Cardiotonic steroids, such as ouabain, are specific inhibitors of the Na/K pump. We exploited the marine toxin, palytoxin, to probe both the ion translocation pathway through the Na/K pump and the site of its interaction with ouabain. Palytoxin uncouples the pump’s gates, which normally open strictly alternately, thus allowing both gates to sometimes be open, so transforming the pump into an ion channel. Palytoxin therefore permits electrophysiological analysis of even a single Na/K pump. We used outside-out patch recording of Xenopus α1β3 Na/K pumps, which were made ouabain-resistant by point mutation, after expressing them in Xenopus oocytes. Endogenous, ouabain-sensitive, Xenopus α1β3 Na/K pumps were silenced by continuous exposure to ouabain. We found that side-chain charge of two residues at either end of the α subunit’s first extracellular loop, known to make a major contribution to ouabain affinity, strongly influenced conductance of single palytoxin-bound pump-channels by an electrostatic mechanism. The effects were mimicked by modification of cysteines introduced at those two positions with variously charged methanethiosulfonate reagents. The consequences of these modifications demonstrate that both residues lie in a wide vestibule near the mouth of the pump’s ion pathway. Bound ouabain protects the site with the strongest influence on conductance from methanethiosulfonate modification, while leaving the site with the weaker influence unprotected. The results suggest a method for mapping the footprint of bound cardiotonic steroid on the extracellular surface of the Na/K pump.
Keywords: Na,K-ATPase; palytoxin; single-channel conductance; cysteine modification; electrostatic mechanism
The Na,K-ATPase (Na/K pump) maintains the concentration gradients of Na and K ions across the surface membrane of animal cells by exporting 3 Na and importing 2 K at the expense of hydrolysis of a single ATP, up to 100 times each second. The cycle of potentially reversible steps that describes this cation exchange (1, 2) may be envisaged as that of a transmembrane ion channel with two gates, one on either side of the ion-binding sites (3, 4). ATP binding and phosphorylation-dephosphorylation reactions drive conformational changes that alternately open and close each gate to exchange ions at the cell interior or exterior while ensuring that both are never open (3–6).
Much is known about kinetic and energetic properties of major reaction steps of the Na,K-ATPase, but further understanding of their mechanisms requires detailed structural information. Important clues have come from recently determined crystal structures of several conformations of a related P type ATPase, the sarcoplasmic and endoplasmic reticular Ca-ATPase (7, 8). Nevertheless, reliable homology models of the Na,K-ATPase are limited to transmembrane domains and conserved cytoplasmic sequences of its α subunit, because of sequence divergence between extracellular loops of the Ca-ATPase and Na,K-ATPase α subunit, their similar topology notwithstanding (9), and because the Na,K-ATPase, unlike the Ca-ATPase, has a β subunit.
To probe the pathway negotiated by the Na and K ions transported by the Na/K pump, we exploit the ability of palytoxin (PTX), a marine toxin (10), to transform an Na/K pump into an ion channel (11), apparently by allowing both of the pump’s two gates to sometimes be open (6, 12). Use of PTX permits high-resolution electrical recording to be combined with mutagenesis and chemical modification to map the Na/K pump’s ion pathway. Cysteine-scanning mutagenesis of transmembrane segments believed to encompass binding sites for the transported Na and K ions suggests that PTX-bound “pump-channels” offer access to the pump’s ion translocation pathway (13).
A prerequisite for studying mutant Na/K pumps expressed in animal cells is a way to distinguish them from the cell’s own Na/K pumps. Because the latter are generally highly sensitive to cardiotonic steroids (e.g., digoxin or ouabain), a family of specific Na/K pump inhibitors, a suitable strategy is to express mutant ouabain-resistant pumps and work with enough ouabain to eliminate responses from endogenous Na/K pumps (for review, see refs. 14 and 15). Rat Na,K-ATPase α1 subunits form naturally ouabain-resistant Na/K pumps because of an Arg at the NH2-terminal end and an Asp at the COOH-terminal end of the predicted first extracellular loop that, if introduced into naturally ouabain-sensitive sheep Na/K pumps, are sufficient to render them ouabain resistant (16); ouabain resistance of sheep Na/K pumps is even greater if the order of the two introduced charged residues is switched (i.e., D-R instead of R-D; ref. 17). In testing ouabain-resistant Na/K pumps, we noted a striking difference between the single-channel conductances of PTX-bound pump-channels depending on whether the order of these residues was R-D or D-R. We show here that this difference reflects a stronger electrostatic influence of the residue at the COOH-terminal end of the loop, which resides in a vestibule near the entrance to the cation pathway. As might be anticipated for a ouabain affinity-determining amino acid, bound ouabain protects against chemical modification (cf. ref. 18) of a residue at that position. These findings suggest a way to map the footprint of ouabain bound to the surface of the Na/K pump.
