

Abstract
Pemphigus vulgaris (PV) is a life-threatening autoimmune blistering skin disease characterized by detachment of keratinocytes (acantholysis). It has been proposed that PV IgG might trigger signaling and that this process may lead to acantholysis. Indeed, we recently identified a rapid and dose-dependent phosphorylation of p38 mitogen-activated protein kinase (p38MAPK) and heat shock protein (HSP) 27 after binding of PV antibodies to cultured keratinocytes. In human keratinocyte cultures, inhibitors of p38MAPK prevented PV IgG-induced phosphorylation of HSP27 and, more importantly, prevented the early cytoskeletal changes associated with loss of cell–cell adhesion. This study was undertaken to (i) determine whether p38MAPK and HSP25, the murine HSP27 homolog, were similarly phosphorylated in an in vivo model of PV and (ii) investigate the potential therapeutic use of p38MAPK inhibition to block blister formation in an animal model of PV. We now report that p38MAPK inhibitors prevented PV blistering disease in vivo. Targeting the end-organ by inhibiting keratinocyte desmosome signaling may be effective for treating desmosome autoimmune blistering disorders.
Keywords: autoimmune, signaling
Pemphigus vulgaris (PV) is a life-threatening autoimmune blistering disease where the autoimmune response targets the epidermis and mucosal epithelia, resulting in flaccid blisters and erosions. The loss of epithelial integrity disrupts the skin barrier function, putting patients at risk for infection as well as fluid and electrolyte imbalance. Before the introduction of systemic corticosteroids, the disease was highly lethal. Although the mortality has been reduced through the use of steroids and potent immunosuppressive drugs, the disease remains lethal, and patients often suffer from and may succumb to the secondary effects of the medications used to treat the disease.
In PV, IgG autoantibodies that bind to the surface of epithelial cells are pathogenic. IgG purified from PV patient sera causes blistering in mouse models (1, 2). In the PV IgG passive transfer model, autoantibodies purified from patient sera bind to keratinocyte desmoglein 3 (dsg3) (3–5) and induce loss of cell–cell adhesion, reproducing the clinical and histological features of the human disease (1, 2). In both the human disease and the PV mouse model, gentle friction of perilesional skin causes sloughing of epidermal sheets (Nikolsky’s sign). Although the model mimics aspects of the disease, the molecular mechanisms of blister formation remain unresolved. In the passive transfer model, the epidermal cell–cell detachment induced by PV autoantibodies is neither Fc- (6), complement- (7), nor plasminogen activator-dependent (8). Thus, the PV passive transfer mouse model represents an end-organ damage model triggered by anti-dsg3 autoantibodies.
It has been proposed that PV IgG might trigger signaling and that this process may lead to acantholysis (9–16). Indeed, we recently identified a rapid and dose-dependent phosphorylation of p38 mitogen-activated protein kinase (p38MAPK) and heat shock protein (HSP) 27 after binding of PV antibodies to cultured keratinocytes (14). PV IgG-induced phosphorylation of p38MAPK and HSP27 was followed by (i) remodeling of the actin cytoskeleton and (ii) keratin intermediate filament retraction. In human keratinocyte cultures, inhibitors of p38MAPK prevented PV IgG-induced phosphorylation of HSP27 and, more importantly, prevented the early cytoskeletal changes associated with loss of cell–cell adhesion (14). We suggested that the observed effects may be important to the mechanism of PV IgG-induced acantholysis because HSP27 has been shown to regulate both actin (17–19) and intermediate filaments (20, 21). Furthermore, p38MAPK-mediated phosphorylation of HSP27 had been shown to regulate the cytoskeleton (22–24). Collectively, these observations suggested that inhibition of this signaling pathway in epidermal epithelia (Fig. 1) could be used to prevent end-organ damage (e.g., blistering) caused by autoantibodies in PV.
Fig. 1.

Model of the molecular mechanism of acantholysis in PV. Autoantibody binding to the desmosome cadherin dsg3 on the surface of epidermal keratinocytes activates sequential phosphorylation of p38MAPK, MAPKAP kinase 2, and HSP27, which is associated with keratin filament retraction, actin cytoskeletal remodeling, and loss of cell–cell adhesion, leading to suprabasilar acantholysis in the skin. Inhibition of keratinocyte p38MAPK blocks these events in tissue culture and blister formation in vivo.
