

Abstract
Genetic instability is a defining feature of human cancer. The main type of genetic instability, chromosomal instability (CIN), enhances the rate of gross chromosomal changes during cell division. CIN is brought about by mutations of CIN genes, i.e. genes that are involved in maintaining the genomic integrity of the cell. A major question in cancer genetics is whether genetic instability is a cause and hence a driving force of tumorigenesis. A mathematical framework for studying the somatic evolution of cancer sheds light onto the causal relations between CIN and human cancer.
Keywords: genetic instability, tumour suppressor genes, somatic evolution of cancer, mathematical modelling
Human tumorigenesis proceeds by the accumulation of inherited and somatic mutations in gatekeepers and caretakers (Vogelstein & Kinzler 2001). Gatekeepers such as oncogenes and tumour suppressor genes (TSGs) encode proteins that function in growth regulatory and differentiation pathways (Knudson 1971, 1993; Friend et al. 1986; Weinberg 1991). Mutations in those genes increase the net reproductive rate of cells and might lead to clonal expansion (Park & Vogelstein 2000). Here, we study cancers that are initiated by the inactivation of one or two TSGs.
Caretakers maintain the genomic integrity of the cell (Lengauer et al. 1998; Loeb 2001). Mutations in caretakers lead to genetic instability, which is found in most human cancers (Vogelstein & Kinzler 2001). In a small fraction of colorectal, endometrial, gastric and some other cancers, inactivation of the mismatch repair pathway leads to an elevated point mutation rate and consequent widespread microsatellite instability (Kinzler & Vogelstein 1996; Perucho 1996). The majority of cancers, however, have chromosomal instability (CIN; Rajagopalan et al. 2003). CIN refers to an increased rate of losing or gaining whole chromosomes or large parts of chromosomes during cell division (Rajagopalan & Lengauer 2004a). The consequence of CIN is an imbalance in chromosome number (aneuploidy) and an increased rate of loss of heterozygosity (LOH). An elevated rate of LOH is an important property of CIN, because it accelerates the inactivation of TSGs.
Note that CIN refers to an enhanced rate of accumulating gross chromosomal changes during cell division; it is not synonymous with the state of aneuploidy that is observed in a static image of the cell's chromosomal content. While CIN is a process that drives most cells to aneuploidy, aneuploidy per se does not prove the existence of CIN. There are several ways in which a cell can become aneuploid in the absence of CIN. The cell could have divided many more times than other cells in the tissue; as gross chromosomal changes (rarely) occur in normal cells, this could lead to aneuploidy without CIN. Alternatively, aneuploidy could be caused by exposure to endogenous or exogenous agents that disrupt spindle formation. It is also possible that cancer cells accumulate gross chromosomal changes at the same rate as normal cells, but that these changes are only lethal in normal cells. This hypothesis is strengthened by the knowledge that mutations in cancer cells disrupt the capacity of cells to undergo apoptosis.
Although CIN is defined as an increased rate of losing or gaining parts of chromosomes or whole chromosomes during cell division, CIN has only been formally demonstrated for whole chromosome losses (Lengauer et al. 1997). There is no assay at present that can reliably measure the rate of formation of changes at the subchromosomal level, such as deletions, insertions, inversions, rearrangements, amplifications, unequal sister chromatid exchange and gene conversion. These changes are at least as common as losses or gains of whole chromosomes.