Results
In outside-out patches excised from uninjected Xenopus oocytes, the presence of 100 μM ouabain effectively prevents current signals from endogenous, ouabain-sensitive, Xenopus Na/K pumps (believed to comprise Xenopus α1 and β3 subunits (19, 20) in response to application of picomolar concentrations of PTX (see Fig. 8, which is published as supporting information on the PNAS web site). In contrast, even in the continuous presence of 100 μM ouabain, picomolar [PTX] readily elicited pump-channel cation currents in patches from oocytes expressing exogenous, ouabain-resistant rat α1, (Fig. 1A) or Xenopus DR-α1 (Fig. 1B), together with Xenopus β3, subunits. The rat α1 subunit forms naturally ouabain-resistant Na/K pumps (largely attributable to residues R-D at the ends of the first extracellular loop; ref. 17), and the Xenopus DR-α1 was rendered ouabain-resistant by the corresponding double mutation Q120D-N131R (Fig. 1D; ref. 17). Because PTX binds tightly to Na/K pumps (6, 12), a nonsaturating concentration was applied until the first pump-channel opened and then withdrawn to avoid transformation of too many pumps into channels (Fig. 1 A and B).
Fig. 1.

Different conductances of alternative ouabain-resistant PTX-bound pump-channels. (A and B) Currents at –50 mV in outside-out patches excised from oocytes expressing rat α1 (A) or Xenopus DR-α1 (B) Na/K pumps in symmetrical 125 mM Na solutions with 100 μM external ouabain but no pipette ATP, briefly exposed to PTX (50 pM in A and 100 pM in B) until the first channel opening was observed. Lower traces show gating transitions of PTX-bound pump-channels at indicated voltages after washout of unbound toxin; dotted lines mark 0, 1, 2, or 3 open channels. (C) Current amplitudes for channels in A (circles) and B (triangles) plotted against voltage; fits (straight lines) between –100 and –20 mV gave channel conductances γ = 7.5 pS for A and γ = 1.8 pS for B. (D) Sequence alignment of WT Xenopus α1, Xenopus DR-α1, rat α1, and sheep α1 Na,K-ATPases, and rabbit SERCA Ca-ATPase; all residues are numbered from Met 1. Loop 1-2 assignment is from cardiotonic steroid-binding studies (e.g., ref. 14), not later SERCA structures.
Amplitudes of single pump-channel currents at several voltages (e.g., Fig. 1 A and B) yielded current-voltage plots (Fig. 1C) whose slopes revealed that the conductance (γ) of PTX-bound pump-channels comprising rat α1 subunits (γ = 8.2 ± 0.2 pS, n = 5) was severalfold larger than that of pump-channels containing Xenopus DR-α1 subunits (γ = 1.9 ± 0.1 pS, n = 6).
We wondered whether the switched positions of the charged residues responsible for ouabain resistance, at opposite ends of the extracellular loop connecting transmembrane helices 1 and 2 (Fig. 1D, loop 1–2), might underlie this difference in conductance. Because the amino acids at positions equivalent to 120 and 131 are Arg and Asp in the rat α1 subunit, but Asp and Arg in the Xenopus DR-α1 subunit, a possible explanation is that residue 131 might lie close to the extracellular mouth of the channel, where its charge could influence the local cation concentration (21, 22). To test this hypothesis, we needed a ouabain-resistant Xenopus α1 subunit (to distinguish it from endogenous Xenopus Na/K pumps) without charged substitutions at positions 120 and 131, so that we could examine their influence independently. This requirement was met by introducing the mutation C113Y in transmembrane helix 1 of Xenopus α1 (23) which, when coexpressed with Xenopus β3 subunits in oocytes, resulted in Na/K pump current that was half-maximally inhibited by ≈20 μM ouabain (data not illustrated). We used these ouabain-resistant C113Y Xenopus Na/K pumps as the background for all subsequent substitutions presented here.
Unitary current measurements in PTX-bound C113Y Na/K pump-channels with various substitutions at position 131 in the Xenopus α1 subunit showed the charge of that residue to be a major determinant of single-channel conductance (Fig. 2). With the native, neutral, Asn (N131; Fig. 2 A and D, circles), the conductance was 3.7 ± 0.2 pS (n = 5), similar to values obtained with other neutral residues, in the mutants N131C (γ = 2.8 ± 0.2 pS, n = 4) and N131Q (γ = 3.8 ± 0.4 pS, n = 2). But γ was larger for the negatively charged substitutes, in mutants N131D (Fig. 2 B and D, triangles; γ = 6.6 ± 0.2 pS, n = 7) and N131E (γ = 6.1 ± 0.5 pS, n = 2), and smaller for the positively charged substitutes, in N131R (Fig. 2 C and D, squares; γ = 0.9 ± 0.1 pS, n = 4) or N131K (γ = 1.0 ± 0.1 pS, n = 2).
Fig. 2.

Charge substitutions at position 131 influence single pump-channel conductance. (A–C) Unitary current transitions of PTX-bound pump-channels at the indicated voltages in outside-out patches expressing C113Y Xenopus α1 Na,K-ATPase mutants with native Asn (A), or with Asp (B) or Arg (C), at position 131. Solutions are as in Fig. 1; brief PTX (100 pM) application is not illustrated in B and C. (D) Current–voltage plots for channels in A (N131; circles, γ = 3.6 pS), B (N131D; triangles, γ = 5.5 pS), and C (N131R; squares, γ = 0.95 pS).