If keratinocyte p38MAPK and HSP27 phosphorylation are part of the acantholytic mechanism, then inhibiting them might prevent PV IgG-induced blistering in an animal model of the disease. This study was undertaken to (i) determine whether p38MAPK and HSP25, the murine HSP27 homolog, were similarly phosphorylated in an in vivo model of PV and (ii) investigate the potential therapeutic use of p38MAPK inhibition to block blister formation in an animal model of PV.
To test whether p38MAPK inhibitors could block blister formation in vivo, we have taken advantage of the PV mouse model developed in our laboratories (1). In this model, the IgG fraction from patients and control nonaffected normal individuals is purified and passively transferred into neonatal mice (1, 25), reproducing the clinical and histological features of the human disease. We now demonstrate that inhibitors of p38MAPK prevent blister formation in this PV mouse model.
Results
Neonatal mice were injected intradermally (i.d.) with PV and normal IgG (1.0 or 1.5 mg/g body weight) as described (6). The p38MAPK inhibitors were administered i.d. in two doses. One of 6.25 μg of SB202190 was given 2 h before the i.d. injection of IgG. The second dose of the same amount of inhibitor was mixed with PV or control IgG and injected i.d. Each animal received a total dose of 12.5 μg of inhibitor. After 18 h, the skin of neonatal mice from the test and control groups was examined clinically and histologically as described (1, 25). Perilesional skin biopsies were examined by direct immunofluorescence (IF) for the presence of PV IgG bound to the epidermal epithelium. Serum samples were collected from the test animals and analyzed for the presence of circulating anti-dsg3 antibodies by ELISA by using the recombinant human dsg3 ectodomain as described (26). Consistent with previous reports, mice injected with PV IgG developed blisters and a positive Nikolsky’s sign (Fig. 2 and Table 1). Histological examination revealed suprabasilar acantholysis (Fig. 3 and Table 2). In contrast, mice treated with the p38MAPK inhibitor SB202190 and pathogenic PV IgG failed to develop blisters, clinically (Fig. 2 and Table 1) and histologically (Fig. 3 and Table 2). Direct IF of nonlesional skin from both PV IgG and PV IgG plus SB202190 mouse skin demonstrated PV antibodies bound to the epidermal keratinocyte cell surface, indicating that the inhibitor did not prevent or alter the binding of PV autoantibodies to the target organ (Fig. 4). Furthermore, analyses of serum samples showed a similar level of anti-dsg3 autoantibodies in the circulation of both PV IgG-treated and PV IgG plus SB202190-treated mice (Fig. 5). These results indicate that the inhibitor did not prevent the diffusion of IgG to the tissue target, i.e., epidermis, nor did they inhibit systemic absorption of the injected IgG. This observation provides further support that the inhibitor was mediating its anti-acantholytic effects by targeting epidermal keratinocytes.
Fig. 2.

Inhibiting p38MAPK prevents clinical blistering in PV passive transfer mice. Neonatal C57BL/6J mice were injected i.d. with either PV IgG (1.5 mg of IgG/g body weight) (Left) or PV IgG (1.5 mg of IgG/g body weight) plus SB202190 (Right). After 18 h, the skin of neonatal mice from the test and control groups was examined clinically. PV IgG-treated mice have a positive Nikolsky’s sign (white arrows), demonstrating loss of epithelial cell–cell adhesion. In contrast, mice treated with the SB202190 and PV IgG have a negative Nikoslky’s sign, indicating that epithelial adhesion remains intact.
Table 1.
Inhibition of clinical disease
*Neonatal mice injected with PV IgG (1.5 mg/g) or PV IgG plus SB202190 and examined 18 h later for Nikolsky’s sign. A total of 24 mice per group were injected. Eleven of 12 mice injected with PV IgG had a positive Nikoslky’s sign, whereas only 1 of the 12 mice injected with PV IgG plus SB202190 had a positive Nikolsky’s sign.
Fig. 3.