A large number of genes have been identified that trigger CIN when mutated in Saccharomyces cerevisiae (Shonn et al. 2000; Kolodner et al. 2002; Nasmyth 2002). These genes are involved in a variety of cellular pathways including chromosome condensation, sister-chromatid cohesion, kinetochore structure and function, microtubule formation and cell-cycle control. Similarly, several genes can cause CIN in Drosophila melanogaster when genetically altered (Fung et al. 2002; Mihaylov et al. 2002). By analogy, we expect hundreds of human CIN genes, but only a few have been identified so far. These genes include hBUB1, MAD2, BRCA1, BRCA2 and hCDC4 (Cahill et al. 1998; Wang et al. 2004). hBUB1 and MAD2 are required for the proper functioning of the spindle assembly checkpoint (Li & Benezra 1996; Pangilinan et al. 1997). This checkpoint modulates the timing of anaphase initiation in mitotic cells containing improperly aligned chromosomes and increases the probability of successful delivery of a correct chromosome set to each daughter cell. BRCA1 and BRCA2 are involved in DNA repair and recombination, checkpoint control of the cell cycle and transcription (Milner et al. 1997; Yarden et al. 2002). Inherited mutations in BRCA1 and BRCA2 lead to high-grade familial breast cancer (Vogelstein & Kinzler 2001). hCDC4 is an E3 ubiquitin ligase that is involved in regulating the G1–S cell-cycle checkpoint by targeting proteins for destruction (Rajagopalan & Lengauer 2004b; Rajagopalan et al. 2004). Inactivation of hCDC4 leads to increased levels of cyclin E, the formation of micronuclei, defects in the execution of anaphase and CIN. Furthermore, the DING gene was found to be somatically mutated in eight out of 23 CIN cancers (Wang et al. 2004). DING is a previously uncharacterized gene whose COOH terminus is homologous with the yeast protein Pds1. Pds1 is an anaphase inhibitor in budding yeast and plays a critical role in the control of anaphase (Nasmyth et al. 2000; Yanagida 2000). Its human orthologue, hSecurin, is essential for the proper function and processing of the separin protease, for separin-dependent cleavage of the cohesin subunit Scc1, and for maintaining chromosome stability (Jallepalli et al. 2001).
The classification of CIN genes is based on the mutational events required to trigger CIN (Michor et al. 2004). Class I CIN genes, such as MAD2, cause CIN if one allele of the gene is mutated or lost. Class II CIN genes, such as hBUB1, trigger CIN if one allele is mutated in a dominant negative fashion. Both classes I and II are ‘single hit’ CIN genes. Class III CIN genes, such as BRCA1 and BRCA2, cause CIN if both alleles are mutated.
A major question in cancer genetics is whether CIN, or any genetic instability, is an early event and hence a driving force of tumorigenesis (Loeb et al. 1974; Breivik & Gaudernack 1999; Tomlinson & Bodmer 1999; Shih et al. 2001; Marx 2002; Rajagopalan et al. 2004). Experimental evidence shows that early colorectal adenomas have allelic imbalance (table 1; Shih et al. 2001), but this provides no proof of underlying CIN. hCDC4 mutations have been shown to occur early in colorectal tumorigenesis (Rajagopalan et al. 2004). Here, we discuss a mathematical framework for the evolutionary dynamics of tumorigenesis offering insights into the causal relations between CIN and curves initiated by inactivation of one or two TSGs.
Table 1.
Experimental evidence of aneuploidy in early tumours.(The table shows the percentage of early colorectal adenomas (size 1–3 mm, total number 32) with allelic imbalance in five different chromosome arms. Ninety per cent of the adenomas have allelic imbalance in any one of the five chromosome arms. Data from Shih et al. (2001).)
| chromosome arm | adenomas with allelic imbalance (%) |
|---|---|
| 1p | 10 |
| 5q | 55 |
| 8p | 19 |
| 15q | 28 |
| 18q | 28 |
| any of these 5 | 90 |
Tissues are organized into small compartments of cells (Mintz 1971; Cairns 1975; Bach et al. 2000; Yatabe et al. 2001). Not all cells of a compartment need be at risk of becoming cancer cells, however, because certain mutations only have an effect if they occur in stem cells (Michor et al. 2003, 2004b). Suppose N0 cells are at risk in any one compartment. Consider a path to cancer initiated by activation of both alleles of a TSG, A. Initially, all cells of the compartment are wild-type with respect to TSG A, A+/+. If the mutation rates inactivating the alleles of A are less than the inverse of N0, then the approximation of homogeneous compartments holds: a mutated cell will either take over the compartment or go extinct before the next mutation arises (Nowak et al. 2002; Komarova et al. 2003; Michor et al. 2004a). Hence the compartment evolves from the state where all cells have two wild-type alleles, A+/+, via the state where all cells have one inactivated allele, A+/−, to the state where all cells have two inactivated alleles, A−/− (figure 1a).
Figure 1.