This simple correlation of conductance with charge of the side chain at residue 131 suggested that the charge might electrostatically influence the local cation concentration near the mouth of the pump-channel (24). In that case, appropriate experimental changes of external cation concentration would be expected to overcome the effects of the charged mutants on pump-channel conductance. The dependence on external Na activity of single-channel conductance of mutants bearing different charges (Fig. 3) confirmed this expectation. Michaelis–Menten fits of the conductance-activity curves gave maximal single-channel conductance: γmax = 18 ± 2 pS and K0.5 = 330 ± 55 mM Na activity for the neutral side chain (N131; circles), γmax = 21 ± 1 pS and K0.5 = 189 ± 12 mM Na activity for the negatively charged side chain (N131D; triangles), and, with the constraint γmax ≤ 22 pS, K0.5 = 1,899 ± 1,187 mM for the positively charged side chain (N131R; squares). Thus, as evident in Fig. 3, the increase in single-channel conductance resulting from replacement of N131 with the acidic Asp (Fig. 2) could be overcome by a 2- to 3-fold reduction of external Na activity, and the reduction in conductance caused by introduction of the basic Arg could be overcome by a severalfold increase of external Na activity.
Fig. 3.
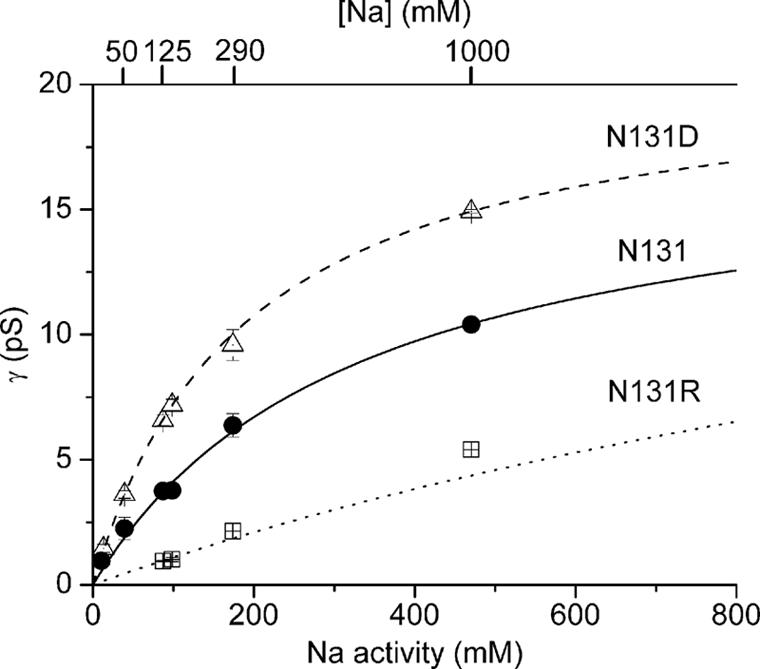
Dependence on Na activity of conductance of single pump-channels with neutral N131 (circles), negative N131D (triangles), or positive N131R (squares) residue at position 131. Experiments are as in Figs. 1 and 2 but with varying external (and internal; Materials and Methods) Na concentration (upper abscissa). Na activity coefficients (from 0.90 to 0.47 for [Na] 7.5 to 1,000 mM) were estimated from the osmolality of Na-sulfamate solutions (25). Curves show Michaelis–Menten fits (see Results for parameters) to mean data.
Additional evidence that these changes in single-pump-channel conductance simply reflect alteration of the charge at position 131 comes from experiments in which the engineered Cys in N131C Na/K pumps was covalently modified by reaction with similarly sized sulfhydryl-directed methanethiosulfonate (MTS) reagents, either neutral or bearing a single positive or negative charge (Fig. 4). Macroscopic current measurements in outside-out patches monitored, first, the rapid conversion of thousands of Na/K pumps to pump-channels by exposure to PTX (Fig. 4 A–C). When the Cys at position 131 was covalently modified by the cationic 2-trimethylaminoethyl methanethiosulfonate (MTSET+), leaving a positively charged adduct linked via a disulfide bond, the macroscopic conductance was sharply decreased (Fig. 4 A and D). This effect persisted after washout of the MTS reagent but was readily reversed by the reducing agent DTT (10 mM; which also caused a small, poorly understood, reversible current decrease). In similar patches, the neutral 2-aminocarbonylethyl methanethiosulfonate (MTSACE) did not detectably alter PTX-induced conductance (Fig. 4 B and D), despite covalently modifying all of the Cys, demonstrated by the ineffectiveness of subsequently applied MTSET+. The anionic 2-sulfonatoethyl methanethiosulfonate (MTSES−), on the other hand, sharply and persistently increased PTX-bound pump-channel conductance Fig. 4 C and D).
Fig. 4.

Residue 131 is water accessible in PTX-bound pump-channels. (A–C) Macroscopic current recordings of N131C Na/K pumps at −50 mV in outside-out patches in 125 mM Na solutions with 100 μM ouabain and 5 mM pipette MgATP, exposed (as indicated) to 100 nM PTX to open thousands of pump-channels. Brief switches from Na to poorly permeant N-methyl-d-glucamine (NMG) or tetramethylammonium (TMA) served to verify pipette-membrane seal integrity. Repeated triangular disturbances of current records (also Figs. 5 and 6) mark application of 100-ms voltage steps to gather current-voltage data. (A) One millimolar MTSET+ diminished PTX-induced inward current, but 10 mM DTT slowly restored it; DTT also caused a rapid and reversible current decrease, which was not studied further. (B) One millimolar MTSACE did not alter PTX-induced current but prevented MTSET+ from decreasing it. (C) One millimolar MTSES− increased PTX-induced current. (D) Mean steady-state PTX-induced current, normalized to control at −100 mV, plotted vs. voltage, before MTS reaction (control; circles, n = 11) or after MTSET+ (squares, n = 7), MTSACE (stars, n = 2), or MTSES− (triangles, n = 2). Linear fits between −100 and −20 mV yielded slopes (in mV−1) of 0.011 for control, 0.003 after MTSET+, 0.010 after MTSACE, and 0.019 after MTSES−.