Inhibiting p38MAPK prevents histologic blistering in PV passive transfer mice. Skin biopsies of mice treated with control IgG (1 mg of IgG/g body weight) (Upper Left), PV IgG (1 mg of IgG/g body weight) (Upper Right), SB202190 (Lower Left), or SB202190 and then PV IgG (Lower Right) were fixed in formalin and stained with hematoxylin/eosin. Suprabasilar acantholysis leading to blister formation (∗) is seen in PV IgG-treated mice but is blocked in mice treated with SB202190 and PV IgG (PV IgG plus SB202190).
Table 2.
Inhibition of histological disease
*Neonatal mice were injected with PV IgG (1.0 mg/g) and examined 18 h later for presence of blister formation by microscopic examination of H&E-stained skin biopsy sections. A total of 24 mice were injected. Eleven of 12 mice injected with PV IgG developed suprabasilar acantholysis, whereas only 1 of the 12 mice injected with PV IgG plus SB202190 demonstrated histological evidence of blistering.
Fig. 4.

The inhibitor does not prevent the diffusion of IgG to the epidermis. Perilesional skin biopsies from control (A), PV IgG-treated (B), and PV IgG plus SB202190-treated (C) mice were examined for the presence of human anti-dsg3 PV IgG by direct immunofluorescence by using a mouse anti-human Cy-2-conjugated monoclonal antibody. A honeycomb pattern of staining in the epidermis (arrows) is seen in both PV IgG- and PV IgG plus SB202190-treated mice, demonstrating that the inhibitor does not prevent binding of PV autoantibodies to the keratinocyte cell surface.
Fig. 5.
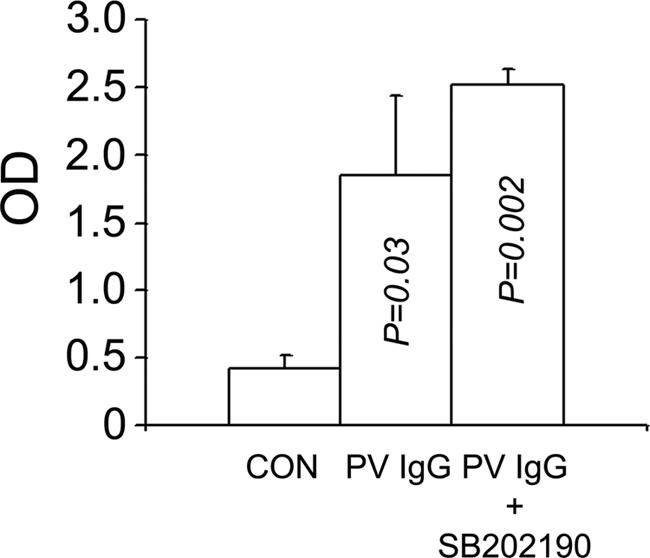
Anti-dsg3 IgG serum levels. The inhibitor does not prevent systemic absorption of the injected IgG. Serum samples obtained from control, PV IgG-treated, and PV IgG plus SB202190-treated mice were examined for the presence of anti-dsg3 autoantibodies by using a dsg3 ectodomain-based ELISA. P value is compared with control; n = 3; SD is shown by error bars. P values were calculated by using the Student t test.
We next examined the phosphorylation state of p38MAPK and HSP25, the murine HSP27 homolog, because our previous observations in human keratinocyte cell cultures demonstrated that both proteins were phosphorylated when keratinocyte cultures were exposed to PV IgG. Consistent with these prior observations, skin extracts from PV IgG-treated mice demonstrated increased phosphorylation of both p38MAPK and HSP25 (Fig. 6). The same amount of total p38MAPK immunoreactivity was present in skin extracts from control, PV IgG, and PV IgG plus inhibitor-treated mice. In contrast, increased phospho-p38MAPK immunoreactivity was observed in skin from PV IgG-treated mice; this PV IgG-activated increase in p38MAPK phosphorylation was prevented by treating mice with the p38MAPK inhibitor SB202190, suggesting a role for p38MAPK autophosphorylation (27) in the acantholytic process. When skin extracts were separated by 2D electrophoresis and immunoblotted with antibodies to HSP25, increased amounts of the most negatively charged HSP25 isoform were observed in PV IgG-treated mice (Fig. 6 C and D). Increased phosphorylation of this HSP25 isoform was inhibited in mice treated with PV IgG and the p38MAPK inhibitor SB202190. Thus, both PV IgG-induced p38MAPK and HSP25 phosphorylation were prevented when mice were treated with SB202190. A second inhibitor of p38MAPK, SB203580, also reduced blister formation in the PV mouse model although the inactive analog SB202474 did not (Fig. 7).
