

Abstract
The mammalian Ste20-like Nck-interacting kinase (NIK) and its orthologs Misshapen in Drosophila and Mig-15 in Caenorhabditis elegans have a conserved function in regulating cell morphology, although through poorly understood mechanisms. We report two previously unrecognized actions of NIK: regulation of lamellipodium formation by growth factors and phosphorylation of the ERM proteins ezrin, radixin, and moesin. ERM proteins regulate cell morphology and plasma membrane dynamics by reversibly anchoring actin filaments to integral plasma membrane proteins. In vitro assays show that NIK interacts directly with ERM proteins, binding their N termini and phosphorylating a conserved C-terminal threonine. In cells, NIK and phosphorylated ERM proteins localize at the distal margins of lamellipodia, and NIK activity is necessary for phosphorylation of ERM proteins induced by EGF and PDGF, but not by thrombin. Lamellipodium extension in response to growth factors is inhibited in cells expressing a kinase-inactive NIK, suppressed for NIK expression with siRNA oligonucleotides, or expressing ezrin T567A that cannot be phosphorylated. These data suggest that direct phosphorylation of ERM proteins by NIK constitutes a signaling mechanism controlling growth factor-induced membrane protrusion and cell morphology.
Keywords: ezrin, moesin, ste20 kinase, membrane protrusion
The Nck-interacting kinase (NIK) is a member of the germinal center kinase subfamily of Ste20/MAP4K serine/threonine kinases (1). Closely related to NIK are the mammalian kinases TNIK (2), MINK (3), and NRK/NESK (4) and orthologs Misshapen (Msn) in Drosophila (5) and MIG-15 in Caenorhabditis elegans (6). NIK and its orthologs share a common function in regulating cell shape and migration. In mice, homozygous knockout of NIK results in early embryonic lethality with defects in mesoderm migration (7), and expression of kinase-inactive NIK attenuates epithelial cell invasion (8). Msn functions in determining epithelial polarity, dorsal closure. and neuronal targeting (5, 9, 10), and MIG-15 controls axonal navigation (6). NIK (1) and Msn (5) also share a conserved activation of the JNK pathway. NIK, however, does not directly phosphorylate JNK, nor do NIK nullizygous embryos precisely phenocopy mice lacking JNK1 or JNK2 (7). Additionally, activation of JNK is not associated with dynamic changes in cell morphology. Hence, NIK substrates that control cell morphogenesis have not been identified.
We now report that the ERM proteins ezrin, radixin, and moesin are substrates for NIK. ERM proteins regulate cell morphology by cross-linking actin filaments to the plasma membrane. The N-terminal FERM (4.1 ERM) domain of ERM proteins binds to integral plasma membrane proteins and the C terminus binds F-actin (11). ERM proteins control cell shape primarily by regulating membrane protrusions and cell-substrate adhesion. In epithelial cells ERM proteins are necessary for the formation of apical microvilli (12–14). In fibroblasts ERM proteins regulate the assembly of focal adhesions (15) and dynamic membrane protrusions, including filopodia and lamellipodia (16, 17).
Inactive ERM proteins are retained in the cytosol in a closed conformation by an intramolecular association of N- and C-terminal domains, which masks a C-terminal F-actin binding site. ERM proteins are coincidence detectors, with activation and release of the N- and C-terminal interaction requiring two sequential regulatory events: binding of phosphatidylinositol 4,5-bisphosphate to the N terminus and phosphorylation of a C-terminal threonine residue (18), conserved in ezrin (T567), radixin (T564), and moesin (T558). Phosphorylation of ERM proteins at specific membrane domains may be regulated by different kinases. ERM proteins are phosphorylated by myotonic dystrophy kinase-related Cdc42-binding kinase in filopodia (19), protein kinase Cα in membrane protrusions (20), and the Rho-associated kinase (ROCK) in microvilli (21), although this latter finding is controversial (22). Kinases mediating phosphorylation of ERM proteins by growth factors to regulate lamellipodium formation, however, have not been identified.
We found that NIK directly phosphorylates the conserved C-terminal threonine in ERM proteins, and that NIK activity and phosphorylation of ezrin T567 are necessary for lamellipodium extension induced by growth factors. NIK binds ERM proteins, localizes with phosphorylated ERM (pERM) proteins at the distal margins of lamellipodia, and is necessary for increased phosphorylation of ERM proteins in response to growth factors. These data suggest that NIK activity and its direct phosphorylation of ERM proteins control growth factor-induced changes in cell morphology.
Results
NIK Activity Is Necessary for Lamellipodium Extension by EGF.
