

Abstract
Replicating large eukaryotic genomes presents the challenge of distinguishing replicated regions of DNA from unreplicated DNA. A heterohexamer of minichromosome maintenance (MCM) proteins is essential for the initiation of DNA replication. MCM proteins are loaded on to unreplicated DNA before replication begins and displaced progressively during replication. Thus, bound MCM proteins license DNA for one, and only one, round of replication and this licence is reissued each time a cell divides. MCM proteins are also the best candidates for the replicative helicases that unwind DNA during replication, but interesting questions arise about how they can perform this role, particularly as they are present on only unreplicated DNA, rather than clustered at replication forks. Although MCM proteins are bound and released cyclically from DNA during the cell cycle, higher eukaryotic cells retain them in the nucleus throughout the cell cycle. In contrast, MCMs are broken down when cells exit the cycle by quiescence or differentiation. We have exploited these observations to develop screening tests for the common carcinomas, starting with an attempt to improve the sensitivity of the smear test for cervical cancer. MCM proteins emerge as exceptionally promising markers for cancer screening and early diagnosis.
Keywords: DNA replication, cancer diagnosis, minichromosome maintenance proteins, replication licencing, cell proliferation
1. Introduction
Cancer will affect one in three of the UK population and at current rates it will cause the death of one in four of them. It is tempting to ask, therefore, why cancer is so common. However, it has been pointed out previously that a more appropriate question is: ‘Why is cancer so rare?’ (Evan & Littlewood 1998; Evan & Vousden 2001). As the human body contains more than 1014 cells and as cancer arises from breakdown of the rules controlling cell population sizes, it is remarkable that these rules fail so rarely. Clearly, less than one in 100 million cells successfully evade the rules that control cell proliferation. These figures become even more startling when they are viewed in the light of genetic stability. Before a cell can divide, it must duplicate its genetic material, DNA. Errors in that process, or failure to correct changes that arise from DNA damage, can result in mutations that contribute to cancer.
Cancer generally arises as a consequence of accumulation of multiple genetic changes that disable or disrupt the mechanisms that regulate cell population sizes. Such changes include:
transformation, enabling a cell to grow in the absence of external growth stimuli;
immortalization, enabling a cell to persist long after normal cells have undergone programmed cell death;
invasiveness, allowing the cancer cell and its progeny to invade surrounding tissues or even remote tissues (metastasis);
ability to attract a new blood supply (angiogenesis), enabling the tumour to grow much larger (reviewed in Hanahan & Weinberg 2000).
Carcinogens, such as tobacco smoke or ionizing radiation, cause cancer by increasing the rate of genetic changes, some of which will affect the genes that mediate these crucial regulatory processes.
In general, the body is designed to minimize the risk of precancerous or cancerous changes. For example, cell immortality is restricted to the minute proportion of cells called stem cells. Other cells die at programmed times. An extraordinary range of mechanisms exists for repairing and correcting damaged DNA (Friedberg 2003). Checkpoints exist to prevent duplication of damaged DNA until the damage has been repaired, so that newly duplicated DNA will have only the original genetic information and not the copies that were corrupted by damage (Zhou & Elledge 2000). The act of duplicating DNA, DNA replication, is extraordinarily accurate. Enzymes that synthesize DNA look back behind them to check the accuracy of their synthesis and to cut out and replace copying errors. Additional ‘mismatch repair’ systems also check the accuracy of the base pairing in newly synthesized DNA, removing mistakes and raising the accuracy of copying to still higher levels of fidelity.
Much of this lecture will be concerned with several aspects of a closely related issue, namely how the cell ensures that it duplicates its DNA once, exactly once and only once, before it divides. Replicating 99.99% of the DNA is not enough. Even a minute unreplicated stretch may lead to a chromosome break at the next cell division. Conversely, it is important that the cell does not over-replicate, overproducing or ‘amplifying’ stretches of DNA. Reinitiation of DNA replication within a single cell cycle, leading to extra copies of local DNA regions, provides the raw material for selection of amplified oncogenes, a phenomenon that is found frequently in human tumours. I will consider the mechanisms that keep track of which regions of DNA have replicated and distinguish them from regions of DNA that remain to be replicated, thus ensuring that all of the DNA is replicated exactly once.
Coupling DNA replication to the cell cycle so that all DNA replicates exactly once is achieved in two ways, involving two groups of proteins. The first group consists of the proteins that make up the prereplication complex or replication licence (Bell & Dutta 2002). These are necessary, but not sufficient, for DNA replication and their collective presence identifies unreplicated DNA. The second group of proteins are the cyclin-dependent kinases that serve as switches to activate sequential phases of the cell cycle (Nurse 2002). Both groups of proteins combine to tightly control the pattern of DNA replication throughout eukaryotic organisms.
Proteins that control DNA replication are interesting for additional reasons. Not only do they have important regulatory roles but they also provide exceptionally promising diagnostic tools for the early detection of cancer. We have used antibodies raised against some of the proteins that regulate human DNA replication to develop screening tests for the common carcinomas. These account for a large part of the cancer burden, yet they are diseases whose treatment is far more successful if they can be detected early in the course of the disease. I shall argue that the markers that we have developed by studying DNA replication may be widely applicable for early detection of most of the most common cancers. These issues will be considered in the final part of the article.
2. Replication origins in higher eukaryotes
When DNA was injected into Xenopus eggs it iniated replication with two surprising details (Harland & Laskey 1980). First, DNA replication did not require specific DNA sequences. For example, bacterial cloning vectors containing no eukaryotic sequences and fragments of animal viral DNA that lacked the viral origin of replication both replicated in Xenopus eggs as efficiently as fragments that contained eukaryotic sequences or viral replication origins (Harland & Laskey 1980; Méchali & Kearsey 1984). The second surprise was that purified DNA microinjected into Xenopus eggs replicated precisely in phase with the endogenous cell cycle clock, even in the absence of an egg nucleus. Remarkably, the DNA replicated once and only once in the equivalent of one embryonic cell cycle, whereas reinitiation of replication occurred if multiple cell cycles were allowed (Harland & Laskey 1980). Tight coupling of DNA replication to the cell cycle was observed in the absence of any detectable origin of replication. Whatever mechanism was preventing reinitiation of replication in a single cell cycle was able to function efficiently in the absence of a specific DNA sequence (Harland & Laskey 1980). This situation contrasted strikingly with the discovery of autonomously replicating sequences (ARS) in yeast, where even point mutation in the conserved autonomously replicating sequence abolished the ability of DNA molecules to replicate (Kearsey 1983, 1984; Celniker et al. 1984). This contrast of results was all the more striking because these two extreme opposites were partly obtained by the same postdoctoral fellow, in the same laboratory, at the same time while working on yeast and Xenopus eggs in parallel (Kearsey 1983, 1984; Méchali & Kearsey 1984).
The observations that injected DNA replicated precisely once every cell cycle, without reinitiating until the next cell cycle, raised two intriguing questions (Harland & Laskey 1980; Laskey & Harland 1981). First, if DNA replication can initiate in a eukaryotic cell under strict cell cycle control, then what are the origins of replication really for? Clearly, they are not essential for the proteins that initiate DNA replication. Subsequent studies looking at patterns of replication of endogenous genes, such as ribosomal RNA genes, have shown that sequence specificity of DNA replication is conferred at later stages of development but that DNA replication is independent of the DNA sequence during the first 12 rapid cell cycles (Hyrien & Méchali 1993; Hyrien et al. 1995).
During the first 12 rapid cell cycles of Xenopus development, the entire S phase of the cell cycle lasts for less than 30 minutes. Rapid chromosome replication is achieved by initiating replication at close intervals on the DNA, replacing this with a more distantly spaced pattern for later cell cycles. Interestingly, initiation is excluded from transcribed regions of the rDNA repeats in later cycles. These observations support the view that patterns of DNA replication are imposed to coordinate replication and transcription traffic on the same template. Without such coordination, head-on collisions between transcription and replication complexes would be frequent and might be difficult to resolve (Brewer 1988). Strong support for this view comes from the observation that there is little, if any, transcription during the first 12 rapid cell cycles. Transcription switches on in the same cell cycle in which replication slows down (Newport & Kirschner 1982). A broadly similar inverse relationship between rates of replication and transcription is seen in the early cell cycles of Drosophila embryos.