Fig. 6.

Inhibition of PV IgG-mediated p38MAPK and HSP27 phosphorylation in skin of PV IgG plus SB202190-treated mice. Neonatal C57BL/6 WT mice were injected i.d. with control IgG (CON; 1 mg of IgG/g body weight), PV IgG (1 mg of IgG/g body weight), or SB202190 and then PV IgG (PV IgG plus SB202190). Skin biopsies were obtained after 18 h of treatment and extracted in IEF lysis buffer. (A) Samples were equally loaded on and separated by SDS/PAGE, transferred to PVDF, and immunoblotted with antibodies to p38MAPK, phospho-p38MAPK, or lactate dehydrogenase (LDH) as a loading control. Blots were developed by enhanced chemiluminescence (ECL) reaction (Amersham Pharmacia). (B) Signal intensity from the ECL reaction for each band was quantified with a GeneGnome scanner (Syngene Bio Imaging) by using GeneSnap software (n = 3, SD shown by error bars). Total levels of p38MAPK are similar in control, PV IgG-treated, and PV IgG plus SB202190-treated mice. Increased amounts of phospho-p38MAPK are present in PV IgG-treated mice (P value compared with control); this increase is blocked in mice treated with SB202190 and PV IgG (no statistically significant difference for p38MAPK phosphorylation in PV IgG plus SB202190 compared with controls, P = 0.45, demonstrating in vivo block of p38MAPK phosphorylation). (C) Increased amounts of the most negatively charged HSP25 isoform (P2) were observed in PV IgG-treated mice and blocked in mice treated with PV IgG plus SB202190. Skin extracts (30 μg) were prepared and separated in the first dimension by using 7 cm, pH 4–7 IPGphor strips (Amersham Pharmacia Biosciences) and in the second dimension by 10% SDS/PAGE, followed by immunoblotting with antibodies to murine HSP25 as described (14). (D) Signal intensity from the ECL reaction for each spot corresponding to the 2D gel HSP25 charge isoforms labeled P0, P1, and P2 were quantified as above with a GeneGnome scanner and GeneSnap software (n = 3; SD shown by error bars) and expressed as a percentage of total HSP25 by using the formula Pn/(P0 + P1 + P2), where Pn corresponds to the signal intensity for n = spot 0, 1, or 2 and P0 + P1 + P2 is the summed signal intensity for all three HSP25 isoforms. An increase in the percentage of the most negatively charged HSP25 isoform, P2, is observed in skin extracts from PV IgG vs. control-treated mice (∗, P = 0.04). This increase is blocked in mice pretreated with SB202190 (∗∗, P = 0.04 compared with PV IgG-treated mice). P values were calculated by using the Student t test.
Fig. 7.

A second p38MAPK inhibitor, SB203580, but not the inactive analog SB202474, blocks blister formation and p38MAPK phosphorylation in PV IgG-treated mice. Skin biopsies of mice treated with PV IgG (1 mg of IgG/g body weight) (A), PV IgG plus SB203580 (B), or PV IgG plus SB202474 (C) were fixed in formalin and stained with hematoxylin/eosin. Suprabasilar acantholysis is seen in PV IgG-treated mice as well as mice pretreated with the inactive analog SB202474 but is blocked in mice pretreated with SB203580. (D) Skin biopsies were extracted in IEF lysis buffer, and extracts (30 μg) were equally loaded on and separated by SDS/PAGE, transferred to PVDF, and immunoblotted with antibodies to p38MAPK (p38) or phospho-p38MAPK (phospho-p38). Blots were developed by ECL reaction.