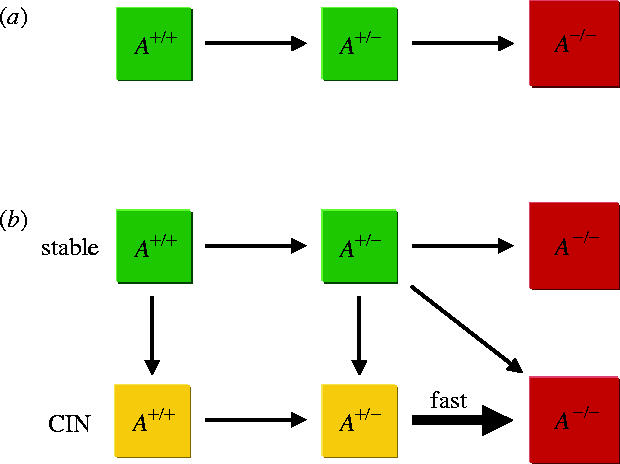
Inactivation dynamics of a tumour suppressor gene (TSG), A, without and with chromosomal instability (CIN) in a compartment of cells. (a) Initially, all cells are wild-type with respect to A, A+/+. Once a cell with one inactivated allele of A emerges and reaches fixation in the compartment, the compartment moves to the state where all cells have one inactivated allele of A, A+/−. Only then does a cell with two inactivated alleles of A emerge and reach fixation in the compartment; the compartment moves to the state where all cells have two inactivated alleles of A, A−/−. (b) CIN can emerge at any stage of tumorigenesis because of mutation of a CIN gene (single hit CIN gene shown here) and increases the rate at which the second allele of a TSG is inactivated. If an A+/− cell with CIN produces an A−/− cell with CIN before reaching fixation in the compartment, a stochastic tunnel arises (diagonal arrow): the compartment moves from state A+/− without CIN to A−/− with CIN without ever visiting A+/− with CIN.
CIN can emerge at any stage of tumorigenesis (figure 1b). First, assume that CIN is dominant, i.e. triggered by mutation of a class I or II CIN gene (Nowak et al. 2002; Michor et al. 2004a). The crucial effect of CIN is to increase the rate of LOH, thereby accelerating the transition from A+/− to A−/−. CIN can have a cost for the cell by increasing the rate of accumulating lethal mutations and triggering apoptosis. However, CIN can also be advantageous by reducing the duration of the cell cycle or by providing increased evolvability in detrimental environments (Bardelli et al. 2001; Breivik 2001). We therefore consider costly, neutral and advantageous CIN phenotypes. If CIN is costly, then the phenomenon of ‘stochastic tunnelling’ arises (Nowak et al. 2002; Komarova et al. 2003; Iwasa et al. 2004): the compartment moves from A+/− without CIN to A−/− with CIN without ever visiting A+/− with CIN.
Suppose that two TSGs, A and B, have to be inactivated consecutively for tumorigenesis (figure 2). The compartment evolves from A+/+B+/+ via A+/−B+/+ to A−/−B+/+, and subsequently to A−/−B+/− and A−/−B−/−. CIN can emerge at any stage of tumorigenesis owing to mutations of class I, II or III CIN genes. Once CIN has emerged, it accelerates the transitions from A+/− to A−/− and from B+/− to B−/−. A stochastic tunnel arises if the compartment moves from A+/−B+/+ without CIN to A−/−B+/+ with CIN without ever visiting A+/−B+/+ with CIN. Inactivation of the first TSG can induce neoplastic growth. We assume that the A−/− compartment gives rise to a small lesion of N1 cells. In this lesion, TSG B has to be inactivated for further tumour progression. Owing to the increased compartment size, the evolutionary pathway might tunnel from A−/−B+/+ directly to A−/−B−/−. Mathematical procedures are outlined in Appendix A.
Figure 2.

Inactivation dynamics of two tumour suppressor genes (TSGs), A and B, without and with chromosomal instability (CIN) in a compartment of N0 cells. Initially, all cells are wild-type. Suppose TSG A has to be inactivated first. Then the compartment evolves from the state where all cells have two wild-type alleles of A, A+/+, via the state where all cells have one inactivated allele of A, A+/−, to the state where all cells have two inactivated alleles of A, A−/−. Inactivation of A leads to clonal expansion of the compartment to N1 cells. It is in this small lesion that TSG B is inactivated, and the compartment moves to the state where all cells have two inactivated alleles of both A and B, A−/−B−/−. CIN can emerge at any stage because of mutation of a class I, II or III CIN gene and leads to rapid LOH of the second allele of TSGs A and B.