3. The concept of a ‘replication licence’
The second interesting question raised by the observation that specific DNA sequences are not needed to couple DNA replication to the cell cycle concerns the mechanism that prevents reinitiation of DNA replication within a single cycle. If this mechanism does not require initiation at specific sequences, then marking the sites at which replication initiated cannot be enough to prevent reinitiation as many other sites could be used to initiate a second round of synthesis. This conclusion invited an alternative concept of a ‘replication licence’ as a positive activator of DNA replication which marks unreplicated DNA, and which is displaced or degraded during replication (Laskey et al. 1981; Blow et al. 1987). Thus, unreplicated DNA would be distinguished from replicated DNA by the presence of the hypothetical ‘licence’, which licenses a single round of replication each cell cycle. An alternative explanation appeared less attractive, namely that a pervasive inhibitor of initiation is distributed over all replicated DNA. Unlike the positive licence model, the negative inhibitory model would not be fail-safe. Failure of the positive licensing model would only cause a gap in the initiation pattern and adjacent replication forks could fill this in. However, failure of the negative inhibitor model would lead to over-replication with consequential gene amplification (Laskey et al. 1981; Blow et al. 1987). Proteins that compose replication licences have now been identified from studies of cell-free systems derived from Xenopus eggs and from genetic studies in both the budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe, together with contributions using several other organisms (reviewed in Bell & Dutta 2002). These proteins will be discussed in more detail in the following sections.
4. Minichromosome maintenance or MCM proteins
Genetic screens for plasmid stability in yeast identified a group of genes called minichromosome maintenance or MCM genes (reviewed in Kearsey & Labib 1998; Tye 1999; Forsburg 2004). Mutation of these genes increased the rate of plasmid loss by decreasing the efficiency of plasmid DNA replication. This behaviour suggested that MCM proteins might have a crucial role in the initiation of DNA replication. This is now known to be true for at least seven of the MCM proteins. Six of them, Mcm2–Mcm7, occur as a single complex, functioning as a heterohexamer in DNA replication. Mcm10 also has a later role in DNA replication but not as part of the same hexameric complex. Other MCM proteins such as Mcm1, which is a transcription factor, may have less direct roles, but will not be discussed further here. Most of the remainder of this article will focus on the roles and clinical exploitation of MCM proteins Mcm2–Mcm7. It should be noted that several of these proteins were originally known by other names such as Cdc21, Cdc46, Cdc47, Bm28 and Mis5 among others. Nomenclature has been subsequently standardized, following the discovery that the six Mcms (Mcm2–Mcm7) are highly conserved throughout eukaryotes. The background to the MCMs, including their species conservation and nomenclature, has been extensively reviewed (Kearsey & Labib 1998; Tye 1999; Forsburg 2004).
Genetic studies in yeast, together with immunodepletion experiments in extracts from Xenopus eggs, have established that MCM proteins are essential for initiation of DNA replication. Furthermore, studies in Xenopus and mammalian cells established that MCMs are present on chromatin in G1 phase of the cell cycle, displaced from chromatin during DNA replication and absent from chromatin during G2 phase of the cycle (Chong et al. 1995; Kubota et al. 1995; Madine et al. 1995a,b). The behaviour of MCMs after displacement of chromatin differs between different species. In yeast displaced MCMs are exported from the nucleus to the cytoplasm, but in higher eukaryotes displaced MCMs remain in the nucleoplasm and are not exported. Thus, G1 phase nuclei from mammalian cells are able to replicate in a Xenopus egg extract from which MCMs have been depleted but G2 nuclei are unable to do this as they lack bound MCM proteins on their chromatin (Madine et al. 1995a,b). MCM proteins are behaving exactly as predicted for a replication licence in the original replication licensing model (Laskey et al. 1981; Blow et al. 1987). They conform less well to a later variant of a replication licensing model (Blow & Laskey 1988) which was adapted to account for the observation that an intact nuclear envelope prevented relicencing of chromatin in Xenopus egg extracts. Ironically the behaviour of MCMs in yeasts, namely, export from the nucleus after displacement during replication, conforms much more closely to the second licensing model which invoked the nuclear envelope than the behaviour of MCMs in animal cells, whereas the behaviour of MCMs in higher eukaryotes still conforms closely to the predictions of the original model.
5. Other proteins of the pre-replication complex
Before MCMs can bind to chromatin three other (non-MCM) proteins must bind first. These are the origin recognition complex (ORC), Cdc6 and Cdt1 (figure 1; reviewed in Bell & Dutta 2002). The ORC consists of six subunits and it binds to the sites at which replication will initiate. As explained above these are best characterized in the yeast S. cerevisiae where initiation of replication occurs at short ARS (Campbell & Newlon 1991). In higher eukaryotes, the precise relationship between the sites of ORC binding and initiation of replication are less clear, though in one case coincidence has been observed, at least at the level of light microscopy (Romanowski et al. 1996). Once ORC has bound to chromatin it serves as a landing pad for binding of Cdc6 and Cdt1. The relative roles of these two proteins are unclear but both are required to enable MCMs to bind (reviewed in Bell & Dutta 2002; Forsburg 2004). Thus, the combination of ORC, Cdc6 and Cdt1 produces the landing pad that allows loading of MCMs to the chromatin. Interestingly, MCMs bind in approximately a 10–40 fold excess of the amount of ORC or Cdc6 that are bound. This is consistent with the distribution shown in figure 2, in which MCM staining is found distributed throughout the unreplicated chromatin, but absent from the replicated chromatin in the same nucleus. The function of bound MCMs is considered in a later section together with the problems of their abundance and distribution. The binding of the MCM heterohexamer (Mcm2–Mcm7) forms an MCM ring around DNA and it is followed by binding of other factors such as Cdc45, Mcm10 and GINS, which, in turn, participate in the chain of events that allows binding of DNA polymerases and their accessory factors to chromatin to allow replication to begin (Bell & Dutta 2002; Forsburg 2004).
Figure 1.

Cyclical assembly and disassembly of a prereplication complex or ‘replication licence’ restricts DNA replication to exactly one round between consecutive cell divisions. During the G1 phase of the cell cycle sequential binding of proteins called Orc, Cdc6 and Cdt1 create a landing pad to recruit an excess of the hexameric minichromosome maintenance (MCM) protein complex. During DNA synthesis (S phase) MCMs are displaced from the replicated chromatin but remain on the unreplicated chromatin until replication is complete in G2 phase. During quiescence, G0, MCMs are rapidly displaced from chromatin and more slowly degraded. Modified from the work of Madine et al. (2000).
Figure 2.

Minichromosome maintenance protein MCM3 is not concentrated at sites of DNA replication whereas replication protein A(RPA) is. Panel (a) shows a nucleus of a Xenopus cultured cell pulse labelled to reveal newly replicated DNA (red) and immunostained for MCM3 (green). Panel (b) shows part of a Xenopus sperm nucleus replicating in Xenopus egg extract stained for newly synthesized DNA (red) and MCM3 (green). Panel (c) shows part of a Xenopus sperm nucleus replicating in Xenopus egg extract under identical conditions to those used in panel (b), but stained for newly synthesized DNA (red) and RPA (green) for comparison because RPA is known to be located at replication forks. MCM3 staining (green) is not co-localized with newly replicated DNA (red) in panel (a) and panel (b) whereas RPA staining is co-localized, producing yellow spots in panel (c). Reproduced with permission from Laskey & Madine (2003).