Discussion
The cadherin family of cell adhesion molecules is important in developmental biology (28), cell adhesion, neoplasia (29), and human autoimmunity (30). Interestingly, in squamous epithelium, the desmosomal cadherins dsg1 and dsg3 mediate cell surface–cytoskeleton interactions that result in adhesive forces that maintain the integrity of these tissues (31). Cell signaling may play a critical role in these interactions. It has been known that patients with pemphigus foliaceus (PF) and PV, two organ-specific autoimmune diseases of squamous epithelium, manufacture anti-dsg1 and anti-dsg3 autoantibodies, which are pathogenic by passive transfer experiments into recipient animals. These autoantibodies bind epitopes located on the desmoglein ectodomain and trigger cell signaling that is associated with collapse of the cytoskeleton and cell detachment in vitro (14).
Exposure of human keratinocytes to pathogenic PV IgG results in the phosphorylation of a number of intracellular proteins. We observed increased p38MAPK and HSP27 phosphorylation in human keratinocyte tissue cultures exposed to PV IgG. PV IgG-mediated phosphorylation of p38MAPK and HSP27 was rapid, time- and dose-dependent events, occurring within 15 min of addition of PV IgG (14). The rapid onset of these events in tissue culture suggested that they may represent some of the earliest events induced by PV IgG before loss of cell–cell adhesion. These initial tissue culture studies suggested a central role for p38MAPK in the mechanism of acantholysis; however, in vivo testing in an animal model of PV was necessary to demonstrate a role for p38MAPK in the acantholytic mechanism of the disease.
We observed that inhibitors of p38MAPK blocked blister formation in vivo. Two different inhibitors of p38MAPK, SB202190 and SB203580, inhibited PV IgG-induced blister formation in the passive transfer mouse model although this effect was more pronounced with SB202190. Importantly, the inhibitor did not prevent systemic absorption of the PV IgG nor did it prevent the binding of PV IgG to the keratinocyte cell surface, indicating that the biologic effect of the inhibitor was occurring downstream of antibody binding to keratinocyte dsg3.
HSP27 and the related murine protein HSP25 function in signal transduction where they may regulate the actin (17–19) and intermediate filament (20, 21) cytoskeleton. Additionally, p38MAPK-mediated phosphorylation of HSP27 modulates its regulation of the cytoskeleton (22–24). Phosphorylation of p38MAPK and HSP25 was observed in the skin of PV IgG-treated mice, confirming that the same phosphorylation events we had observed in tissue culture also occurred in vivo. Furthermore, the PV IgG-induced phosphorylation of p38MAPK and HSP25 was blocked in the skin of mice treated SB202190, and this inhibition of phosphorylation correlated with the inhibition of blister formation. The in vivo inhibition of PV IgG-mediated p38MAPK phosphorylation by SB202190 suggests that p38MAPK autophosphorylation (27) may be part of the acantholytic process.
Taken together, our observations in the PV IgG passive transfer model demonstrate that p38MAPK inhibitors can prevent skin blistering by inhibiting PV IgG-activated signaling in epidermal cells targeted by PV autoantibodies. In the passive transfer mouse model, a single dose of pathogenic human autoantibodies is administered to the test animal. In the human disease, there is ongoing production of autoantibodies. Thus, in PV patients, continuous dosing with inhibitor will be required to block antibody-induced acantholysis. Although the compounds used in this study are effective at blocking keratinocyte p38MAPK and antibody-induced acantholysis, they are not appropriate for clinical use in patients suffering from pemphigus due to toxic side effects. Several newer generation p38MAPK inhibitors are currently in clinical trials for inflammatory joint disease (32). The use of p38MAPK inhibitors in PV may be a particularly attractive and practical approach for treating the life-threatening autoimmune skin disease should these newer compounds prove safe in people.
Methods
Materials.
Rabbit polyclonal anti-HSP25 antibodies were from StressGen (Victoria, BC, Canada), rabbit polyclonal anti-p38MAPK antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), monoclonal anti-phospho-p38MAPK antibodies were from Cell Signaling Technology (Beverly, MA), and polyclonal anti-lactate dehydrogenase V (LDH) antibodies were from Cortex Biochem (San Leandro, CA). The p38MAPK inhibitors SB202190 and SB203580 and the inactive analog SB202474 were from Calbiochem (La Jolla, CA).
IgG Preparation.