The importance of early CIN in tumorigenesis depends on the number of possible CIN mutations, which in turn depends on the number of CIN genes in the human genome. We can calculate the minimum number of CIN genes in the genome that are needed to ensure that in a significant fraction of tumours, a CIN mutation precedes the inactivation of one or two TSGs. Table 2 provides examples for the critical number of class I, II and III CIN genes (n1, n2 and n3, respectively) depending on the relative fitness value of CIN cells (r) and the compartment size after inactivation of TSG A (N1). Table 3 shows the fraction of cancers initiated by a CIN mutation depending on r and n1, n2 and n3.
Table 2.
Minimum number of class I, II and II CIN genes (n1, n2 and n3, respectively) required for emergence of CIN before the inactivation of the first tumour suppressor gene (TSG). (The relative fitness value of CIN cells is denoted by r. Parameter values are N0=4, u=10−7, p0=10−6, p=10−2, τ0=10 days and τ1=1 day.)
| CIN before 1 TSG | CIN before 2 TSGs | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| independent of N1 | N1=104 | N1=105 | |||||||
| r | n1 | n2 | n3 | n1 | n2 | n3 | n1 | n2 | n3 |
| 1.0 | 2 | 12 | 89 | 1 | 1 | 16 | 1 | 2 | 37 |
| 0.9 | 2 | 14 | 96 | 1 | 1 | 17 | 1 | 3 | 41 |
| 0.5 | 4 | 42 | 169 | 1 | 2 | 35 | 1 | 8 | 74 |
Table 3.
Percentage of tumours initiated by inactivation of one or two tumour suppressor genes (TSGs) in which CIN emerges before the inactivation of the first TSG.(The number of class I, II and III CIN genes is given by n1, n2 and n3, respectively. The relative fitness value of CIN cells is denoted by r. Parameter values are N0=4, N1=10−4, u=10−7, p0=10−6, p=10−2, τ0=10 days and τ1=1 day.)
| CIN genes | r | CIN before 1 TSG (%) | CIN before 2 TSGs (%) |
|---|---|---|---|
| n1=1 | 1.0 | 50 | 97 |
| 0.9 | 46 | 97 | |
| 0.5 | 21 | 88 | |
| n2=5 | 1.0 | 31 | 94 |
| 0.9 | 28 | 93 | |
| 0.5 | 11 | 76 | |
| n3=10 | 1.0 | 1.4 | 31 |
| 0.9 | 1.2 | 26 | |
| 0.5 | 0.4 | 7.7 |
In summary, one or a few neutral CIN genes are enough to ensure that CIN initiates tumour formation in a pathway where one TSG needs to be eliminated in a rate limiting situation. One or a few costly CIN genes are enough to ensure that CIN initiates tumour formation in a pathway where two TSGs need to be eliminated in rate limiting situations.
Acknowledgments
The program for Evolutionary Dynamics is supported by Jeffrey E. Epstein.
Appendix A
The stochastic process illustrated in figures 1b and 2 can be described by differential equations. Denote by X0, X1, X2 and X4 the probabilities that a compartment is in state A+/+B+/+, A+/−B+/+, A−/−B+/+ and A−/−B−/−, respectively, without chromosomal instability (CIN). Denote by Y0, Y1, Y2 and Y4 the probabilities that a compartment is in state A+/+B+/+, A+/−B+/+, A−/−B+/+ and A−/−B−/−, respectively, with CIN. The differential equations are given by
| (A 1) |
The transition rates between the states are given by r0=2u/τ0, r1=N0(u+p0)/τ0, , c0=N0ucρ/τ0, , s0=r0, s1=N0p/τ0 and (Nowak et al. 2002; Komarova et al. 2003; Michor et al. 2004a). Cells with genotype A+/+B+/+ and A+/−B+/+ divide every τ0 days, while cells with genotype A−/−B+/+ and A−/−B+/− divide every τ1 days.