Because NIK acts downstream of receptor tyrosine kinases (23, 24), we asked whether its kinase activity regulates dynamic changes in cell morphology in response to growth factors. In rat mammary epithelial MTLn3 cells, which extend broad lamellipodia with addition of EGF (25), NIK activity was necessary for increased membrane protrusion by EGF. In MTLn3 cells infected with control adenovirus (Ad) vector, EGF (25 nM) rapidly induced lamellipodia extending around the cell body (Fig. 1A Top and see Movie 1, which is published as supporting information on the PNAS web site). The area of lamellipodium protrusion with EGF increased significantly by 2 min (P < 0.1; n = 6), reaching a maximum ≈3-fold increase at 10 min (Fig. 1B). Delayed cell spreading after 5 min resulted in a modest (<15%) but reproducible increase in the size of the cell body. In cells infected with Ad containing WT NIK (Ad-NIK) lamellipodium extension with EGF was more rapid than in control cells, with a significant increase in area at 1 min (P < 0.1; n = 6) and a maximum ≈3-fold increase at 5 min (Fig. 1 A Middle and B; see Movie 2, which is published as supporting information on the PNAS web site). Cell spreading occurred after 5 min, resulting in ≈25% increase in the size of the cell body. Cells infected with Ad containing a kinase-inactive NIK-D152N (Ad-NIK-D152N) were more spread than Ad and Ad-NIK cells (Fig. 1A Bottom) in the absence of EGF, consistent with previous findings that expression of kinase-inactive NIK (8) and the related MINKβ (3) increases cell adhesion and spreading. These cells had constitutive small lamellipodia and ruffles in the absence of EGF but lamellipodium area did not increase with EGF (Fig. 1 A Bottom and B; see Movie 3, which is published as supporting information on the PNAS web site). Kymograph analysis of time-lapse videos (Fig. 1C) indicated that with EGF lamellipodium extension was maximum in Ad cells at 5–6 min and in Ad-NIK cells at 3–4 min. Kymograph analysis also confirmed that in Ad-NIK-D152N cells there was constitutive membrane ruffling but no increase in lamellipodium area with EGF.
Fig. 1.

NIK activity is necessary for extension of lamellipodium by EGF. (A) Time-lapse images of MTLn3 cells infected with empty Ad or Ad expressing WT NIK (Ad-NIK) or NIK-D152N (Ad-NIK-D152N) and treated with EGF (25 nM) are shown for the indicated times. See also Movies 1–3. White lines drawn around the periphery of cell bodies and the distal margins of membrane protrusions were used to calculate lamellipodium area. (B) Means ± SEM of lamellipodia area obtained from six cells in three cell preparations are shown. (C) Representative kymographs from cells infected with the indicated Ad and treated with EGF are shown. Arrows indicate maximum membrane protrusion. (Scale bar: 10 μm.)
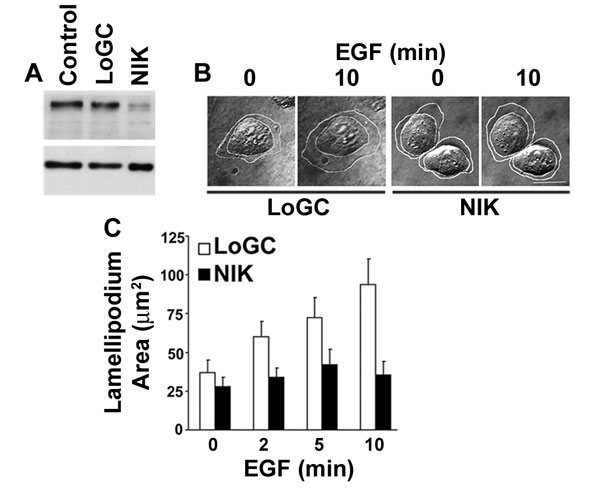
Attenuated lamellipodium extension also was seen with suppression of endogenous NIK by transfecting MTLn3 cells with siRNA oligonucleotides. NIK expression in cell lysates decreased ≈70% at 72 h after transfection with NIK siRNA oligonucleotides compared with untransfected cells (control) or transfection with low GC content oligonucleotides (see Fig. 7A, which is published as supporting information on the PNAS web site). The percent suppression reflected transfection efficiency, as determined by cotransfection with siGLO RNA-induced silencing complex-free siRNA. Time-lapse images of cells positive for siGLO fluorescence indicated that cells transfected with low GC content oligonucleotides, but not with NIK siRNA oligonucleotides, extended broad lamellipodia in response to EGF (see Fig. 7 B and C). Differences in the area of quiescent and stimulated lamellipodia compared with Ad-infected cells at 24 h likely reflect the 72-h cultures used with oligonuleotide transfection. Unlike cells expressing NIK-D152N, however, cells with suppressed NIK expression were not more spread compared with control cells. Hence, the C-terminal regulatory domain of NIK may affect cell-substrate adhesion independent of the N-terminal kinase domain.
NIK Directly Phosphorylates ERM Proteins.
In considering NIK substrates that might regulate membrane protrusion, we tested ERM proteins because the Drosophila Ste20-like kinase Slik phosphorylates Drosophila moesin to maintain epithelial morphology (26). In vitro kinase assays showed that WT NIK tagged with a Myc epitope expressed in COS-7 cells and immunoprecipitated phosphorylated a GST fusion of the C terminus of moesin (M287-577) but not the N terminus (M1-310) or GST (Fig. 2A). M287-577 was not phosphorylated by kinase-inactive NIK-D152N-Myc expressed and immunoprecipitated from COS-7 cells (Fig. 2A). Additionally, NIK, but not NIK-D152N, phosphorylated GST fusions of the C terminus of radixin (R373-585) and ezrin (E280-586) (Fig. 2B). As a positive control we confirmed that NIK phosphorylated a GST fusion of the C terminus of the Na-H exchanger NHE1, as described (24) (Fig. 2B). Mass spectrometry of moesin M287-577 phosphorylated by NIK revealed a single monophosphorylated peptide (pTLRQIRQGNTK, residues 558–568) with a phosphorylation peak at T558 (data not shown). In vitro kinase assays confirmed that NIK expressed and immunoprecipitated from COS-7 cells phosphorylated full-length moesin WT but not containing an Asp substitution for T558 (M-T558D) (Fig. 2C). Substitution with Asp was used to mimic phosphorylation and induce an open conformation. Direct phosphorylation of moesin T558 was confirmed by using the NIK domain (22-374) purified from Sf9 cells, which phosphorylated a GST fusion of moesin WT C terminus (M414-577) and a C terminus containing an Ala substitution for T567 (M-T567A), but not containing an Ala substitution for T558 (M-T558A) (Fig. 2D). Purified NIK domain also phosphorylated ezrin T567, as determined by using an in vitro-translated C terminus of ezrin (E280-586) as a substrate and immunoblotting with phospho-specific antibodies for the C-terminal phosphorylated threonine conserved in all three ERM proteins (Fig. 2E). These data indicate that NIK directly phosphorylates the C terminus of all three ERM proteins and selectively phosphorylates moesin T558 and ezrin T567, which is a phosphorylation site conserved in radixin.