The current-voltage relationships in Fig. 4D show that macroscopic conductance was unchanged by neutral MTSACE, decreased ≈3-fold by cationic MTSET+, and roughly doubled by anionic MTSES−, closely similar to the effects on single-channel conductance of mutations that replaced N131 with amino acids containing neutral, basic, or acidic side chains (e.g., Fig. 2D). These findings admit two conclusions. First, the rate of extracellular Na ion entry into a PTX-bound pump-channel can be modulated by an apparently electrostatic mechanism involving the charge of the moiety at position 131, indicating that this residue lies near the entrance to the cation pathway. Second, pump-channel conductance responds to the charge, but not the bulk, of the moiety at that position, because all three MTS reagents leave an adduct ≈5 × 6 Å. The amino acid at position 131 therefore must reside in a space near the channel mouth wide enough to accommodate the neutral MTSACE adduct without sterically impeding Na ion flow into the channel.
Its rapid reaction with the hydrophilic MTS reagents (Fig. 4) demonstrates that position 131 is water accessible in the presence of PTX. But is that accessibility a consequence of PTX action? We exploited the ability of DTT to reverse the effect of MTSET+ to demonstrate that MTSET+ reacts with Cys at 131 before PTX application (Fig. 5A). We first exposed N131C Na/K pumps to 2 mM MTSET+ for 5 min and then washed it out, after which PTX caused only a small conductance increase. Moreover, reexposure to MTSET+ then had no effect (Fig. 5A), suggesting that all Cys already had been modified during the first MTSET+ exposure before PTX. That modification was confirmed by the large conductance increase caused by DTT and a subsequent large decrease on reexposure to MTSET+ (Fig. 5A). We conclude that PTX is not required for the residue at position 131 to be accessible to an MTS reagent.
Fig. 5.

Residue 131 is water accessible without PTX. (A) Outside-out patch with N131C Na/K pumps were held at –50 mV and exposed for 5 min to 2 mM MTSET+ in the presence of 1 μM ouabain. After [ouabain] was raised to 100 μM, PTX (100 nM) elicited a small inward current that was unaltered by reexposure to MTSET+ but was greatly increased by 20 mM DTT; a third exposure (≈20 s) to MTSET+ abolished that current. (B) Similar to A except that [ouabain] was much higher, 10 mM, during the 5-min exposure to 2 mM MTSET+. After lowering [ouabain] to 100 μM, PTX (100 nM) induced a large inward current that was rapidly decreased by a second exposure (≈20 s) to MTSET+.
The apparent affinity with which ouabain impairs Na/K pump function is strongly influenced by the nature of the residue at position 131 (17). So we tested whether the presence of bound ouabain would alter accessibility of the Cys in N131C Na/K pumps to MTSET+. We again applied MTSET+ for 5 min, but this time in the continuous presence of 10 mM ouabain (Fig. 5B), and then washed it out and applied PTX, which induced the usual large conductance increase. Reexposure to the MTSET+ promptly reduced that conductance by approximately two-thirds (as in Figs. 4 A and D), and that fraction was fully restored by DTT, indicating that none of the Cys had been modified by MTSET+ during the simultaneous exposure to ouabain. Ouabain bound to the pumps therefore had prevented MTSET+ from reaching the otherwise accessible sulfhydryl side chain of the Cys at ouabain affinity-determining position 131.
Although the Arg at position 131 in Xenopus DR-α1 Na/K pump subunits therefore can account, at least qualitatively, for the smaller conductance of the resulting PTX-bound pump-channels compared with those of rat α1 (RD) Na/K pumps, we wondered whether the charge at the NH2-terminal end of that first extracellular loop, at position 120, also influences channel conductance. Ouabain apparent affinity is lowered by substitutions that introduce a charged residue at that position (16, 17), which is a Gln in native ouabain-sensitive Xenopus α1 Na/K pumps (Fig. 1D), but an Arg in rat α1 and an Asp in Xenopus DR-α1, both ouabain-resistant Na/K pump subunits. We therefore characterized modification by MTS reagents of the engineered Cys in Q120C Na/K pumps (Fig. 6). The normalized current–voltage curves before and after modification show that, as in N131C Na/K pumps (Fig. 4D), positively charged MTSET+ decreased, whereas negatively charged MTSES− increased, PTX-induced conductance (Fig. 6A), consistent with an electrostatic action. But these effects at position 120, conductance reduction by approximately one-third or increase by approximately one-quarter (Fig. 6A), were much smaller than those at position 131 (Fig. 4D), suggesting that position 120 might be more distant from the channel mouth. Further distinguishing position 120 from 131, bound ouabain could not protect the Cys at position 120 from modification by MTSET+ (Fig. 6B). After exposure of Q120C Na/K pumps to MTSET+ in the presence of 10 mM ouabain, and then washout of MTSET+, PTX application opened ≈1,000 pump-channels, which were unaffected by reexposure to MTSET+. The Cys at position 120 all must have been modified during the first exposure to MTSET+, in the presence of 10 mM ouabain, which means that bound ouabain did not prevent the MTS reagent from reaching those Cys residues.