CIN can arise at any stage of tumorigenesis and causes very fast LOH. The mutation rate triggering CIN, uc, depends on the number of class I, II and III CIN genes in the human genome, n1, n2 and n3, respectively. The mutation rates of class I and II CIN genes are uc=2n1(u+p0) and uc=2n2u, respectively. The mutation rate of the first allele of class III CIN genes is 2n3u, and the mutation rate of the second allele of class III CIN genes is u=p0. The probability of fixation of a CIN cell depends on its somatic fitness, r, and is given by (Nowak et al. 2003).
Equation (A 1) is a system of linear differential equations which can be solved analytically using standard techniques.
Footnotes
One contribution of 17 to a Discussion Meeting Issue `Chromosome segregation'.
References
- Bach S.P, Renehan A.G, Potten C.S. Stem cells: the intestinal stem cell as a paradigm. Carcinogenesis. 2000;21:469–476. doi: 10.1093/carcin/21.3.469. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Cahill D.P, Lederer G, Speicher M.R, Kinzler K.W, Vogelstein B, Lengauer C. Carcinogen-specific induction of genetic instability. Proc. Natl Acad. Sci. USA. 2001;98:5770–5775. doi: 10.1073/pnas.081082898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivik J. Don't stop for repairs in a war zone: Darwinian evolution unites genes and environment in cancer development. Proc. Natl Acad. Sci. USA. 2001;98:5379–5381. doi: 10.1073/pnas.101137698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivik J, Gaudernack G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin. Cancer Biol. 1999;9:245–254. doi: 10.1006/scbi.1999.0123. [DOI] [PubMed] [Google Scholar]
- Cahill D.P, Lengauer C, Yu J, Riggins G.J, Willson J.K.V, Markowitz S.D, Kinzler K.W, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- Friend S.H, Bernards R, Rogelj S, Weinberg R.A, Rapaport J.M, Albert D.M, Dryja T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- Fung S.M, Ramsay G, Katzen A.L. Mutations in Drosophila myb lead to centrosome amplification and genomic instability. Development. 2002;129:347–359. doi: 10.1242/dev.129.2.347. [DOI] [PubMed] [Google Scholar]
- Iwasa Y, Michor F, Nowak M.A. Stochastic tunnels in evolutionary dynamics. Genetics. 2004;166:1571–1579. doi: 10.1534/genetics.166.3.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli P.V, Waizenegger I.C, Bunz F, Langer S, Speicher M.R, Peters J.M, Kinzler K.W, Vogelstein B, Lengauer C. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445–457. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Kinzler K.W, Vogelstein B. Lessons from hereditary colon cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Knudson A.G. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson A.G. Antioncogenes and human cancer. Proc. Natl Acad. Sci. USA. 1993;90:10 914–10 921. doi: 10.1073/pnas.90.23.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner R.D, Putnam C.D, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- Komarova N.L, Sengupta A, Nowak M.A. Mutation-selection networks of cancer initiation: tumor suppressor genes and chromosomal instability. J. Theor. Biol. 2003;223:433–450. doi: 10.1016/s0022-5193(03)00120-6. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler K.W, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler K.W, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- Loeb L.A. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- Loeb L.A, Springgate C.F, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- Marx J. Debate surges over the origins of genomic defects in cancer. Science. 2002;297:544–546. doi: 10.1126/science.297.5581.544. [DOI] [PubMed] [Google Scholar]
- Michor F, Nowak M.A, Frank S.A, Iwasa Y. Stochastic elimination of cancer cells. Proc. R. Soc. Lond. B. 2003;270:2017–2024. doi: 10.1098/rspb.2003.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michor F, Iwasa Y, Nowak M.A. Dynamics of cancer progression. Nature Rev. Cancer. 2004a;4:197–206. doi: 10.1038/nrc1295. [DOI] [PubMed] [Google Scholar]
- Michor F, Iwasa Y, Rajagopalan H, Lengauer C, Nowak M.A. Linear model of colon caner initiation. Cell Cycle. 2004b;3:358–362. [PubMed] [Google Scholar]