Fig. 2.

NIK directly phosphorylates the C terminus of ERM proteins. (A) Autoradiograms of kinase reaction (Upper) and Coomassie-stained fusion proteins (Lower) are shown. Myc-tagged WT NIK or NIK-D152N was expressed in COS-7 cells, immunoprecipitated, and used for in vitro kinase assays with GST or GST-fusion proteins containing moesin C terminus (M287-577) or N terminus (M1-310). The high-molecular-mass band at 160 kDa corresponds to autophosphorylated NIK. (B) Autoradiograms of kinase reaction (Upper) and Coomassie-stained fusion proteins (Lower) are shown. WT NIK or NIK-D152N was expressed, immunoprecipitated, and reacted in vitro with GST fusion proteins containing the C terminus of NHE1, radixin (R373-585), or ezrin (E280-586). (C) Autoradiograms of kinase reaction with WT NIK as in A and B (Upper) and Coomassie-stained fusion proteins (Lower) are shown. GST and fusion proteins containing full-length moesin WT (M-WT) or moesin with T558 substituted with aspartic acid (M-T558D) were used as substrates. (D Upper) Autoradiogram of kinase reaction with purified NIK domain (amino acids 22–374) and fusion proteins of the C terminus of moesin WT (M414–577) or containing single substitutions of Thr-558 or Thr-567 with alanine (M-T558A and M-T567A, respectively) is shown. (D Lower) Coomassie-stained fusion proteins used for kinase reaction are shown. (E) Immunoblots (IB) with antiphospho-ERM antibodies (Upper) and antiezrin antibodies (Lower) of C-terminal ezrin translated in vitro and incubated with purified NIK domain active or inactivated by boiling are shown.
NIK Activity Is Necessary for Phosphorylation of ERM Proteins by EGF in Epithelial Cells.
The abundance of pERM proteins increased in MTLn3 cells infected with Ad-NIK compared with cells infected with Ad or kinase-inactive NIK-D152N (Fig. 3A Upper). Ad-NIK and Ad-NIK-D152N had no effect on the expression of ezrin (Fig. 3A Lower) or moesin (data not shown) compared with control Ad. In Ad-infected cells the abundance of pERM proteins increased with EGF, reaching a maximum at 5 min (Fig. 3B). In cells expressing Ad-NIK-D152N the abundance of pERM proteins was similar to that in control Ad cells in the absence of EGF but did not increase with EGF (Fig. 3B). The EGF-induced increase in pERM proteins also was attenuated in cells transfected with NIK siRNA oligonucleotides compared with cells transfected with control siRNA (see Fig. 8, which is published as supporting information on the PNAS web site). Although suppression of endogenous NIK had no effect on the abundance of total ERM proteins (data not shown), the abundance of pERM proteins in quiescent cells was reduced compared with cells expressing kinase-inactive NIK. This difference might reflect the longer time with NIK siRNA (72 h) compared with Ad-NIK-D152N (24 h), or perhaps the regulatory domain of NIK is necessary to maintain the steady-state phosphorylation of ERM proteins. Immunolabeling indicated an increase in pERM proteins predominantly in membrane protrusions of Ad cells (Fig. 3C). In cells infected with Ad-NIK-D152N, immunolabeling of pERM proteins was markedly less at 5 min with EGF compared with Ad cells and was more uniformly distributed along the cell membrane (Fig. 3C). Immunolabeling MTLn3 cells expressing NIK-Myc or NIK-D152N-Myc with anti-Myc antibodies and maintained in growth medium indicated diffuse cytosolic fluorescence and a sharp band of label at the distal margins of membrane protrusions (Fig. 3D). Membrane protrusions were markedly longer and broader in cells expressing NIK-Myc compared with those expressing NIK-D152N-Myc. Hence, kinase activity of NIK is not necessary for its localization at the plasma membrane but is necessary for EGF-induced increase in pERM proteins and lamellipodium extension.
Fig. 3.

NIK activity is necessary for phosphorylation of ERM proteins by EGF. (A) Immunoblots for pERM proteins (Upper) and ezrin (Lower) in lysates of quiescent MTLn3 cells infected with Ad, Ad-NIK, or Ad-NIK-D152N are shown. (B) Time-dependent phosphorylation of ERM proteins with EGF is indicated by immunoblotting for pERM proteins and ezrin in lysates of MTLn3 cells infected with Ad or Ad-NIK-D152N. (C) Immunolabeling with anti-pERM antibodies of MTLn3 cells infected with Ad or Ad-NIK-D152N in the absence (−EGF) or presence of 25 nM EGF for 5 min (+EGF) indicates pERM proteins are predominantly in lamellipodia in Ad-infected cells with EGF. (D) GFP fluorescence (Top) and anti-Myc immunolabeling of cells expressing NIK-Myc (Middle) or NIK-D152N-Myc (Bottom) are shown. (Scale bar: 10 μm.)