PV sera (mucocutaneous) have been described (33). Data presented are from IgG purified from a single PV patient whose serum was available in sufficient quantities to carry out the described studies (the activity of this serum was determined by indirect immunofluorescence on sectioned monkey esophagus with a titer of 1:640). Two additional sera were tested and demonstrated similar results. The PV IgG were purified from PV patient sera by ammonium sulfate precipitation followed by affinity chromatography on Protein G (HiTrap; Amersham Pharmacia, Piscataway, NJ) as described (14). IgG fractions were dialyzed against PBS and sterile filtered. Purity was confirmed by SDS/PAGE, and activity was assayed by indirect IF and ELISA. Control IgG (no activity by indirect IF) were prepared in parallel from normal human sera.
Passive Transfer Mouse Model.
Breeding pairs of C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained at the University of North Carolina Division of Laboratory Animal Medicine Facility in accordance with International Animal Care and Use Committee protocols. Neonatal mice (24–36 h old with body weights between 1.4 and 1.6 g) were used for passive transfer experiments. Neonates were injected i.d. with a sterile solution of either control IgG or PV IgG as described (1, 34, 35). For direct clinical examination, mice were injected with PV or control IgG at 1.5 mg/g body weight in a total volume of 50 μl of PBS. This dose of PV IgG resulted in gross sloughing of the skin. The skin of neonatal mice from the test and control groups was examined 18 h after the injection of IgG for the presence of Nikolsky’s sign, in which gentle friction of perilesional skin causes sheet-like sloughing of the epidermis. A second group of animals received a lower dose of PV IgG (1.0 mg/g body weight in 50 μl of PBS) to preserve the cutaneous architecture lost by epithelial sloughing at the higher dose. After clinical examination, the animals were killed, and skin and serum specimens were obtained for routine histological examination by using light microscopy (hematoxylin/eosin staining) and direct IF assays to detect keratinocyte cell surface-bound pemphigus IgG. Serum samples were assayed for the presence of circulating human anti-dsg3 by IgG ELISA against baculovirus-expressed ectodomains of human dsg3 as described (26). Additional skin samples were harvested to prepare protein extracts for SDS/PAGE and 2D gel electrophoresis. After transfer to PVDF, membranes were probed by immunoblot for proteins of interest. For in vivo inhibitor studies, mice were preinjected with 6.25 μg of SB202190 in 50 μl i.d., and then reinjected i.d. with 6.25 μg of SB202190 plus PV IgG/50 μl (total of 12.5 μg of SB202190 at 2 h). Various doses of inhibitor were tested for their ability to block PV IgG-induced acantholysis. The dose of 12.5 μg of SB202190 was selected based on its ability to consistently block acantholysis. Data representative of the dose response are presented in Fig. 8, which is published as supporting information on the PNAS web site. No increased mortality was observed in the inhibitor vs. control mice. As a control, a second group of mice (n = 3) was pretreated with the inactive analog SB202474 (total of 12.5 μg of SB202470) (36) following the split dose protocol used for SB202190. Additionally, a third group of mice (n = 3) was pretreated with the p38MAPK inhibitor SB203580 (total of 25 μg of SB202470) following the split dose protocol used for SB202190. Blister formation was reduced with SB203580; however, the effect was more pronounced with SB202190.
2D Gel Electrophoresis.
Extracts were prepared from skin biopsies by Dounce homogenization in IEF lysis buffer (8 M urea/4% CHAPS/2.5 mM DTT/40 mM Tris/10 μM pepstatin/100 μM leupeptin/10 μM E-64/1 mM PMSF). Protein concentration was by modified Bradford as described (37). IPG buffer (pH 4–7; Amersham Pharmacia) was added to each sample to a final concentration of 0.5% before isolelectric focusing. Samples were separated in the first dimension by using 7 cm, pH 4–7, linear IPGphor strips (Amersham Pharmacia) and in the second dimension by 10% SDS/PAGE. Gels were transferred to PVDF membranes for immunoblot analysis. Western blots were developed by ECL reaction, and the signal intensity was quantified by scanning chemiluminescence on a GeneGnome scanner (Syngene Bio Imaging, Frederick, MD) by using GeneSnap software. The signal intensity for each HSP25 isoform was expressed as a percentage of total HSP25 by using the formula Pn/(P0 + P1 + P2), where n corresponds to the signal intensity for spot 0, 1, or 2 and P0 + P1 + P2 is the summed signal intensity for all three HSP25 isoforms. Statistical significance was determined by using the Student t test.