6. Preventing reinitiation within a single cell cycle
Coordinating multiple initiations of replication within eukaryotic chromosomes causes logistical problems. With over 1000 initiation events in each human chromosome, how does the cell keep track of which regions of DNA it has already replicated in order to distinguish them from those regions which await replication? Incomplete replication would result in chromosome breakage at the next division, increasing the probability of chromosome rearrangement, which in turn can lead to accelerated carcinogenesis. Conversely, over-replication, that is, replicating the same stretch of DNA twice, can contribute to gene amplification which is the raw material for oncogene amplification, which again contributes to accelerated carcinogenesis. Hence, it is vitally important that cells replicate their DNA once, exactly once and only once in each cell cycle. The proteins of the prereplication complex contribute to the coupling of DNA replication to the cell cycle in such a way that all of the DNA is replicated exactly once (figure 1). Thus, the prereplication complex, or replication licence, is assembled only during G1 before replication starts. It is dismantled during DNA replication in S phase and only reassembled after the cell has passed through mitosis. Several different mechanisms combine to prevent reinitiation of replication within a single cycle (reviewed in Bell & Dutta 2002; Shreeram & Blow 2003). Different combinations may be found between different eukaryotic species. Examples of mechanisms that prevent reinitiation include the phosphorylation of MCM proteins when they are displaced from chromatin during replication and phosphorylations which appear to prevent rebinding until dephosphorylation takes place at mitosis. Other regulatory mechanisms include the export from the nucleus or degradation of Cdc6, ensuring that no more Cdc6 is available to load the MCM proteins. Similarly, Cdt1 activity can be inhibited by another small protein, called geminin, which is made during S phase of the cycle and which prevents Cdt1 from allowing further MCM binding (Wohlschlegel et al. 2000; Tada et al. 2001). Several other variations of these themes have been reported (reviewed in Bell & Dutta 2002; Forsburg 2004).
In addition to regulated behaviour of the proteins of the prereplication complex, a second major group of proteins, which play important roles in preventing reinitiation of replication, are the cyclin-dependent kinases. The direction of the progression through consecutive phases of the cell cycle is determined by synthesis and sudden degradation of the cyclin partners of cyclin-dependent kinases. Sudden degradation is an irreversible mechanism for ensuring that the direction of cycle phase progression cannot be reversed. The G2 phase cannot follow the M phase because the G2 cyclins are suddenly destroyed as cells pass through mitosis. There can be no going back, only progression to the correct next phase. For example, in yeasts, activation of S phase by specific cyclins and the cyclin-dependent kinase Cdc28 inhibits the assembly of pre-initiation complexes (reviewed in Nurse 2002). Once pre-existing complexes have been activated, no more can be formed. The same also appears to be true in mammalian cells, in which both cyclin E and cyclin A have been implicated in the initiation of DNA replication. We have developed several cell-free systems from mammalian cells which initiate DNA replication in vitro (Krude et al. 1997; Stoeber et al. 1998; Krude 2000; Coverley et al. 2002) and used them to investigate the relative roles of cyclins and cdks. One variant of these systems involves incubation of G1 phase nuclei in G1 phase extract and it depends on the addition of recombinant cyclins and cyclin-dependent kinases for initiation of replication (figure 3; Coverley et al. 2002). In that system, cyclin E/Cdk2 was found to stimulate prereplication complex assembly and specifically MCM binding. Cyclin A/Cdk2, on the other hand, was found to stimulate replication from existing prereplication complexes and inhibit the formation of any further complexes. Thus cyclin E/Cdk2 appears to open a window of opportunity for MCM binding as cells escape from quiescence, while cyclin A/Cdk2 appears to close that window, ensuring that once DNA replication has started no new prereplication complexes can form, thus minimizing the risk of reinitiation of replication on replicated DNA. This role for cyclin E/Cdk2 of stimulating prereplication complex assembly as cells escape from quiescence has been confirmed in fibroblasts from cyclin E null mice. They escape from quiescence more slowly and fail to load MCMs efficiently during escape from quiescence (Geng et al. 2003).
Figure 3.

Cyclin E promotes prereplication complex assembly after cells are released from quiescence, whereas cyclin A initiates DNA synthesis but inhibits assembly of any further prereplication complexes once DNA synthesis has started. (a) Replication of G1 nuclei in G1 cytosol when cyclin CDK complexes are added sequentially to the incubation. (i) and (ii) show the microscope images obtained from the first and third bars of the histogram. Unreplicated nuclei are stained red; replicated nuclei are stained both red and green and thus appear yellow. (b) Summary scheme of the different roles of cyclin E-Cdk2 and cyclin A-Cdk2 in the control of DNA synthesis as cells re-enter the cell cycle from quiescence. Reproduced with permission from Coverley et al. (2002).
7. Are MCMs the replicative helicases?
The MCM proteins are highly conserved from yeasts to higher plants to humans (Kearsey & Labib 1998; Tye 1999; Forsburg 2004). In each of these cases they form a heterohexamer consisting of one subunit each of Mcm2–Mcm7. MCMs are also present in archaea, though in the form of a homohexamer (or possibly heptamer), i.e. six (or possibly seven) copies of the same subunit. Archaeal MCMs and human MCMs have been reported to show DNA helicase activity unwinding the two strands of the double helix, and MCMs are the best candidates for the DNA helicases that unwind DNA at replication forks (Ishimi 1997; Forsburg 2004). This would be compatible with the fact that they are required for initiation of DNA replication in eukaryotes and archaea. However, there is a paradox. The distribution of MCM proteins seen in the cells of higher eukaryotes is not that expected of replicative helicases. Viral and bacterial helicases are found at the site of the replication fork, yet the MCMs of higher eukaryotes are found on unreplicated DNA and are apparently not concentrated at replication forks. In yeast, MCM proteins have been detected close to replication forks by cross-linking (Aparicio et al. 1997), though it is not clear that MCMs are preferentially located there, rather than more easily cross-linked to other proteins present at the fork. There is abundant evidence in animal cells that MCM proteins are not preferentially localized at sites of DNA replication (Madine et al. 1995a,b, Krude et al. 1996; Dimitrova et al. 1999). Figure 2 contrasts the distribution of MCMs at sites of unreplicated DNA with the distribution of a protein that is known to be concentrated at replication forks, namely the essential replication protein RPA. This binds single-stranded DNA at replication forks and co-localizes with sites of DNA replication throughout S phase (figure 2c). The distribution of RPA is seen to be exactly coincident with that of newly replicated DNA in a short pulse, resulting in the yellow colour from superposition of DNA replication (red) and RPA (green) in figure 2c.
If MCMs are distributed throughout unreplicated DNA, rather than concentrated at replication forks, then the paradox arises of how they can act as replicative helicases yet fail to coincide spatially with sites of DNA replication. This paradox is enhanced by the observations that MCMs are present in a 10–40 fold excess of the ORC which is required for MCM loading. Another interesting question is: ‘If MCMs require ORC for loading, how do they come to be distributed at sites that are remote from ORC? Do they spread away from their loading sites?’ Studies of the S. cerevisiae MCM complex by Schwacha & Bell (2001) led us to propose a possible reconciliation of these observations (Laskey & Madine 2003). Schwacha & Bell (2001) observed that the ATPase activities of the MCM hexamer are strikingly similar to those of the mitochondrial F1 ATPase in both kinetics and subunit behaviour. Both complexes are ring-like hexamers composed of three ATPase catalytic subunits (Mcm4, 6 and 7) interspersed with three auxiliary subunits (Mcm2, 3 and 5). Schwacha & Bell (2001) showed that these two ATPases also show strikingly similar responses to specific mutations in their Walker A boxes. As the F1 ATPase of mitochondria resembles a rotary motor or dynamo, rotating with respect to the gamma subunit in the central channel, the possibility arises that the MCM complex is also a rotary motor that rotates a spindle in the central hole. The size of the hole in the central channel of the MCM complex is 2 nm (Sato et al. 2000), sufficient to accommodate double-, or single-stranded DNA, raising the possibility that MCMs are rotary motors that rotate DNA. We have proposed (Laskey & Madine 2003) that the MCMs might be members of a growing class of hexameric ATPases that translocate DNA longitudinally by helical rotation. Table 1 lists precedents of ATPases that pump DNA longitudinally through a central cavity by coupling ATP hydrolysis with rotational translocation of DNA. They include bacteriophage Φ29 gp 10, which has been proposed to pump DNA into the bacteriophage capsid (Simpson et al. 2000), Ruv B, which is responsible for Holliday junction branch migration in Escherichia coli, and FtsK, a protein involved in linking chromosome segregation and cell division in E. coli (Aussel et al. 2002).