Phosphorylation of Ezrin T567 Is Necessary for EGF-Induced Membrane Protrusion.
C-terminal phosphorylation of ERM proteins is necessary for their function in regulating dynamic membrane structures. We therefore asked whether phosphorylation of T567 in ezrin is necessary for EGF-induced membrane protrusion by expressing a mutant ezrin T567A in MTLn3 cells. Cotransfection with GFP was used to mark transfected cells. In cells expressing GFP alone, lamellipodium area increased with EGF (Fig. 4 A Top and B; see Movie 4, which is published as supporting information on the PNAS web site). Compared with Ad-infected cells (Fig. 1), cells expressing GFP had a more flattened morphology, 2-fold greater lamellipodium area in the absence of EGF, and a smaller fold-increase after 10 min with EGF (≈1.7 with GFP and ≈3.0 with Ad). The reduced robustness of the response in GFP-positive cells compared with Ad infection might be caused by the effects of nucleofectin on membrane fluidity or membrane dynamics because the increase in lamellipodium area in untransfected MTLn3 cells treated with EGF was similar to that of Ad cells (data not shown). In MTLn3 cells transiently expressing WT ezrin, the morphology was similar to that of cells expressing GFP alone, but lamellipodium area was greater in the absence and presence of EGF (Fig. 4 A Middle and B; see Movie 5, which is published as supporting information on the PNAS web site). Cells expressing ezrin T567A (Fig. 4A Bottom, white lines) were flatter and more spread, like cells expressing NIK-D152N (Fig. 1A Bottom), but unlike Ad-NIK-D152N cells, they did not have increased lamellipodium area in the absence of EGF. Adjacent untransfected cells (Fig. 4A Bottom, red lines) also were more spread compared with cells transfected with GFP or WT ezrin, perhaps because ezrin T567A induced changes in cell–cell adhesion. In cells expressing ezrin T567A the increase in lamellipodium area with EGF was delayed and attenuated (Fig. 4 A Bottom and B; see Movie 6, which is published as supporting information on the PNAS web site). These data indicate that inactive ezrin, like inactive NIK, inhibits EGF-induced lamellipodia.
Fig. 4.
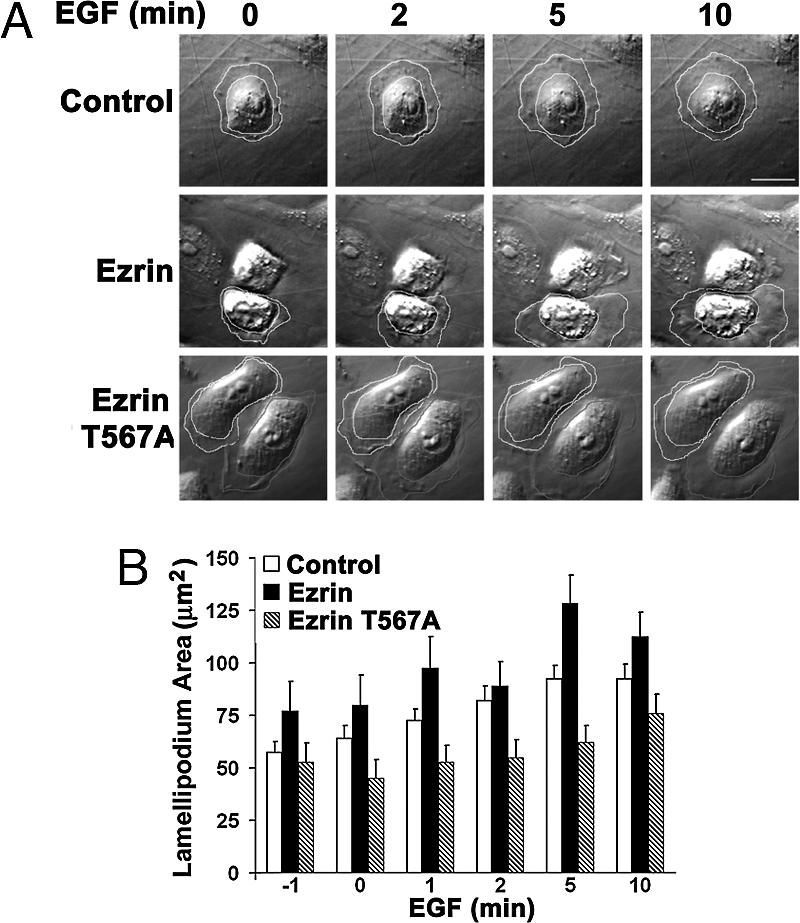
Phosphorylation of T567 in ezrin is necessary for lamellipodium extension with EGF. (A) Time-lapse images of MTLn3 cells transfected with empty vector (Control) or vector encoding WT ezrin (Ezrin) or ezrin T567A (EzrinT567A) and treated with EGF (25 nM) are shown for the indicated times. See also Movies 4–6. Transfected cells (outlined in white) were identified by coexpression of GFP. Lamellipodium area was determined as in Fig. 1. (B) Mean ± SEM of lamellipodium area of six cells in three cell preparations is shown. (Scale bar: 10 μm.)
NIK Activity Is Necessary for Phosphorylation of ERM Proteins by PDGF but Not Thrombin in Fibroblasts.