Supplementary Material
Acknowledgments
We thank Drs. Lowell Goldsmith and Kevin McGowan for their relevant discussions of this manuscript. This work was supported by National Institutes of Health Grants RO1 AI49427 (to D.S.R.), AI40768 (to Z.L.), and AR30281, AR32599, and T32 AR07369 (to L.A.D.).
Abbreviations
- dsg
desmoglein
- HSP
heat shock protein
- p38MAPK
p38 mitogen-activated protein kinase
- PV
pemphigus vulgaris
- i.d.
intradermal
- IF
immunofluorescence.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office. L.S. is a guest editor invited by the Editorial Board.
References
- 1.Anhalt G. J., Labib R. S., Voorhees J. J., Beals T. F., Diaz L. A. N. Engl. J. Med. 1982;306:1189–1196. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi Y., Patel H. P., Labib R. S., Diaz L. A., Anhalt G. J. J. Invest. Dermatol. 1985;84:41–46. doi: 10.1111/1523-1747.ep12274679. [DOI] [PubMed] [Google Scholar]
- 3.Amagai M., Klaus-Kovtun V., Stanley J. R. Cell. 1991;67:869–877. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- 4.Amagai M., Karpati S., Prussick R., Klaus-Kovtun V., Stanley J. R. J. Clin. Invest. 1992;90:919–926. doi: 10.1172/JCI115968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eyre R. W., Stanley J. R. J. Clin. Invest. 1988;81:807–812. doi: 10.1172/JCI113387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mascaro J. M., Jr., Espana A., Liu Z., Ding X., Swartz S. J., Fairley J. A., Diaz L. A. Clin. Immunol. Immunopathol. 1997;85:90–96. doi: 10.1006/clin.1997.4408. [DOI] [PubMed] [Google Scholar]
- 7.Anhalt G. J., Till G. O., Diaz L. A., Labib R. S., Patel H. P., Eaglstein N. F. J. Immunol. 1986;137:2835–2840. [PubMed] [Google Scholar]
- 8.Mahoney M. G., Wang Z. H., Stanley J. R. J. Invest. Dermatol. 1999;113:22–25. doi: 10.1046/j.1523-1747.1999.00632.x. [DOI] [PubMed] [Google Scholar]
- 9.Seishima M., Esaki C., Osada K., Mori S., Hashimoto T., Kitajima Y. J. Invest. Dermatol. 1995;104:33–37. doi: 10.1111/1523-1747.ep12613469. [DOI] [PubMed] [Google Scholar]
- 10.Esaki C., Seishima M., Yamada T., Osada K., Kitajima Y. J. Invest. Dermatol. 1995;105:329–333. doi: 10.1111/1523-1747.ep12319948. [DOI] [PubMed] [Google Scholar]
- 11.Osada K., Seishima M., Kitajima Y. J. Invest. Dermatol. 1997;108:482–487. doi: 10.1111/1523-1747.ep12289726. [DOI] [PubMed] [Google Scholar]
- 12.Caldelari R., de Bruin A., Baumann D., Suter M. M., Bierkamp C., Balmer V., Muller E. J. Cell Biol. 2001;153:823–834. doi: 10.1083/jcb.153.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen V. T., Ndoye A., Shultz L. D., Pittelkow M. R., Grando S. A. J. Clin. Invest. 2000;106:1467–1479. doi: 10.1172/JCI10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkowitz P., Hu P., Liu Z., Diaz L. A., Enghild J. J., Chua M. P., Rubenstein D. S. J. Biol. Chem. 2005;280:23778–23784. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- 15.Aoyama Y., Owada M. K., Kitajima Y. Eur. J. Immunol. 1999;29:2233–2240. doi: 10.1002/(SICI)1521-4141(199907)29:07<2233::AID-IMMU2233>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen V. T., Arredondo J., Chernyavsky A. I., Kitajima Y., Pittelkow M., Grando S. A. J. Biol. Chem. 2004;279:2135–2146. doi: 10.1074/jbc.M309000200. [DOI] [PubMed] [Google Scholar]