Table 1.
Examples of DNA rotary motors.
| DNA motor | organism | function |
|---|---|---|
| Gp10 | Bacteriophage Φ29 | packaging double-stranded DNA into capsid |
| RuvB | Escherichia coli | Holliday junction branch migration |
| SpoIIIE | Bacillus subtilis | chromosome segregation |
| TrwB | Escherichia coli | conjugation |
| FtsK | Escherichia coli | DNA supercoiling and chromosome segregation |
The model we have proposed to reconcile the MCM paradox is shown in figure 4. The key feature we propose is that the MCM hexamer can move relative to double-stranded DNA. We envisage that this occurs by helical rotation along DNA in the same way that a nut would move along a threaded bolt. We envisage two phases. First, in G1 phase of the cell cycle MCMs loaded at the origin by ORC, Cdc6 and Cdt1 would translocate away from the origin by helical rotation along double-stranded DNA, resulting in MCM distribution throughout the unreplicated DNA. In this phase the type of movement we propose is strikingly similar to that of other hexameric ATPases that serve as DNA pumps (Egelman 2001). Second, in S phase of the cell cycle we envisage essentially the same relative movement but with one crucial difference, namely the immobilization of the MCM hexamer. Instead of the MCMs moving along a fixed DNA molecule, immobilization of the MCMs would translocate DNA towards the replication origin and replication forks (figure 4). This could provide a physical mechanism to account for the repeated suggestions that DNA is spooled through fixed sites of DNA replication. A crucial feature of this model is the unwinding of DNA at the replication fork even though the rotary movement is coming from MCM helicases located upstream of the fork. This model is compatible with the distribution of MCM proteins on unreplicated DNA (figure 2) and with the observed excess of MCMs in higher eukaryotes. Interestingly, Kaplan & O'Donnell (2002) have shown that the bacterial replicative helicase DnaB can enclose double-stranded DNA and translocate along it, exactly as proposed for the main driving force in the model shown in figure 4. It will be interesting to see if the MCM ATPase can translocate double-stranded DNA as we propose and if this can result in DNA unwinding at immobilized replication forks, as proposed in figure 4.
Figure 4.

A hypothetical rotary pump model showing two stages in the distribution and function of MCM hexameric ATPase complexes. First, in G1 phase, MCM hexamers would move spirally along the helical groves of unreplicated DNA from their loading site at the origin of replication. Second, in S phase MCMs would become immobilized so that exactly the same rotary mechanism would move the DNA instead of the MCM proteins, resulting in translocation of DNA towards the replication forks. As DNA is twisted by fixed MCMs in S phase, it would become unwound at the distant replication fork. Reproduced with permission from Laskey & Madine (2003).
8. MCM acetylation and the control of DNA replication
MCM proteins undergo post-translational modification. Specifically, MCMs become hyper-phosphorylated as they are displaced from DNA during replication (Todorov et al. 1995). Furthermore, it has been shown that inhibition of protein phosphorylation in G2 phase is sufficient for MCM proteins to rebind to chromatin, though it is not clear that this is directly due to MCM dephosphorylation (Coverley et al. 1998). In addition to MCM phosphorylation, we have discovered an acetyltransferase that specifically and preferentially acetylates MCM3 (Takei et al. 2001). The Mcm3 acetylase (Mcm3AP) shows homology to the acetyl CoA binding sites of other known acetyltransferases. Mutation of one of these conserved motifs abolishes acetyltransferase activity (Takei et al. 2001).
When the wild-type acetylase is overexpressed in human cells by transfection, the proportion of cells in S phase falls. Transfection with the mutant that has lost acetyltransferase activity does not decrease the proportion of cells in S phase, suggesting that activity of the Mcm3 acetylase inhibits entry into S phase (Takei et al. 2001). This conclusion is supported and extended by experiments in which G1 phase nuclei are added to extracts from transfected or mock-transfected cells in vitro (Takei et al. 2001). Extracts from asynchronous mock-transfected cells induce late G1 nuclei to enter S phase. In contrast, extracts from cells transfected with the wild-type Mcm3 acetylase, but not extracts from cells transfected with the inactive mutant enzyme, fail to enter S phase in vitro even though the same extracts are capable of supporting elongation of replication, indicating that the acetylase is inhibiting initiation rather than elongation of replication. A further mutation was constructed which inhibited binding of Mcm3 to the acetylase (Takei et al. 2002). This mutant also fails to inhibit DNA replication, either in vivo by transfection or in vitro, indicating that both acetylase activity and ability to bind to Mcm3 are required to inhibit replication. Taken together these results suggest that a function of Mcm3 acetylation is the inhibition of initiation of DNA replication. We are currently investigating the signalling pathways that activate expression of Mcm3 acetylase.
9. MCM proteins as diagnostic markers for cancer screening
Although MCM proteins bind to chromatin before DNA replication and are displaced from chromatin during DNA replication, they remain in the nuclei of animal cells throughout the proliferating cell cycle (reviewed in Forsburg 2004). They show only a cycle of binding to chromatin and release, rather than a cycle of synthesis and degradation. While this is true of actively cycling cells, MCMs are broken down when cells drop out of cycle either through quiescence or through differentiation (Musahl et al. 1998; Todarov et al. 1998; Madine et al. 2000). Their breakdown takes place over several days, so that fully quiescent or differentiated cells lack detectable MCM proteins. These observations led us and others to investigate the value of antibodies raised against human MCM proteins as markers of proliferating cells in histological and cytological pathology specimens (Hiraiwa et al. 1997; Todarov et al.1998; Williams et al. 1998 and reviewed in Tachibana et al. 2005). Specifically, we wished to know if malignant and pre-malignant cells would re-express MCM proteins, distinguishing them from the differentiated and quiescent cells that are in the vast majority in normal tissues. Although other proliferation markers are known, such as Ki67 or proliferating cell nuclear antigen (PCNA), their performance has not been ideal for reasons discussed below.
The first clinical problem we approached using antibodies against MCM proteins is the reliability of the Papanicolaou smear test for cancer of the uterine cervix, the Pap smear or cervical smear test. This is an extremely important public health measure that saves over 1000 lives a year in the UK alone, yet it repeatedly receives negative publicity arising from the fact that it is a very difficult test to perform reliably. A major source of this difficulty lies in the problem of discriminating a small number of precancerous cells from a large majority of normal cells when both normal and abnormal cells are stained the same colour and show only subtle differences in their structures. In the conventional smear test, cells are scraped from the surface of the cervix and either smeared on to a glass slide or, increasingly, suspended in liquid and collected as a monolayer on the slide. They are then stained with histochemical dyes to reveal subtle structural differences between differentiated keratinocytes from normal tissue and less mature premalignant or malignant cells. Figure 5a shows a conventional cervical smear preparation stained with the Papanicolaou stain. It contains a cluster of abnormal cells in the centre of the field. Figure 5b shows a monolayer preparation from the same sample in which the cells have been stained with an antibody against Mcm2, in addition to the Papanicolaou stain. Premalignant cells contain the MCM antigens and are stained brown. It becomes much easier to detect the abnormal cells when they are stained a different colour, such as by MCM staining.
Figure 5.

Development of an immuno-enhanced Papanicolaou smear test for cancer of the cervix. Both images were produced from one cytology sample containing high-grade premalignant cells (centre of panel a). In a second preparation from the same sample (b), an antibody against MCM2 allows the pre-malignant cells to be stained a different colour from the normal cells (in this case brown), thereby enabling them to be identified more easily. Reproduced from Baldwin et al. (2003).