Previous studies found that growth factors induce Tyr phosphorylation of ezrin (27, 28), and thrombin induces phosphorylation of the C-terminal conserved threonine in ERM proteins (29) we identified as a NIK phosphorylation site. To further confirm the action and specificity of NIK activity we examined the kinetics of phosphorylation of the conserved threonine in ERM proteins in CCL39 fibroblasts, which express receptors for PDGF and thrombin (24). PDGF induced a rapid increase in the abundance of pERM proteins in CCL39 cells infected with control Ad (Fig. 5A). In contrast to the sustained increase in pERM proteins in MTLn3 cells with EGF at 10 min (Fig. 3B), the response in CCL39 cells was more transient, returning to unstimulated levels at 5 min. The abundance of pERM proteins in quiescent cells expressing Ad-NIK-D152N was similar to that in Ad controls but did not increase with PDGF (Fig. 5A). Thrombin also increased phosphorylation of the C-terminal conserved threonine of ERM proteins in CCL39 cells infected with control Ad that was sustained at 5 min, and this increase was not attenuated in cells infected with Ad-NIK-D152N (Fig. 5B). NIK activity also was not necessary for activation of ERK by PDGF. The abundance of phosphorylated ERK, as indicated by immunoblotting cell lysates with phospho-specific antibodies for Thr-202/Tyr-204 of ERK1 and ERK2, was similar in quiescent and PDGF-stimulated Ad and Ad-NIK-D152N cells (Fig. 5C). These data indicate that NIK activity is necessary for increased phosphorylation of ERM proteins by growth factors (EGF and PDGF) but not by thrombin and suggest that NIK mediates the PDGF response by either acting downstream or independently of an ERK-mediated pathway.
Fig. 5.

NIK activity is necessary for phosphorylation of ERM proteins by PDGF but not by thrombin. Representative immunoblots of lysates prepared from CCL39 fibroblasts infected with Ad or Ad-NIK-D152N and treated for the indicated times with PDGF (50 ng/ml) (A Upper and C) or thrombin (30 nM) (B Upper) are shown. Blots were probed with anti-pERM antibodies (A and B), antiphospho-ERK antibodies (C), or antiezrin antibodies (A–C). Data normalized from five (A Lower) and three (B Lower) cell preparations are expressed as the mean ± SEM of fold-change relative to abundance of pERM proteins in Ad-infected cells at time 0. The immunoblot in C is representative of five cell preparations.
NIK Binds the N Terminus of Moesin.
The localization of NIK and pERM proteins at the distal margins of lamellipodia and the ability of NIK to phosphorylate full-length moesin in vitro (Fig. 2C), which is predicted to be in closed conformation, suggest that NIK might bind ERM proteins. An in vitro binding assay confirmed that in vitro-translated 35S-labeled NIK bound a GST fusion of the N terminus of moesin (M1-310) but not the C terminus (M287-577) or GST (Fig. 6A). NIK also bound the N terminus of ezrin (data not shown), and although less strongly, full-length moesin WT and T558D (Fig. 6B). To determine the moesin-binding site in NIK, we incubated GST-M1-310 with in vitro-translated NIK containing truncations in the C-terminal regulatory domain. Binding equivalent to that with full-length NIK was found for truncated NIK containing residues 1–321. However, NIK (residues 1–288), which contains only the kinase domain, did not bind GST-M1-310 (Fig. 6C). These data indicate that the N-terminal 33 aa of the regulatory domain of NIK are necessary for binding ERM proteins.
Fig. 6.

NIK 1-310 binds the FERM domain of moesin and is sufficient to phosphorylate ERM proteins in cells. (A Upper) Autoradiogram of binding assay with full-length NIK translated in vitro in the presence of [35S]methionine and GST and GST-moesin FERM domain (M1-310) and C terminus (M287-577) is shown. (A Lower) Coomassie-stained fusion proteins are shown. (B) NIK binding to the indicated fusion proteins relative to binding GST-M1-310 is shown. (C) Schematic representation of full-length (resides 1–1289) and C-terminal-truncated NIK used in binding assays is shown. ++, domains with comparable binding to GST-moesin 1–310. (D) Expression of an EGFP fusion of truncated NIK-1-340 is sufficient to increase phosphorylation of endogenous ERM proteins in CCL39 fibroblasts. Phosphorylation of ERM proteins was determined by immunolabeling with anti-pERM antibody. *, cells expressing NIK-1-340-EGFP are identified by fluorescence. (E) Ezrin coprecipitates with Myc-tagged kinase-inactive NIK (mNIK-D152N). (Top and Middle) Myc immunoprecipitates from lysates of CCL39 fibroblasts transfected with empty vector (−) or vector containing Myc-NIK-D152N (+) were probed with antibodies against Myc (Top) and ezrin (Middle). (Bottom) Ezrin in cell lysates was determined by immunoblotting. IP, immunoprecipitation; IB, immunoblotting.
Transient expression of NIK1-340 in CCL39 fibroblasts was sufficient to increase phosphorylation of ERM proteins at the plasma membrane (Fig. 6D). NIK1-340 does not contain binding sites for Nck (proline-rich motifs between residues 392 and 618) and MEKK1 (amino acids 908-1233), which suggests that NIK binding to Nck or MEKK1 is not necessary for NIK recruitment to the plasma membrane to phosphorylate ERM proteins. Because the attachment of cells expressing only the kinase domain of NIK (residues 1–288) was markedly impaired, we were unable to determine whether the moesin-binding domain was necessary to increase phosphorylation of ERM proteins in cells. We also confirmed that NIK associates with endogenous ezrin. However, ezrin coprecipitated with transiently expressed NIK-D152N (Fig. 6E) but not with WT NIK (data not shown), suggesting that ERM proteins may be allosterically regulated by NIK-induced phosphorylation.