Fig. 6.

MTS modification of Q120C Na/K pump-channels. (A) Mean steady-state PTX-induced current–voltage plots, normalized to control current at −90 mV; experiment and solutions as in Fig. 4. Relative to control (circles, n = 8), MTSET+ decreased current ≈30% (squares, n = 4) and MTSES− increased current ≈25% (triangles, n = 4). Linear fits between −100 and −20 mV gave slopes 0.011 mV−1 before MTS application, 0.008 mV−1 after MTSET+, and 0.016 mV−1 after MTSES−. (B) Bound ouabain (10 mM) does not protect Q120C Na/K pumps from modification by MTSET+; experiment and solutions as in Fig. 5B. After lowering [ouabain] to 100 μM, PTX induced current that was unaffected by reexposure to MTSET+, indicating that all Cys had been modified by MTSET+ in 10 mM ouabain.
Discussion
Our results show that a cysteine introduced at ouabain affinity-determining position 131 is accessible to MTS reagents in functioning Na/K pumps in the absence of PTX (Fig. 5A), that the cysteine resides in a space large enough to accept a ≈5-Å × 6-Å neutral adduct without impeding cation flow through PTX-bound pump-channels (Fig. 4), but that position 131 lies close enough to the ion-pathway entrance for its side-chain charge to strongly influence cation flow (Figs. 2–4). Because PTX-bound pump-channels are almost always open under the macroscopic recording conditions of Fig. 4 (presence of ATP and absence of K; refs. 6 and 12), the similar effects on macroscopic and single-channel conductance of charge (positive or negative) deposition at position 131, by MTS reagent and mutagenesis, respectively, argue that they result from through-space electrostatic forces. That raising or lowering bulk external Na activity can compensate for the conductance changes due to altered charge at position 131, and that maximal conductance seems little affected (Fig. 3), argues that the electrostatic forces reduce or increase the local activity of Na ions at the pump-channel mouth, as reported for other cation (e.g., refs. 21 and 22) or anion (e.g., refs. 26 and 27) channels. The apparent independence of these effects on conductance from the bulk of the introduced side chain or reaction adduct, and the ready accessibility of sulfhydryls at position 131 (regardless of PTX), suggests that extracellular cations approach and leave the translocation pathway via a wide vestibule.
These findings mesh well with expectations based on structures of the related sarcoplasmic reticular Ca-ATPase (7, 8). Because the Na/K pump α subunit and Ca-ATPase sequences are sufficiently homologous within their transmembrane domains, other than in extracellular loops (but cf. ref. 9), the locus of Na,K-ATPase α- and β-subunit interactions, Ca-ATPase structures may be used to interpret Na,K-ATPase data (28, 29). No Ca-ATPase structure so far shows an open luminal ion translocation pathway (7, 8, 30), but structural alterations of the ion-binding pockets between principal conformations led to the proposal that Ca ions reach the sarcoplasmic reticulum via a path suggested by a crevice between transmembrane (TM) segments TM4 and TM6 in MgF-complexed Ca-ATPase (PDB ID code, 1WPG; ref. 7). At the outer end of TM6, marked by Ca-ATPase residue P789, the proposed pathway broadens, leading released ions past the TM1-TM2 loop toward the lumen of the sarcoplasmic reticulum (Fig. 7).
Fig. 7.

Luminal view of E2P-related Ca-ATPase structure (PDB ID code 1WPG; ref. 7). The backbone carbons of E79 and A87 (equivalent to Q120 and N131 in Xenopus α1 Na,K-ATPase and to R118 and D129 in rat α1 Na,K-ATPase; see Fig. 1D) are 15.9 Å and 10.8 Å, respectively, distant from P789 (equivalent to T806 in Xenopus α1). Figure prepared with PyMOL (www.pymol.org).
The residue equivalent to P789 is a Thr in all Na,K-ATPase α subunits, in which it has been suggested to play a role in regulating access of external cations to their binding sites (31). In other work (32), we have found that this Thr marks a point of narrowing of the ion pathway of PTX-bound Na/K pump-channels, reinforcing structural similarity between the Na,K-ATPase α subunit and Ca-ATPase in this region. Fig. 7 offers a luminal view of the latter and highlights residues A87 and E79, corresponding to N131 and Q120 in the Xenopus Na,K-ATPase, giving their distances from P789 as rough estimates of their proximity to the narrow channel entrance. These distances, ≈11Å and 16 Å, are consistent (even allowing for uncertainties of up to ≈6 Å due to unknown side-chain or MTS-adduct orientation) with our observed strong electrostatic influence on channel conductance of charge at position 131 and weaker, although similar, effect at position 120. Our results therefore would support similar distances separating the corresponding residues in the Na/K pump.