Figure 6 (top two panels) shows sections through normal cervix (top left) and a high grade premalignant lesion of the cervix (top right) of the type that the smear test is designed to detect. Only the basal and suprabasal layers of the normal tissue are stained with the antibody. This is the expected pattern, as these are the known sites of cell proliferation in the normal ectocervix. By the time cells have matured and migrated to the surface, where they would be sampled in the smear test, they have lost the MCM antigen. In contrast, in the premalignant lesion highly stained cells are visible at the surface of the lesion and therefore available for sampling in the smear test. One small clinical trial of this technique has been published (Williams et al. 1998) and much larger trials are in progress. The key feature of this approach rests on the fact that many carcinomas pass through lengthy pre-invasive stages. If they can be caught and treated during the pre-invasive stage then the probability of successful treatment is greatly increased. This is graphically illustrated by the success of the existing Pap smear test in reducing deaths from cervical cancer, preventing over 1000 each year in Britain alone.
Figure 6.

Patterns of proliferating cells in normal and malignant tissue from the cervix or colon. The left hand panels show sections through normal cervix or normal colon tissue immunostained with antibodies against MCM5 (brown) which is confined to the nuclei of cells in precise proliferating zones and is lost as cells progress towards the surface of the epithelium. In contrast, premalignant and malignant lesions continue to express the MCM5 marker right up to the surface and beyond. Reproduced with permission from Freeman et al. (1999).
The benefits of early detection equally apply to many of the other most common cancers. Histological sections through normal and malignant cervix and colon are shown in figure 6. A recurring feature in these images is the lack of MCM-stained cells at the surface of normal tissues. By the time cells have progressed to the surface and are ready to be shed from the body surface, they have broken down the MCM antigen (figure 6; left panels). In contrast, MCM-stained cells are found at the surface of premalignant or malignant lesions in a wide range of common carcinomas and their premalignant counterparts (figure 6; right panels). This raises the possibility of screening strategies for early detection of many of the common carcinomas by detecting the MCM antigen in cells exfoliated from the tissue surfaces. In many cases cells will be exfoliated into body fluids, such as urine, faeces, or sputum from the lungs. Therefore, we have attempted to recover cells from a range of body fluids to see if detection of the MCM antigen in exfoliated cells can be used as a screening strategy for the early, pre-invasive stages of several of the most common cancers (Williams et al. 1998; Freeman et al. 1999; Stoeber et al. 1999; Davies et al. 2002; Sirieix et al. 2003 and reviewed in Tachibana et al. 2005). Figure 7 illustrates the results of staining exfoliated cells from normal (left panels) or malignant/premalignant (right panels) tissues. Clinical trials are now in progress to assess the value of MCM antibodies for detecting several of the common carcinomas while they are still in the pre-invasive stages and therefore more easily treated. Current trials include cervical, lung, colon and bladder cancers. Although there is encouraging evidence that MCM antibodies can detect malignant and premalignant cells in body fluids, further studies are needed to determine how effectively they work in the day-to-day clinical setting, where samples cannot always be handled immediately or optimally. Nevertheless, the results so far provide a strong incentive to extend these studies in the hope that they can improve early detection and hence treatment of the common carcinomas.
Figure 7.
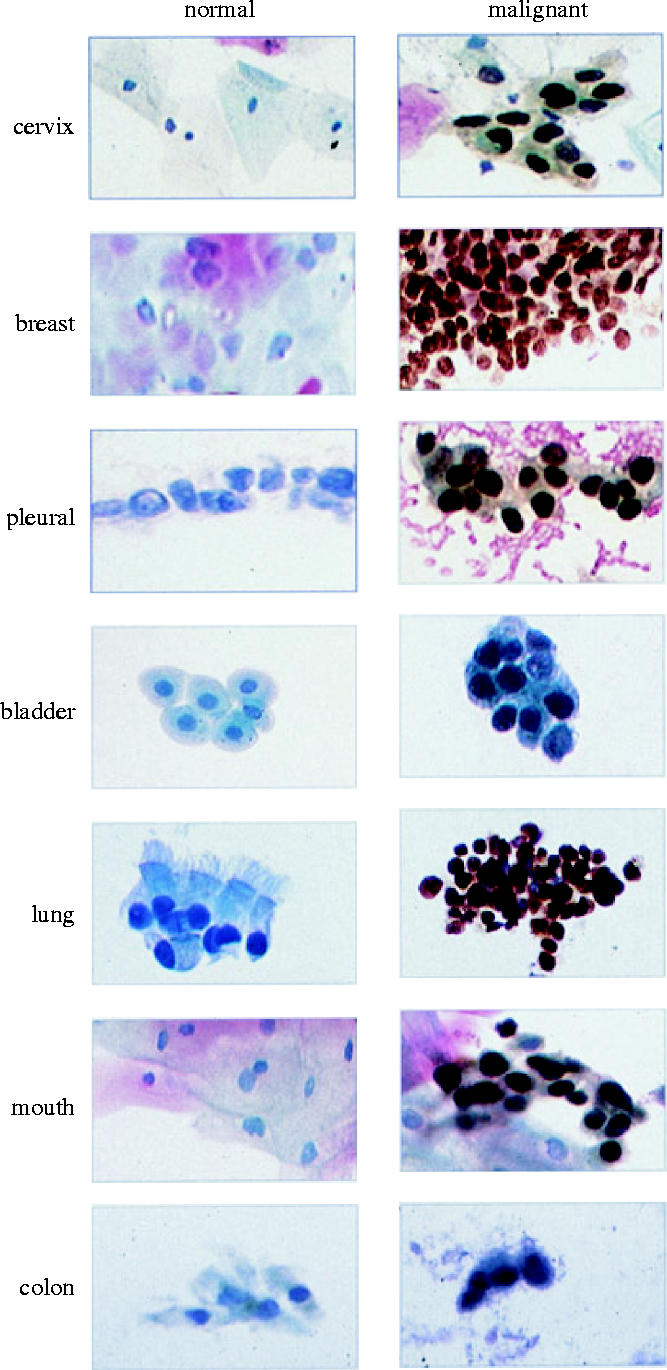
Comparison of normal and malignant cells recovered from body fluids and stained with antibodies against MCM cell proliferation markers. The left-hand column shows cells recovered from body fluids of normal volunteers whereas the right-hand column shows cells recovered from patients with malignant or premalignant diseases of the equivalent site (brown nuclei). Images provided by L. Morris and N. Coleman and reproduced from Laskey (2003) with permission.
It is reasonable to ask why MCMs should be used for this purpose? Why not use the previous proliferation markers, Ki67 and PCNA, or why not use antibodies against the wide range of intensely studied oncogenes and tumour-suppressor genes? Throughout our studies with MCM antibodies we have compared their performance to Ki67 or PCNA. The performance of PCNA as a proliferation marker is problematic because it is not unique to proliferating cells. PCNA is also required for DNA repair, so low levels of PCNA persist in nonproliferating cells. Hence, there is no true background level but the threshold levels set for detection are necessarily semi-arbitrary. Threshold levels which present a low background in nonproliferating tissues result in positive staining of only a subset of the proliferating cells (Williams et al. 1998). The performance of Ki67 is rather different. Ki67 does give low backgrounds in nonproliferating cells; however, it consistently stains only a subset of the proliferating cells that are stained with MCM antibodies. While almost all the cells of a malignant or premalignant lesion are stained with antibodies against MCMs, only about two-thirds of these are stained with Ki67 (Williams et al. 1998; Freeman et al. 1999). Furthermore, the role of Ki67 in the cell is not clear but a nucleolar function has been proposed, in which case it is possible that Ki67 is more closely associated with cell growth rather than cell proliferation (Scholzen & Gerdes 2000).