Discussion
NIK and ERM proteins have been shown independently to regulate cell morphology, but their functional interaction in controlling plasma membrane dynamics was not previously recognized. We found that ERM proteins are substrates for NIK, NIK and pERM proteins localize at the distal margins of lamellipodia, and NIK activity is necessary for phosphorylation of ERM proteins in response to EGF and PDGF. Additionally, NIK activity and phosphorylation of ezrin T567 are necessary for increased lamellipodium extension by growth factors. Collectively, these data suggest that phosphorylation of ERM proteins by NIK may constitute a signaling mechanism controlling growth factor-induced membrane protrusion and cell morphology.
Substrates for NIK and its orthologs in pathways regulating cell morphology remain poorly understood. NIK and its Drosophila ortholog Msn increase phosphorylation of JNK (1) and BSK (30), respectively. Phosphorylation of JNK by NIK, however, is not direct but mediated through NIK binding to a scaffolding complex with MEKK1. Msn also increases phosphorylation of Bicaudal-D (31) to regulate epithelial planar polarity (9); however, whether Bicaudal-D is directly phosphorylated by Msn is unknown. Moesin, the single ERM protein in Drosophila, also is necessary to maintain epithelial integrity (32), and it will be interesting to determine whether Dmoesin is a substrate for Msn. HGK, the human homolog of NIK, acts downstream of receptors for hepatocyte growth factor to increase phosphorylation of STAT3 (8). Although STAT3 coprecipitates with HGK, its direct phosphorylation is undetermined. In mammalian fibroblasts NIK binds to the Na-H exchanger NHE1 and directly phosphorylates NHE1 to increase H+ efflux in response to PDGF (24). ERM proteins also bind to NHE1 (33), and by anchoring actin filaments ERM proteins maintain the localization of NHE1 at the leading edge of migrating fibroblasts. Because a leading-edge H+ efflux by NHE1 is necessary for membrane protrusion and directed migration (34), the possibility of a structural and functional link between NIK, ERM proteins, and NHE1 in promoting membrane protrusion is intriguing.
Activation of ERM proteins requires two sequential steps (18). First, N-terminal binding to phosphatidylinositol 4,5-bisphosphate recruits ERM proteins from the cytosol to the plasma membrane and permits N-terminal binding to transmembrane proteins. ERM proteins can be retained at the plasma membrane but not bound to F-actin (33). A second activation step, C-terminal phosphorylation, is necessary for F-actin binding. The required second activation step of phosphoryation may restrict actin tethering by ERM proteins to specialized membrane domains, such as protruding membranes. Although several kinases, including myotonic dystrophy kinase-related Cdc42-binding kinase (19), protein kinase Cα (20), and Rho-associated kinase (ROCK) (21), phosphorylate the conserved C-terminal threonine in ERM proteins, they have not been shown to mediate growth factor-induced phosphorylation of ERM proteins at lamellipodia.
Because activation of ERM proteins promotes F-actin anchoring to the plasma membrane, their phosphorylation by NIK likely stabilizes extending lamellipodia. However, we predict that NIK also regulates membrane dynamics through mechanisms independent of ERM proteins. MTLn3 cells expressing NIK-D152N, but not ezrin T567A, had constitutive, albeit small, ruffles. Substrates, including NHE1 (24), and possibly gelsolin or cofilin, which are phosphorylated by the closely related kinases TNIK (2) and NRK (4), respectively, might contribute to NIK-dependent membrane protrusion. Additionally, NIK phosphorylation of ERM proteins or other substrates might act coordinately with Nck to promote or stabilize membrane protrusions. Our finding that NIK activity is necessary to phosphorylate ERM proteins in response to EGF and PDGF, but not to thrombin, is consistent with NIK binding to the Src homology 3 domain of Nck (1), an adaptor protein associated with receptor tyrosine kinases, and with Msn binding to DOCK (10), the Drosophila ortholog of Nck. Nck also binds and activates the Wiskott-Aldrich syndrome protein WASP (35) and the WASP family verprolin homologous protein WAVE (36), which promote actin assembly by the Arp2/3 complex and membrane protrusion. Although NIK may act coordinately with Nck to regulate membrane dynamics, its phosphorylation of ERM proteins can occur independently of Nck because truncated NIK 1-321 lacking the C-terminal Nck-binding domain was sufficient to increase phosphorylation of ERM proteins in quiescent cells. Additionally, kinase inactive NIK-D152N did not block activation of ERK1/ERK2 by PDGF, suggesting that NIK regulates ERM protein phosphorylation downstream or independently of an ERK-mediated pathway, the latter possibility being consistent with NIK acting independently of Nck.
Our findings indicate that activation of ERM proteins by NIK is a cellular mechanism to promote local alterations in cell morphology in response to growth factors. This mechanism is likely important in migrating cells because NIK activity is necessary for growth factor-induced phosphorylation of ERM proteins in lamellipodia. Because activation of NIK (8) and ezrin (37) is implicated in processes related to tumor cell dissemination with aberrant growth factor signaling, a functional interaction between NIK and ERM proteins might play a previously unrecognized role in tumor cell metastasis.