Most importantly, these results help to define further the binding site on the Na/K pump for cardiotonic steroids, which for centuries have been administered to treat congestive heart failure (33). Cardiotonic steroids target the pump’s extracellular surface. Mutagenesis has identified residues in the outermost segments of TM1, TM2, TM4, TM5, TM6, and TM7, and their connecting loops, that influence apparent affinity of ouabain binding or action (14, 17, 23, 34–37). Although evidence for direct interaction of these residues with cardiotonic steroids is lacking, the consensus interpretation pictures the steroid docking on the Na/K pump’s external surface, interacting with many of the identified amino acids (14, 38–40). Identification of the conserved Thr atop TM6 (34) and neighboring residues in the TM5-TM6 hairpin loop (35) as key determinants of ouabain apparent affinity led to the suggestion that cardiotonic steroids might act by preventing motion of the TM5-TM6 hairpin loop, thereby impeding ion passage to or from the deeper binding sites between TM4, TM5, and TM6 (35). In accord with such a mechanism, binding of ouabain traps two Na ions inside the phosphorylated Na/K pump (41), and cardiotonic steroid can bind to, and shut, PTX-bound Na/K pump-channels (see Fig. 8; also ref. 12).
Although these effects could all be explained by ouabain binding elsewhere, it is intriguing that omeprazole inhibits gastric H/K pumps, close relatives of Na/K pumps, by reacting directly with the Cys analogous to the Na/K pump’s conserved TM6-Thr (42). And mutating that Thr to Cys renders the Na/K pump susceptible to inhibition by omeprazole (31), while making it less sensitive to inhibition by ouabain (31, 36). This correlation, together with similarities between omeprazole inhibition of H/K pumps and cardiotonic steroid inhibition of Na/K pumps, supports the idea that cardiotonic steroids dock in the space above TM6 shown in Fig. 7. Indeed, two recent models posit that ouabain docks in that cavity, albeit with very different orientations (39, 40). Our observation that bound ouabain prevents access of MTS reagent to a Cys substituted at position 131 (Fig. 5B) but fails to protect a Cys at position 120 (Fig. 6B) does not allow us to distinguish between those two proposed orientations. But, taken together, our results suggest that cardiotonic steroids bind near the ion-translocation pathway in a manner that leaves residue 120 uncovered while impeding MTS access to position 131 (Figs. 5B, 6B, and 8). This finding provides the basis for a method of mapping the footprint of a bound steroid, and, hence, of distinguishing between docking models (39, 40), by evaluating the steroid’s ability to protect introduced Cys from modification by hydrophilic suflhydryl reagents, such as MTSET+.
Materials and Methods
Molecular Biology.
Substitutions in the Xenopus α1 subunit (gift from R. F. Rakowski, Ohio University, Athens, OH) were made by QuikChange (Stratagene, La Jolla, CA) and verified by automated sequencing. The double mutation Q120D/N131R (17) or the single substitution C113Y (23) both yielded ouabain-resistant Xenopus α1 subunits; the latter also was insensitive to the MTS reagents used here and so formed the template for other mutations. cDNA in pSD5 vector was transcribed by using mMessage machine SP6 (Ambion, Austin, TX). Xenopus laevis oocytes were injected with a 50-nl mixture of 10 ng of Xenopus β3 (20) cRNA and 30 ng of ouabain-resistant α1 cRNA (native rat α1 or mutated Xenopus α1). Injected oocytes were incubated at 18°C for 1–3 days before recording.
Electrophysiology.
Macroscopic or microscopic currents in outside-out patches (43) at 21–24°C were filtered at 1 KHz, recorded with an Axopatch 200B, and sampled at 10 KHz with a digidata 1322A A/D board by using pClamp 9. Data also were continuously acquired at lower rates with a Minidigi 1A (Axon Instruments, Union City, CA) and were analyzed with Clampfit 9 and Origin 7.0 (MicroCal, Originlab Corp., Northampton, MA). Borosilicate glass (PG52151-4; WPI, Sarasota, FL) pipettes pulled on a vertical puller (PP-83; Narishige, Tokyo, Japan) were coated with Sylgard for microscopic measurements. Filled with standard internal solution, pipette resistance was 10–30 MΩ for microscopic and 2–6 MΩ for macroscopic measurements (for which 80–90% of series resistance was compensated). Microscopic currents were filtered at 50–150 Hz to resolve the smallest events (e.g., Fig. 2C Bottom). After correction for small baseline drifts, all-points histograms were fitted with sums of Gaussians; average differences between adjacent peaks plotted against voltage yielded single-channel current–voltage relationships, and linear fits between −100 and −20 mV gave single-channel conductance. Data are given as mean ± SEM.
Solutions.