The argument for preferring MCMs as markers for early detection of cancers over the wide range of oncogenes and tumour-suppressor genes is illustrated in figure 8. The cellular signalling pathways contributed to by oncogenes and tumour-suppressor genes are complex and redundant (reviewed by Hanahan & Weinberg 2000). Many different growth factors can activate cell proliferation via many different growth factor receptors which in turn activate many intracellular signal transduction pathways. Conversely, several different tumour-suppressor genes restrain cell proliferation by opposing these pathways at multiple different points. Hence, the diversity and redundancy of oncogenes and tumour-suppressor genes in regulating cell proliferation render them unsuitable as general markers for detection of a wide range of tumours. In contrast, as illustrated in figure 8, the MCM proteins lie on an apparently unique and conserved pathway of initiation of DNA replication, conserved from yeasts to humans and even to archaea (Kearsey & Labib 1998; Tye 1999; Forsburg 2004). Hence, there is no known way that a eukaryotic cell can replicate its DNA in the absence of MCM proteins and a tumour cell that is incapable of replicating its DNA is unlikely to pose a serious health threat. The properties of MCM proteins make them almost ideal markers for early detection of cancer. They are abundant and stable antigens and they are present in all proliferating cells in all the proliferating stages of the cell cycle: G1, S, G2 and M phases. They are essentially absent from nonproliferating cells and their detection by antibodies is simple and reliable. The clinical trials that are in progress should establish whether these encouraging early results can be sustained in clinical practice to improve the early detection and hence the treatment of many forms of cancer.
Figure 8.
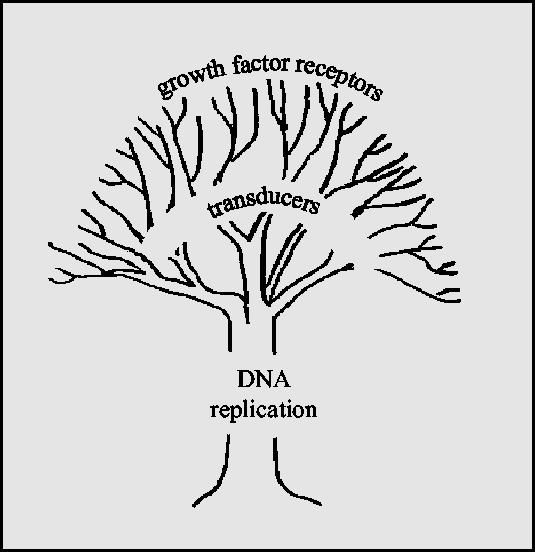
Schematic justification for using replication proteins for cancer screening. While oncogenes and tumour-suppressor genes act on complex and often parallel pathways there is a single, highly conserved mechanism for controlling DNA replication, which is highly suitable as a source of markers to detect malignant or premalignant cells replicating at the surface of tissues.
10. Detection of S phase cells in tissue biopsies by in situ DNA replication or geminin staining
Antibodies against MCM proteins can detect cells which are in the proliferating cycle and distinguish them from cells which are terminally differentiated or quiescent as explained above. However, they do not reliably identify the rate of proliferation of a tumour. For example, if cells are lingering or arrested in the G1 or G2 phases of the cell cycle they will proliferate much more slowly than the cells which pass rapidly through these phases. This difference could be monitored with the aid of a further marker which is specific for cells making DNA. For cell culture this is achieved easily by incubating cells in the presence of a radioactive nucleotide or a modified nucleotide such as bromodeoxyuridine (BrdU); but it is not desirable to inject patients with either tritiated thymidine or BrdU. Therefore, there is a need for a method to determine which cells are in S phase of the cell cycle within a tissue biopsy from patients. We have developed a simple method of achieving this based on the control incubations used to detect S phase contaminants in nuclear preparations for our mammalian cell-free systems (Mills et al. 2000). The method is extremely simple and it can be performed rapidly. Biopsies are quickly frozen in liquid nitrogen; frozen sections are cut on a cryotome, placed on a microscope slide and thawed by addition of a simple buffer containing ribonucleoside triphosphates and deoxyribonucleoside triphosphates, at least one of which is labelled to allow fluorescent detection of DNA synthesis. The slide is incubated in the detection buffer for between 15 min and 2 h so that nuclei, which were already making DNA in vivo, continue to incorporate the labelled precursor in vitro. Pre-existing replication forks continue to synthesize DNA on the microscope slide so that an S phase nucleus becomes fluorescently labelled, while unreplicating nuclei remain unlabelled. Figure 9 shows the types of images obtained from normal or malignant tissues. Reconstruction experiments using cultured cells which had been pre-labelled for DNA synthesis in culture have confirmed that the nuclei which label in vitro on the slide are the same nuclei which were replicating in vivo (Mills et al. 2000). This technique should be able to add prognostic information on the growth rate of tumours and it can be performed both easily and rapidly.
Figure 9.

Identification of S phase cells in frozen tissue sections by virtue of their DNA replication in situ is potentially useful in diagnosing cancer. The top panel shows the method used. Tissue is frozen and sectioned while frozen. The section is quick thawed and flooded with incubation mix on a microscope slide and incubated for 15–120 min in the presence of fluorescent precursors of DNA synthesis. Unreplicated nuclei are stained red, whereas replicated nuclei are stained red plus green and thus appear yellow. Bottom left panel shows a section through a normal human colon. Bottom right panel shows a section through an adenocarcinoma of the colon. Top panel reproduced from Mills et al. (2000).
When used optimally, in situ DNA replication provides a simple method for detecting S phase cells in tissue biopsies. However, its success depends critically on rapid freezing of fresh tissue immediately after biopsy. Delayed freezing can result in sample degradation and inactive assays. Therefore, we have sought an alternative method which can be used successfully on formalin-fixed, paraffin-embedded material. A small regulatory protein, geminin, which inhibits MCM loading on to chromatin provides an effective marker for this purpose (Gonzalez et al. 2004). Antibodies against geminin stain cells that are in S phase or G2 of the cell cycle but not cells that are in G1, the most variable phase of the cycle. Geminin is broken down suddenly at the metaphase–anaphase transition during mitosis and resynthesized at the G1/S phase boundary. We have shown that essentially all cells that stain positive in the in situ DNA replication assay of cultured cells also stain positive for geminin (Gonzalez et al. 2004). Furthermore, the percentage of cells expressing geminin in a breast tumour predicts the clinical outcome and time to distant metastases. Thus, tumours with less than 6% of geminin-stained cells show markedly better survival than tumours containing more than 6% of geminin-stained cells. The advantage of geminin staining over in situ DNA replication is its robustness. Geminin staining can be performed on formalin-fixed, paraffin-embedded material efficiently, whereas in situ replication critically depends on rapid freezing for its success.
11. Conclusion and perspectives
In this lecture I have argued that the strict conservation of the eukaryotic replication machinery offers excellent opportunities for its exploitation in clinical practice. I have also argued that there are many tantalizing problems remaining to be solved in the field of eukaryotic DNA replication. These include the perplexing flexibility in patterns of initiation of DNA replication within higher eukaryotes, as well as further details of the mechanisms that couple DNA replication to the cell cycle to ensure complete synthesis of exactly one round each cycle. We now know that sequential binding and release of MCM proteins to and from DNA, under the control of cyclin-dependent kinases, provides a marker to distinguish unreplicated DNA from replicated DNA. However, many questions remain, including:
What are the roles of ORC, Cdc6 and Cdt1 in loading MCMs on to the template? What is the role of cyclin E/Cdk2 in facilitating MCM loading as cells emerge from quiescence?
Are MCMs the main eukaryotic DNA helicases in replication and, if so, is their unexpected distribution and abundance due to a rotary pumping function as suggested in figure 4?
What determines MCM degradation when cells enter quiescence and how does phosphorylation prevent their rebinding during displacement through replication?
Can the range of diagnostic tests based on the presence of MCM proteins in proliferating cells be used to increase the reliability of the cervical smear test and to develop similar tests for the other common carcinomas?