Materials and Methods
Materials and methods for cell culture, transfections, in vitro translation, in vitro kinase and binding assays, immunoblotting, immunolabeling, and immunoprecipitation have been described (8, 24, 25, 33). All methods are described in detail in Supporting Text, which is published as supporting information on the PNAS web site.
Ad and siRNA Oligonucleotides.
To generate NIK and NIK-D152N Ad, full-length WT mouse NIK and NIK-D152N (24) were excised by HindIII/NotI digestion from pCRII-TOPO, ligated into the same sites in pAD-Track-CMV, and amplified in Escherichia coli XL1-blue. Positive clones were identified by analytical restriction enzyme digestion, and plasmids from these clones were linearized by Pme1 digestion, gel-purified, and used to transform the BJ5183-AD-1 strain of E. coli (Stratagene, La Jolla, CA) carrying the pADEasy-1 plasmid that encodes the Ad-5 genome. Plasmid DNA was purified from kanamycin-resistant colonies and analyzed by PacI digestion. 293Ad cells were transfected with recombined pTrack-CMV and pADEasy-1, and 8 days after transfection spreading Ad within the 293Ad cell monolayer was determined by plaque formation and fluorescence of GFP was encoded by pADEasy-1. Ad was collected by repetitive freeze-thaw cycles followed by centrifugation at 12,000 × g for 10 min at 4°C and used for subsequent infection of 293Ad cells. Optimal titers for cell infection reached 90–100% infection after 48 h, determined by GFP fluorescence.
The siRNA oligonucleotide duplex (5′-UAA UGA AAG CAC CAU AGU AUG UUG C-3′) to rat NIK was synthesized by Invitrogen, Carlsbad, CA (Stealth Select RNAi). Stealth RNAi Negative Control Low GC (low GC content; Invitrogen) was used as a control. siGLO RNA-induced silencing complex-free siRNA (Dharmacon, Lafayette, CO) was used to monitor transfection efficiency. MTLn3 cells were cotransfected with 200 nm NIK or control siRNA and 150 nm siGLO by using Oligofectamine (Invitrogen) according to the manufacturer's protocol. After 48 h cells were replated and maintained in the absence of FBS for 4 h before imaging or harvesting at 72 h posttransfection.
Video Microscopy.
MTLn3 cells in six-well plates were placed on a preheated, climate-regulated stage mounted on a Zeiss (Thornwood, NY) Axiovert S-100 microscope. Images were captured every 20 s, beginning 2 min before adding EGF and ending 10 min after adding EGF (25 nM; Invitrogen) by using a Spot CCD camera through a ×40 Hoffmann modulation objective and operated with Openlab software. Lamellipodium area was designated as the region from the cell body to the periphery of the plasma membrane. Area calculations performed with Openlab software were made by subtracting the area of the cell body from the total cell area.
Supplementary Material
Acknowledgments
We thank Hitesh Patel for assistance with video microscopy; Neetu Gupta for help with transfection of MTLn3 cells; Torsten Wittmann for help with kymography; and Brandon LaMere for technical assistance. This work was supported by a Swiss National Science Foundation fellowship (to M.B.) and National Institutes of Health (NIH) Grants T32 DE07204 (to A.L.S.) and GM47413 (to D.L.B.). This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant C06 RR16490 from the National Center for Research Resources (NIH). Work conducted at Sugen was funded by the Pfizer Corporation.
Abbreviations
- NIK
Nck-interacting kinase
- Msn
Misshapen
- ERM
ezrin, radixin, moesin
- pERM
phosphorylated ERM
- Ad
adenovirus
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Su Y. C., Han J., Xu S., Cobb M., Skolnik E. Y. EMBO J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu C. A., Shen M., Huang B. C., Lasaga J., Payan D. G., Luo Y. J. Biol. Chem. 1999;274:30729–30737. doi: 10.1074/jbc.274.43.30729. [DOI] [PubMed] [Google Scholar]
- 3.Hu Y., Leo C., Yu S., Huang B. C., Wang H., Shen M., Luo Y., Daniel-Issakani S., Payan D. G., Xu X. J. Biol. Chem. 2004;279:54387–54397. doi: 10.1074/jbc.M404497200. [DOI] [PubMed] [Google Scholar]
- 4.Nakano K., Kanai-Azuma M., Kanai Y., Moriyama K., Yazaki K., Hayashi Y., Kitamura N. Exp. Cell Res. 2003;287:219–227. doi: 10.1016/s0014-4827(03)00136-8. [DOI] [PubMed] [Google Scholar]
- 5.Su Y. C., Maurel-Zaffran C., Treisman J. E., Skolnik E. Y. Mol. Cell Biol. 2000;20:4736–4744. doi: 10.1128/mcb.20.13.4736-4744.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poinat P., De Arcangelis A., Sookhareea S., Zhu X., Hedgecock E. M., Labouesse M., Georges-Labouesse E. Curr. Biol. 2002;12:622–631. doi: 10.1016/s0960-9822(02)00764-9. [DOI] [PubMed] [Google Scholar]
- 7.Xue Y., Wang X., Li Z., Gotoh N., Chapman D., Skolnik E. Y. Development (Cambridge, U.K.) 2001;128:1559–1572. doi: 10.1242/dev.128.9.1559. [DOI] [PubMed] [Google Scholar]