Standard external solution contained 125 mM NaOH, 120 mM sulfamic acid, 10 mM Hepes, 5 mM BaCl2, 1 mM MgCl2 and 0.5 mM CaCl2 (pH 7.4, 245–255 mOsm/kg). Standard internal (pipette) solution contained 125 mM NaOH, 100 mM sulfamic acid, 20 mM HCl, 10 mM Hepes, 1 mM MgCl2, and 1 mM EGTA, (pH 7.4, 245–250 mOsm/kg). For macroscopic current measurements, internal solution also included 5 mM MgATP. For measurements of the [Na] dependence of single pump-channel conductance (Fig. 3), the 5 mM Ba was omitted from external solutions to preclude possible charge-screening effects at low external [Na]; single-channel conductance with 125 mM symmetrical [Na] was the same with or without 5 mM Ba. The high [Na] external solution contained 1,000 mM NaOH, 990 mM sulfamic acid, 10 mM Hepes, 1 mM MgCl2 and 0.5 mM CaCl2 (pH 7.4, 1,800 mOsm/kg) and was used with internal solution containing 1,040 mM NaOH, 990 mM sulfamic acid, 10 mM Hepes, 10 mM tetraethylammonium Cl, 1 mM MgCl2, and 10 mM EGTA (pH 7.4, 1,800 mOsm/kg). Intermediate [Na] external solutions containing 145 or 290 mM Na were obtained by mixing the 125 mM Na (without Ba) and 1M Na external solutions and were used with the standard 125 mM Na internal solution. Low [Na] external solutions contained 15 or 50 mM NaOH, 10 mM Hepes, 1 mM MgCl2, 0.5 mM CaCl2, plus sulfamic acid to pH 7.4 and sucrose to 250 mOsm/kg, and were used with internal solution containing 7.5 mM NaOH, 230 mM sucrose, 10 mM Hepes, and 1 mM MgCl2, (pH 7.4 with HCl, 255 mOsm/kg): This concentration of internal sucrose did not affect pump-channel conductance, because, with 145 mM external Na, single-channel conductance at negative potentials was the same with standard 125 mM Na internal solution as with internal solution containing 7.5 mM Na and 230 mM sucrose.
All external solutions included 100 μM (unless otherwise indicated) ouabain to avoid signals from endogenous Na/K pumps. PTX (Calbiochem) was diluted into external solutions from a 100 μM aqueous stock solution. All PTX-containing external solutions included 0.001% BSA and 1 mM Na-borate (44). MTS reagents (Toronto Research Biochemicals, North York, ON, Canada) were kept on ice as 100 mM aqueous stock solutions until dilution into external solutions immediately before use. During prolonged (≥2-min) MTS applications, MTS-containing external solution reservoirs were refreshed at 1.5-min intervals to maintain reactivity (45).
Supplementary Material
Acknowledgments
We thank Nazim Fataliev for technical assistance, Paola Vergani for help with initial mutagenesis, Martin Mense and Daniel Peluffo for comments on an early draft, Nicolás Reyes for discussion, and R. F. Rakowski for providing Xenopus α1 Na,K-ATPase cDNAs. The work was supported by National Institutes of Health Grant HL36783 (to D.C.G.) and a fellowship from the Vicente Trust (to P.A.).
Glossary
Abbreviations
- MTS
methanethiosulfonate
- MTSACE
2-aminocarbonylethyl methanethiosulfonate
- MTSES−
2-sulfonatoethyl methanethiosulfonate
- MTSET+
2-trimethylaminoethyl methanethiosulfonate
- PTX
palytoxin
- TM
transmembrane.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Post R., Sen A. M., Rosenthal A. S. J. Biol. Chem. 1965;240:1437–1445. [PubMed] [Google Scholar]
- 2.Albers R. W. Annu. Rev. Biochem. 1967;36:727–756. doi: 10.1146/annurev.bi.36.070167.003455. [DOI] [PubMed] [Google Scholar]
- 3.Patlak C. S. Bull. Math. Biophys. 1957;19:209–235. [Google Scholar]
- 4.Vidaver G. A. J. Theor. Biol. 1966;10:301–306. doi: 10.1016/0022-5193(66)90128-7. [DOI] [PubMed] [Google Scholar]
- 5.Laüger P. Biochim. Biophys. Acta. 1979;552:143–161. doi: 10.1016/0005-2736(79)90253-0. [DOI] [PubMed] [Google Scholar]
- 6.Artigas P., Gadsby D. C. Proc. Natl. Acad. Sci. USA. 2003;100:501–505. doi: 10.1073/pnas.0135849100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toyoshima C., Nomura H., Tsuda T. Nature. 2004;432:361–368. doi: 10.1038/nature02981. [DOI] [PubMed] [Google Scholar]
- 8.Moller J. V., Nissen P., Sorensen T. L., Maire M. Curr. Opin. Struct. Biol. 2005;15:387–393. doi: 10.1016/j.sbi.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y. K., Kaplan J. H. J. Biol. Chem. 2000;275:19185–19191. doi: 10.1074/jbc.M000641200. [DOI] [PubMed] [Google Scholar]
- 10.Moore R. E., Scheuer P. J. Science. 1971;172:495–498. doi: 10.1126/science.172.3982.495. [DOI] [PubMed] [Google Scholar]
- 11.Scheiner-Bobis G., Meyer zu Heringdorf D., Christ M., Habermann E. Mol. Pharmacol. 1994;45:1132–1136. [PubMed] [Google Scholar]