Acknowledgments
I am deeply grateful to the Royal Society for the encouragement that the award of the Croonian Lecture provides and I apologize for the delayed submission of the written version of the lecture, which was initially due to a health problem. I am particularly grateful to past and present members of the research laboratory, especially Mark Madine and Tony Mills for their longstanding contributions to the topics discussed in this lecture. All of the diagnostic work described here has been the result of an enjoyable collaboration with Dr Nick Coleman and members of his laboratory. I also wish to thank Sally Hames, Jackie Marr and Linda Ko Ferrigno for help. Finally, I am grateful to Cancer Research UK and the Medical Research Council for long-term support and to the Louis Jeantet Foundation for providing the funds which allowed us to initiate the translational and clinical research without delay.
References
- Aparicio O.J, Weinstein D.M, Bell S.P. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Aussel L, Barre F.X, Aroyo M, Stasiak A, Stasiak A.Z, Sherratt D. FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell. 2002;108:195–205. doi: 10.1016/s0092-8674(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Baldwin P, Laskey R, Coleman N. Translational approaches to improving cervical screening. Nat. Rev. Cancer. 2003;3:217–226. doi: 10.1038/nrc1010. [DOI] [PubMed] [Google Scholar]
- Bell S.P, Dutta A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Blow J.J, Dilworth S.M, Dingwall C, Mills A.D, Laskey R.A. Chromosome replication in cell-free systems from Xenopus eggs. Phil. Trans. R. Soc. B. 1987;317:483–494. doi: 10.1098/rstb.1987.0075. [DOI] [PubMed] [Google Scholar]
- Blow J.J, Laskey R.A. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature. 1988;332:546–548. doi: 10.1038/332546a0. [DOI] [PubMed] [Google Scholar]
- Brewer B.J. When polymerases collide: replication and the transcriptional organisation of the E. coli chromosome. Cell. 1988;53:679–686. doi: 10.1016/0092-8674(88)90086-4. [DOI] [PubMed] [Google Scholar]
- Campbell J.L, Newlon C.S. Chromosomal DNA replication. In: Broach J.R, Pringle J.R, editors. The molecular and cellular biology of the yeast Saccharomyces cerevisiae. vol. 1. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1991. pp. 41–146. [Google Scholar]
- Celniker S.J, Sweder K, Srienc F, Bailey J.E, Campbell J.L. Deletion mutations affecting autonomously replicating sequence Ars1 of Saccharomyces cerevisiae. Mol. Cell. Biol. 1984;4:2455–2466. doi: 10.1128/mcb.4.11.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J.P.J, Mahbubani H.M, Khoo C.-Y, Blow J. Purification of an Mcm-containing complex as a component of the DNA replication licensing system. Nature. 1995;375:418–421. doi: 10.1038/375418a0. [DOI] [PubMed] [Google Scholar]
- Coverley D, Wilkinson H, Madine M.A, Mills A, Laskey R.A. Protein kinase inhibition in G2 causes mammalian MCM proteins to reassociate with chromatin and restores ability to replicate. Exp. Cell. Res. 1998;238:63–69. doi: 10.1006/excr.1997.3829. [DOI] [PubMed] [Google Scholar]
- Coverley D, Laman H, Laskey R.A. Distinct roles for cyclins E and A during DNA replication. Nat. Cell. Biol. 2002;4:523–528. doi: 10.1038/ncb813. [DOI] [PubMed] [Google Scholar]
- Davies J.R, Freeman A, Morris L.S, Bingham S, Dilworth S, Scott I, Laskey R.A, Miller R, Coleman N. Analysis of minichromosome maintenance proteins as a novel method for detection of colorectal cancer in stool. Lancet. 2002;359:1917–1919. doi: 10.1016/S0140-6736(02)08739-1. [DOI] [PubMed] [Google Scholar]
- Dimitrova D.S, Todorov I.T, Melendy T, Gilbert D.M. Mcm2, but not RPA, is a component of the mammalian early G1-phase pre-replication complex. J. Cell. Biol. 1999;146:709–722. doi: 10.1083/jcb.146.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman E. Pumping DNA. Nature. 2001;409:573–575. doi: 10.1038/35054652. [DOI] [PubMed] [Google Scholar]
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- Evan G.I, Vousden K.H. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Forsburg S.L. Eukaryotic MCM proteins: beyond replication initiation. Microbiol. Mol. Biol. Rev. 2004;68:109–131. doi: 10.1128/MMBR.68.1.109-131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman A, Morris L.S, Mills A.D, Stoeber K, Laskey R.A, Williams G.H, Coleman N. Minichromosome maintenance proteins as biological markers of dysplasia and malignancy. Clin. Cancer Res. 1999;5:2121–2132. [PubMed] [Google Scholar]
- Freidberg E.C. DNA damage and repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- Geng Y, et al. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez M.A, Tachibana K.K, Chin S.-F, Callagy G, Madine M.A, Vowler S.L, Pinder S.E, Laskey R.A, Coleman N. Geminin predicts adverse clinical outcome in breast cancer by reflecting cell cycle progression. J. Pathol. 2004;204:121–130. doi: 10.1002/path.1625. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harland R.M, Laskey R.A. Regulated replication of DNA microinjected into eggs of Xenopus laevis. Cell. 1980;21:761–771. doi: 10.1016/0092-8674(80)90439-0. [DOI] [PubMed] [Google Scholar]
- Hiraiwa A, Fujita M, Nagasaka T, Adachi A, Ohashi M, Ishibashi M. Immunolocalisation of hCDC47 protein in normal and neoplastic human tissues and its relation to growth. Int. J. Cancer. 1997;74:180–184. doi: 10.1002/(sici)1097-0215(19970422)74:2<180::aid-ijc7>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Hyrien O, Méchali M. Chromosomal replication initiates and terminates at random sequences but at regular intervals in the ribosomal DNA of Xenopus early embryos. EMBO J. 1993;12:4511–4520. doi: 10.1002/j.1460-2075.1993.tb06140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O, Maric C, Méchali M. Transition in specification of embryonic metazoan DNA replication origins. Science. 1995;270:994–997. doi: 10.1126/science.270.5238.994. [DOI] [PubMed] [Google Scholar]
- Ishimi Y. A DNA helicase activity is associated with an MCM4, -6 and -7 protein complex. J. Biol. Chem. 1997;272:24508–24513. doi: 10.1074/jbc.272.39.24508. [DOI] [PubMed] [Google Scholar]
- Kaplan D.L, O'Donnell M. DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell. 2002;10:647–657. doi: 10.1016/s1097-2765(02)00642-1. [DOI] [PubMed] [Google Scholar]
- Kearsey S.E. Analysis of sequences conferring autonomous replication in baker's yeast. EMBO J. 1983;2:1571–1575. doi: 10.1002/j.1460-2075.1983.tb01626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey S.E. Structural requirements for the function of a yeast chromosomal replicator. Cell. 1984;37:299–307. doi: 10.1016/0092-8674(84)90326-x. [DOI] [PubMed] [Google Scholar]
- Kearsey S.E, Labib K. MCM proteins: evolution, properties and role in DNA replication. Biochim. Biophys. Acta. 1998;1398:113–136. doi: 10.1016/s0167-4781(98)00033-5. [DOI] [PubMed] [Google Scholar]
- Krude T. Initiation of human DNA replication in vitro using nuclei from cells arrested at an initiation-competent state. J. Biol. Chem. 2000;275:13 699–13 707. doi: 10.1074/jbc.275.18.13699. [DOI] [PubMed] [Google Scholar]
- Krude T, Musahl C, Laskey R.A, Knippers R. Human replication proteins hcdc21, hcdc46 and P1mcm3 bind chromatin uniformly before S-phase and are displaced locally during DNA replication. J. Cell. Sci. 1996;109:309–318. doi: 10.1242/jcs.109.2.309. [DOI] [PubMed] [Google Scholar]