- Mihaylov I.S, et al. Control of DNA replication and chromosome ploidy by geminin and cyclin A. Mol. Cell. Biol. 2002;22:1868–1880. doi: 10.1128/MCB.22.6.1868-1880.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner J, Ponder B, Hughes-Davies L, Seltmann M, Kouzarides T. Transcriptional activation functions in BRCA2. Nature. 1997;386:772–773. doi: 10.1038/386772a0. [DOI] [PubMed] [Google Scholar]
- Mintz B. Clonal basis of mammalian differentiation. Symp. Soc. Exp. Biol. 1971;25:345–370. [PubMed] [Google Scholar]
- Nasmyth K. Segregating sister genomes: the molecular biology of chromosome separation. Science. 2002;297:559–565. doi: 10.1126/science.1074757. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Peters J.M, Uhlmann F. Splitting the chromosome: cutting the ties that bind sister chromatids. Science. 2000;288:1379–1385. doi: 10.1126/science.288.5470.1379. [DOI] [PubMed] [Google Scholar]
- Nowak M.A, Komarova N.L, Sengupta A, Jallepalli P.V, Shih IeM, Vogelstein B, Lengauer C. The role of chromosomal instability in tumor initiation. Proc. Natl Acad. Sci. USA. 2002;99:16 226–16 231. doi: 10.1073/pnas.202617399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M.A, Michor F, Iwasa Y. The linear process of somatic evolution. Proc. Natl Acad. Sci. USA. 2003;100:14 966–14 969. doi: 10.1073/pnas.2535419100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangilinan F, Li Q, Weaver T, Lewis B.C, Dang C.V, Spencer F. Mammalian BUBI protein kinases: map positions and in vivo expression. Genomics. 1997;46:379–388. doi: 10.1006/geno.1997.5068. [DOI] [PubMed] [Google Scholar]
- Park B.H, Vogelstein B. Clinical oncology. In: Abeloff M.D, editor. 2nd edn. Churchill Livingston; London: 2000. [Google Scholar]
- Perucho M. Cancer of the microsatellite phenotype. Biol. Chem. 1996;377:675–684. [PubMed] [Google Scholar]
- Shih I.M, Zhou W, Goodman S.N, Lengauer C, Kinzler K.W, Vogelstein B. Evidence that genetic instability occurs at an early state of colorectal tumorigenesis. Cancer Res. 2001;61:818–822. [PubMed] [Google Scholar]
- Shonn M.A, McCarroll R, Murray A.W. Requirement of the spindle checkpoint for proper chromosome segregation in budding yeast meiosis. Science. 2000;289:300–303. doi: 10.1126/science.289.5477.300. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Lengauer C. CIN-ful cancers. Cancer Chemother. Pharmacol. 2004a;54:S65–S68. doi: 10.1007/s00280-004-0889-8. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Lengauer C. hCDC4 and genetic instability in cancer. Cell Cycle. 2004b;3:693–694. doi: 10.4161/cc.3.6.940. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Nowak M.A, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nature Rev. Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli P.V, Rago C, Velculescu V.E, Kinzler K.W, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- Tomlinson I, Bodmer W. Selection, the mutation rata and cancer: ensuring that the tail does not wag the dog. Nature Med. 1999;5:11–12. doi: 10.1038/4687. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler K.W. 2nd edn. McGraw-Hill; Toronto: 2001. The genetic basis of human cancer. [Google Scholar]
- Wang Z, et al. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004;64:2998–3001. doi: 10.1158/0008-5472.can-04-0587. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. Tumor suppressor genes. Science. 1991;254:1138–1146. doi: 10.1126/science.1659741. [DOI] [PubMed] [Google Scholar]
- Yanagida M. Cell cycle mechanisms of sister chromatid separation; roles of Cut1/separin and Cut2/-securin. Genes Cells. 2000;5:1–8. doi: 10.1046/j.1365-2443.2000.00306.x. [DOI] [PubMed] [Google Scholar]
- Yarden R.I, Pardo-Reoyo S, Sgagias M, Cowan K.H, Brody L.C. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nature Genet. 2002;30:265–269. doi: 10.1038/ng837. [DOI] [PubMed] [Google Scholar]
- Yatabe Y, Tavare S, Shibata D. Investigating stem cells in human colon by using methylation patterns. Proc. Natl Acad. Sci. USA. 2001;98:10 839–10 844. doi: 10.1073/pnas.191225998. [DOI] [PMC free article] [PubMed] [Google Scholar]