- 8.Wright J. H., Wang X., Manning G., LaMere B. J., Le P., Zhu S., Khatry D., Flanagan P. M., Buckley S. D., Whyte D. B., et al. Mol. Cell Biol. 2003;23:2068–2082. doi: 10.1128/MCB.23.6.2068-2082.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paricio N., Feiguin F., Boutros M., Eaton S., Mlodzik M. EMBO J. 1999;18:4669–4678. doi: 10.1093/emboj/18.17.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruan W., Pang P., Rao Y. Neuron. 1999;24:595–605. doi: 10.1016/s0896-6273(00)81115-0. [DOI] [PubMed] [Google Scholar]
- 11.Bretscher A., Edwards K., Fehon R. G. Nat. Rev. Mol. Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi K., Sato N., Kasahara H., Funayama N., Nagafuchi A., Yonemura S., Tsukita S. J. Cell Biol. 1994;125:1371–1384. doi: 10.1083/jcb.125.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crepaldi T., Gautreau A., Comoglio P. M., Louvard D., Arpin M. J. Cell Biol. 1997;138:423–434. doi: 10.1083/jcb.138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saotome I., Curto M., McClatchey A. I. Dev. Cell. 2004;6:855–864. doi: 10.1016/j.devcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Mackay D. J., Esch F., Furthmayr H., Hall A. J. Cell Biol. 1997;138:927–938. doi: 10.1083/jcb.138.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamb R. F., Ozanne B. W., Roy C., McGarry L., Stipp C., Mangeat P., Jay D. G. Curr. Biol. 1997;7:682–688. doi: 10.1016/s0960-9822(06)00295-8. [DOI] [PubMed] [Google Scholar]
- 17.Castelo L., Jay D. G. Mol. Biol. Cell. 1999;10:1511–1520. doi: 10.1091/mbc.10.5.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fievet B. T., Gautreau A., Roy C., Del Maestro L., Mangeat P., Louvard D., Arpin M. J. Cell Biol. 2004;164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura N., Oshiro N., Fukata Y., Amano M., Fukata M., Kuroda S., Matsuura Y., Leung T., Lim L., Kaibuchi K. Genes Cells. 2000;5:571–581. doi: 10.1046/j.1365-2443.2000.00348.x. [DOI] [PubMed] [Google Scholar]
- 20.Ng T., Parsons M., Hughes W. E., Monypenny J., Zicha D., Gautreau A., Arpin M., Gschmeissner S., Verveer P. J., Bastiaens P. I., Parker P. J. EMBO J. 2001;20:2723–2741. doi: 10.1093/emboj/20.11.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oshiro N., Fukata Y., Kaibuchi K. J. Biol. Chem. 1998;273:34663–34666. doi: 10.1074/jbc.273.52.34663. [DOI] [PubMed] [Google Scholar]
- 22.Matsui T., Yonemura S., Tsukita S. Curr. Biol. 1999;9:1259–1262. doi: 10.1016/s0960-9822(99)80508-9. [DOI] [PubMed] [Google Scholar]
- 23.Becker E., Huynh-Do U., Holland S., Pawson T., Daniel T. O., Skolnik E. Y. Mol. Cell Biol. 2000;20:1537–1545. doi: 10.1128/mcb.20.5.1537-1545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan W., Nehrke K., Choi J., Barber D. L. J. Biol. Chem. 2001;276:31349–31356. doi: 10.1074/jbc.M102679200. [DOI] [PubMed] [Google Scholar]
- 25.Chan A. Y., Raft S., Bailly M., Wyckoff J. B., Segall J. E., Condeelis J. S. J. Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- 26.Hipfner D. R., Keller N., Cohen S. M. Genes Dev. 2004;18:2243–2248. doi: 10.1101/gad.303304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bretscher A. J. Cell Biol. 1989;108:921–930. doi: 10.1083/jcb.108.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava J., Elliott B. E., Louvard D., Arpin M. Mol. Biol. Cell. 2005;16:1481–1490. doi: 10.1091/mbc.E04-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura F., Huang L., Pestonjamasp K., Luna E. J., Furthmayr H. Mol. Biol. Cell. 1999;10:2669–2685. doi: 10.1091/mbc.10.8.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su Y. C., Treisman J. E., Skolnik E. Y. Genes Dev. 1998;12:2371–2380. doi: 10.1101/gad.12.15.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houalla T., Hien Vuong D., Ruan W., Suter B., Rao Y. Mech. Dev. 2005;122:97–108. doi: 10.1016/j.mod.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Speck O., Hughes S. C., Noren N. K., Kulikauskas R. M., Fehon R. G. Nature. 2003;421:83–87. doi: 10.1038/nature01295. [DOI] [PubMed] [Google Scholar]
- 33.Denker S. P., Huang D. C., Orlowski J., Furthmayr H., Barber D. L. Mol. Cell. 2000;6:1425–1436. doi: 10.1016/s1097-2765(00)00139-8. [DOI] [PubMed] [Google Scholar]
- 34.Denker S. P., Barber D. L. J. Cell Biol. 2002;159:1087–1096. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohatgi R., Nollau P., Ho H. Y., Kirschner M. W., Mayer B. J. J. Biol. Chem. 2001;276:26448–26452. doi: 10.1074/jbc.M103856200. [DOI] [PubMed] [Google Scholar]
- 36.Eden S., Rohatgi R., Podtelejnikov A. V., Mann M., Kirschner M. W. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- 37.Yu Y., Khan J., Khanna C., Helman L., Meltzer P. S., Merlino G. Nat. Med. 2004;10:175–181. doi: 10.1038/nm966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}