- 12.Artigas P., Gadsby D. C. J. Gen. Physiol. 2004;123:357–376. doi: 10.1085/jgp.200308964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horisberger J. D. Physiology. 2004;19:377–387. doi: 10.1152/physiol.00013.2004. [DOI] [PubMed] [Google Scholar]
- 14.Lingrel J. B., Argüello J. M., van Huysse J., Kuntzweiler T. A. Ann. N.Y. Acad. Sci. 1997;834:194–206. doi: 10.1111/j.1749-6632.1997.tb52251.x. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan J. Annu. Rev. Biochem. 2002;71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 16.Price E. M, Lingrel J. B. Biochemistry. 1988;27:8400–8408. doi: 10.1021/bi00422a016. [DOI] [PubMed] [Google Scholar]
- 17.Price E. M., Rice D. A., Lingrel J. B. J. Biol. Chem. 1990;265:6638–6641. [PubMed] [Google Scholar]
- 18.Pedemonte C., Kaplan J. H. Am. J. Physiol. 1990;258:C1–C23. doi: 10.1152/ajpcell.1990.258.1.C1. [DOI] [PubMed] [Google Scholar]
- 19.Verrey F., Kairouz P., Schaerer E., Fuentes P., Geering K., Rossier B. C., Kraehenbuhl J. P. Am. J. Physiol. 1989;256:F1034–F1043. doi: 10.1152/ajprenal.1989.256.6.F1034. [DOI] [PubMed] [Google Scholar]
- 20.Good P. J., Richter K., Dawid I. B. Proc. Natl. Acad. Sci. USA. 1990;87:9088–9092. doi: 10.1073/pnas.87.23.9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imoto K., Busch C., Sakmann B., Mishina M., Konno T., Nakai J., Bujo H., Mori Y., Fukuda K., Numa S. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- 22.MacKinnon R., Latorre R., Miller C. Biochemistry. 1989;28:8092–8099. doi: 10.1021/bi00446a020. [DOI] [PubMed] [Google Scholar]
- 23.Canessa C. M., Horisberger J. D., Louvard D., Rossier B. C. EMBO J. 1992;11:1681–1687. doi: 10.1002/j.1460-2075.1992.tb05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green W. N., Andersen O. S. Annu. Rev. Physiol. 1991;53:341–359. doi: 10.1146/annurev.ph.53.030191.002013. [DOI] [PubMed] [Google Scholar]
- 25.Bonicamp J. M., Loflin A., Clark R. W. J. Chem. Educ. 2001;78:1541–1543. [Google Scholar]
- 26.Middleton R. E., Pheasant D. J., Miller C. Nature. 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- 27.Chen M. F., Chen T. Y. J. Gen. Physiol. 2003;122:133–145. doi: 10.1085/jgp.200308844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sweadner K., Donnet C. Biochem. J. 2001;356:685–704. doi: 10.1042/0264-6021:3560685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogawa H., Toyoshima C. Proc. Natl. Acad. Sci. USA. 2002;99:15977–15982. doi: 10.1073/pnas.202622299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picard M., Toyoshima C., Champeil P. J. Biol. Chem. 2006;281:3360–3369. doi: 10.1074/jbc.M511385200. [DOI] [PubMed] [Google Scholar]
- 31.Capendeguy O., Horisberger J. D. J. Physiol. 2005;565:207–218. doi: 10.1113/jphysiol.2004.080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reyes N., Gadsby D. C. Nature, 2006 doi: 10.1038/nature05129. in press. [DOI] [PubMed] [Google Scholar]
- 33.Blaustein M. P., Robinson S. W., Gottlieb S. S., Balke C. W., Hamlyn J. M. Mol. Interv. 2003;3:68–72. doi: 10.1124/mi.3.2.68. [DOI] [PubMed] [Google Scholar]
- 34.Burns E. L., Price E. M. J. Biol. Chem. 1993;268:25632–25635. [PubMed] [Google Scholar]
- 35.Palasis M., Kuntzweiler T. A., Arguello J. M., Lingrel J. B. J. Biol. Chem. 1996;271:14176–14182. doi: 10.1074/jbc.271.24.14176. [DOI] [PubMed] [Google Scholar]
- 36.Qiu L. Y., Koenderink J. B., Swarts H. G., Willems P. H., De Pont J. J. J. Biol. Chem. 2003;278:47240–47244. doi: 10.1074/jbc.M308833200. [DOI] [PubMed] [Google Scholar]
- 37.Crambert G., Schaer D., Roy S., Geering K. Mol. Pharmacol. 2004;65:335–341. doi: 10.1124/mol.65.2.335. [DOI] [PubMed] [Google Scholar]
- 38.Middleton D. A., Rankin S., Esmann M., Watts A. Proc. Natl. Acad. Sci. USA. 2000;97:13602–13607. doi: 10.1073/pnas.250471997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu L. Y., Krieger E., Schaftenaar G., Swarts H. G., Willems P. H., De Pont J. J., Koenderink J. B. J. Biol. Chem. 2005;280:32349–32355. doi: 10.1074/jbc.M505168200. [DOI] [PubMed] [Google Scholar]
- 40.Keenan S. M., DeLisle R. K., Welsh W. J., Paula S., Ball W. J., Jr. J. Mol. Graph. Model. 2005;23:465–475. doi: 10.1016/j.jmgm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Stürmer W, Apell H. J. FEBS Lett. 1992;300:1–4. doi: 10.1016/0014-5793(92)80151-6. [DOI] [PubMed] [Google Scholar]
- 42.Besancon M., Simon A., Sachs G., Shin J. M. J. Biol. Chem. 1997;272:22438–22446. doi: 10.1074/jbc.272.36.22438. [DOI] [PubMed] [Google Scholar]
- 43.Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 44.Bottinger H, Beress L., Habermann E. Biochim. Biophys. Acta. 1986;861:165–176. doi: 10.1016/0005-2736(86)90576-6. [DOI] [PubMed] [Google Scholar]
- 45.Karlin A., Akabas M. H. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}