- Krude T, Jackman M, Pines J, Laskey R.A. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell. 1997;88:109–119. doi: 10.1016/s0092-8674(00)81863-2. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Mimura S, Nishimoto S.-I, Takisawa H, Nojima H. Identification of the yeast MCM3 related protein as a component of Xenopus DNA replication licensing factor. Cell. 1995;81:601–609. doi: 10.1016/0092-8674(95)90081-0. [DOI] [PubMed] [Google Scholar]
- Laskey R.A. DNA and cancer. In: Krude T, editor. DNA: Changing Science and Society. Cambridge University Press; 2003. pp. 88–106. [Google Scholar]
- Laskey R.A, Harland R.M. Replication origins in the eukaryotic chromosome. Cell. 1981;24:283–284. doi: 10.1016/0092-8674(81)90316-0. [DOI] [PubMed] [Google Scholar]
- Laskey R.A, Madine M.A. A rotary pumping model for helicase function of MCM proteins at a distance from replication forks. EMBO Rep. 2003;4:26–30. doi: 10.1038/sj.embor.embor706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskey R.A, Harland R.M, Earnshaw W.C, Dingwall C. Chromatin assembly and the co-ordination of DNA replication in the eukaryotic chromosome. In: Schweiger H, editor. International cell biology. Springer; Heidelberg: 1981. pp. 162–167. [Google Scholar]
- Madine M.A, Khoo C.-Y, Mills A.D, Laskey R.A. MCM3 complex required for cell cycle regulation of DNA replication in vertebrate cells. Nature. 1995;375:421–424. doi: 10.1038/375421a0. [DOI] [PubMed] [Google Scholar]
- Madine M, Khoo C.-Y, Mills A.D, Musahl C, Laskey R.A. The nuclear envelope prevents reinitiation of replication by regulating the binding of MCM3 to chromatin in Xenopus egg extracts. Curr. Biol. 1995;5:1270–1279. doi: 10.1016/s0960-9822(95)00253-3. [DOI] [PubMed] [Google Scholar]
- Madine M.A, Swietlik M, Pelizon C, Romanowski P, Mills A.D, Laskey R.A. The roles of the MCM, Orc and Cdc6 proteins in determining the replication competence of chromatin in quiescent cells. J. Struct. Biol. 2000;129:198–210. doi: 10.1006/jsbi.2000.4218. [DOI] [PubMed] [Google Scholar]
- Méchali M, Kearsey S.E. Lack of specific sequence requirement for DNA replication in Xenopus eggs compared with high sequence specificity in yeast. Cell. 1984;38:55–64. doi: 10.1016/0092-8674(84)90526-9. [DOI] [PubMed] [Google Scholar]
- Mills A.D, Coleman N, Morris L.S, Laskey R.A. Detection of S-phase cells in tissue sections by in situ DNA replication. Nat. Cell. Biol. 2000;2:244–245. doi: 10.1038/35008670. [DOI] [PubMed] [Google Scholar]
- Musahl C, Holthoff H.P, Lesch R, Knippers R. Stability of the replicative Mcm3 protein in proliferating and differentiating human cells. Exp. Cell. Res. 1998;241:260–264. doi: 10.1006/excr.1998.4041. [DOI] [PubMed] [Google Scholar]
- Newport J, Kirschner M. A major developmental transition in early Xenopus embryos. I. Characterization and timing of cellular changes at the midblastula stage. Cell. 1982;30:675–686. doi: 10.1016/0092-8674(82)90272-0. [DOI] [PubMed] [Google Scholar]
- Nurse P.M. Cyclin dependent kinases and cell cycle control. Chembiochem. 2002;3:596–603. doi: 10.1002/1439-7633(20020703)3:7<596::AID-CBIC596>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Romanowski P, Madine M.A, Rowles A, Blow J.J, Laskey R.A. The Xenopus origin recognition complex is essential for DNA replication and MCM binding to chromatin. Curr. Biol. 1996;6:1416–1425. doi: 10.1016/s0960-9822(96)00746-4. [DOI] [PubMed] [Google Scholar]
- Sato M, Gotow R, You Z, Komamura-Kohno Y, Uchiyama Y, Yabuta N, Nojima H, Ishimi Y. Electron microscopic observation and single-stranded DNA binding activity of the Mcm4, 6, 7 complex. J. Mol. Biol. 2000;300:421–431. doi: 10.1006/jmbi.2000.3865. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. Ki-67 protein from the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Schwacha A, Bell S.P. Interactions between two catalytically distinct MCM sub-groups are essential for co-ordinated ATP hydrolysis and DNA replication. Mol. Cell. 2001;8:1093–1104. doi: 10.1016/s1097-2765(01)00389-6. [DOI] [PubMed] [Google Scholar]
- Shreeram S, Blow J.J. The role of the replication licensing system in cell proliferation and cancer. Prog. Cell Cycle Res. 2003;5:287–293. [PMC free article] [PubMed] [Google Scholar]
- Simpson A.A, et al. Structure of the bacteriophage Φ29 DNA packaging motor. Nature. 2000;408:745–750. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirieix P.A, O'Donovan M, Brown J, Save V, Coleman N, Fitzgerald R.C. Surface expression of mini-chromosome maintenance proteins provides a novel method for detecting patients at risk for developing adenocarcinoma in Barrett's esophagus. Clin. Cancer Res. 2003;9:2560–2566. [PubMed] [Google Scholar]
- Stoeber K, Mills A.D, Kubota Y, Krude T, Romanowski P, Marheineke K, Laskey R.A, Williams G.H. Cdc6 protein causes premature entry into S phase in a mammalian cell-free system. EMBO J. 1998;17:7219–7229. doi: 10.1093/emboj/17.24.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeber K, et al. Immunoassay for urothelial cancers that detects DNA-replication protein Mcm5 in urine. Lancet. 1999;354:1524–1525. doi: 10.1016/S0140-6736(99)04265-8. [DOI] [PubMed] [Google Scholar]
- Tachibana, K. K., Gonzalez, M. A. & Coleman, N. 2005 Cell-cycle-dependent regulation of DNA replication and its relevance to cancer pathology. J. Pathol.205, 123–129. [DOI] [PubMed]
- Tada S, Li A, Maiorano D, Méchali M, Blow J.J. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat. Cell. Biol. 2001;3:107–113. doi: 10.1038/35055000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Swietlik M, Tanoue A, Tsujimoto J, Kouzarides T, Laskey R. MCM3AP, a novel acetyltransferase that acetylates replication protein MCM3. EMBO Rep. 2001;2:119–123. doi: 10.1093/embo-reports/kve026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Assenberg M, Tsujimoto G, Laskey R. The MCM3 acetylase MCM3AP inhibits initiation, but not elongation of DNA replication via interaction with MCM3. J. Biol. Chem. 2002;277:43 121–43 125. doi: 10.1074/jbc.C200442200. [DOI] [PubMed] [Google Scholar]
- Todorov I.T, Attaran A, Kearsey S.E. BM28, a human member of the Mcm2-3-5 family is displaced from chromatin during DNA replication. J. Cell. Biol. 1995;129:1433–1445. doi: 10.1083/jcb.129.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov I.T, Werness B.A, Wang H.Q, Buddharajul N, Todorova P.D, Slocum H.K, Brooks J.S, Huberman J.A. HsMcm2/BM28: a novel proliferation marker for human tumours and normal tissues. Lab. Invest. 1998;78:73–78. [PubMed] [Google Scholar]
- Tye B.K. MCM proteins in DNA replication. Annu. Rev. Biochem. 1999;68:649–686. doi: 10.1146/annurev.biochem.68.1.649. [DOI] [PubMed] [Google Scholar]
- Williams G.H, Romanowski P, Mills A.D, Morris L, Stoeber K, Marr J, Laskey R.A, Coleman N.C. Improved cervical smear assessment using antibodies against proteins that regulate DNA replication. Proc. Natl Acad. Sci. USA. 1998;95:14 932–14 937. doi: 10.1073/pnas.95.25.14932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlegel J.A, Dwyer B.T, Dhar S.K, Cvetic C, Walter J.C, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science. 2000;290:2309–2312. doi: 10.1126/science.290.5500.2309. [DOI] [PubMed] [Google Scholar]
- Zhou B.S, Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]