- 17.Benndorf R., Hayess K., Ryazantsev S., Wieske M., Behlke J., Lutsch G. J. Biol. Chem. 1994;269:20780–20784. [PubMed] [Google Scholar]
- 18.Geum D., Son G. H., Kim K. J. Biol. Chem. 2002;277:19913–19921. doi: 10.1074/jbc.M104396200. [DOI] [PubMed] [Google Scholar]
- 19.Panasenko O. O., Kim M. V., Marston S. B., Gusev N. B. Eur. J. Biochem. 2003;270:892–901. doi: 10.1046/j.1432-1033.2003.03449.x. [DOI] [PubMed] [Google Scholar]
- 20.Perng M. D., Cairns L., van den I. P., Prescott A., Hutcheson A. M., Quinlan R. A. J. Cell Sci. 1999;112:2099–2112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- 21.Evgrafov O. V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C. L., Schagina O., Verpoorten N., Van Impe K., Fedotov V., et al. Nat. Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 22.Lavoie J. N., Hickey E., Weber L. A., Landry J. J. Biol. Chem. 1993;268:24210–24214. [PubMed] [Google Scholar]
- 23.Lavoie J. N., Lambert H., Hickey E., Weber L. A., Landry J. Mol. Cell. Biol. 1995;15:505–516. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guay J., Lambert H., Gingras-Breton G., Lavoie J. N., Huot J., Landry J. J. Cell Sci. 1997;110:357–368. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- 25.Rock B., Labib R. S., Diaz L. A. J. Clin. Invest. 1990;85:296–299. doi: 10.1172/JCI114426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arteaga L. A., Prisayanh P. S., Warren S. J., Liu Z., Diaz L. A., Lin M. S. J. Invest. Dermatol. 2002;118:806–811. doi: 10.1046/j.1523-1747.2002.01743.x. [DOI] [PubMed] [Google Scholar]
- 27.Ge B., Gram H., Di Padova F., Huang B., New L., Ulevitch R. J., Luo Y., Han J. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- 28.Gumbiner B. M. Nat. Rev. Mol. Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 29.Conacci-Sorrell M., Zhurinsky J., Ben-Ze’ev A. J. Clin. Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green K. J., Gaudry C. A. Nat. Rev. Mol. Cell Biol. 2000;1:208–216. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 31.Getsios S., Huen A. C., Green K. J. Nat. Rev. Mol. Cell Biol. 2004;5:271–281. doi: 10.1038/nrm1356. [DOI] [PubMed] [Google Scholar]
- 32.O’Neill L. A. J. Nat. Rev. Drug Discov. 2006;5:549–563. doi: 10.1038/nrd2070. [DOI] [PubMed] [Google Scholar]
- 33.Ding X., Aoki V., Mascaro J. M., Jr., Lopez-Swiderski A., Diaz L. A., Fairley J. A. J. Invest. Dermatol. 1997;109:592–596. doi: 10.1111/1523-1747.ep12337524. [DOI] [PubMed] [Google Scholar]
- 34.Roscoe J. T., Diaz L., Sampaio S. A., Castro R. M., Labib R. S., Takahashi Y., Patel H., Anhalt G. J. J. Invest. Dermatol. 1985;85:538–541. doi: 10.1111/1523-1747.ep12277362. [DOI] [PubMed] [Google Scholar]
- 35.Rock B., Martins C. R., Theofilopoulos A. N., Balderas R. S., Anhalt G. J., Labib R. S., Futamura S., Rivitti E. A., Diaz L. A. N. Engl. J. Med. 1989;320:1463–1469. doi: 10.1056/NEJM198906013202206. [DOI] [PubMed] [Google Scholar]
- 36.Lee J. C., Laydon J. T., McDonnell P. C., Gallagher T. F., Kumar S., Green D., McNulty D., Blumenthal M. J., Heys J. R., Landvatter S. W., et al. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 37.Hu P., O’Keefe E. J., Rubenstein D. S. J. Invest. Dermatol. 2001;117:1059–1067. doi: 10.1046/j.0022-202x.2001.01523.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}