

Abstract
The T cell immunoglobulin mucin (TIM) proteins are type I membrane glycoproteins expressed on T cells and containing common structural motifs. While our understanding on the distribution and functions of TIM family members is still incomplete, data from several recent reports indicate that these proteins, together with T cell receptor and costimulatory signals, regulate the expansion and effector functions of T helper cells. In the current review, we provide evidences indicating that TIM-3 is capable of modulating the function of CD4+CD25+ regulatory T cells and inhibiting aggressive Th1 mediated auto- and allo-immune responses. Similarly, additional data suggest that TIM-2 molecules function by negatively regulating Th2 immune responses. In contrast, TIM-1 appears to be an activation molecule for all T cells, although the mechanisms through which TIM-1 activates T cells remain to be elicited.
Keywords: T cells, tolerance, Th1/Th2 paradigm, transplantation, regulatory T cells
Differentiation and the subsequent clonal expansion of T helper (Th) precursor cells into T effector populations plays an important role in the adaptive immune response and provides protection against intracellular viruses and pathogenic bacteria. However, unrestrained activation of Th effector cells has also been shown to underlie a number of inflammatory disorders. In this context, activation of Th1 effector cells has been implicated in rheumatoid arthritis, inflammatory bowel disease (IBD), autoimmune disorders including diabetes and multiple sclerosis (Romagnani 1994), as well as allograft rejection (Strom et al. 1996; Li et al. 1998). In contrast, Th2 cell activation is believed to play a critical role in the pathogenesis of allergic asthma (Anderson & Coyle 1994) and has been linked, albeit not necessarily causally, to the acquisition of transplant tolerance (Strom et al. 1996; Li et al. 1998). The extent of T cell activation and differentiation is largely determined by the duration and strength of T cell receptor mediated stimulation (Kundig et al. 1996). In addition, a number of costimulatory molecules including the TNF receptor family members (CD154, CD134, CD137) (Locksley et al. 2001), immunoglobulin (Ig) superfamily members (CD28, CTLA4, ICOS, PD-1) (Salomon & Bluestone 2001) and CD2 family members (SLAM, CD2, 2B4), as well as cytokines such as IL-2 and IL-15, regulate the extent of clonal expansion, deletion, and/or energy induction (Refaeli et al. 1998). The T cell immunoglobulin mucin (TIM) proteins represent a previously unidentified group of molecules that act in concert with T cell receptor (TCR) and costimulatory signals to regulate expansion and effector functions of Th1 and Th2 cells. TIM family members are type I membrane glycoproteins expressed on T cells and containing common structural motifs, namely Ig V domain, highly glycosylated mucin domain and cytoplasmic domain. The mouse gene family includes 8 members (encoding TIM-1, TIM-2, TIM-3 and TIM-4 proteins and the putative TIM-5 to TIM-8 proteins), while the human gene family includes 3 members encoding TIM-1, TIM-3 and TIM-4 proteins (Kuchroo et al. 2003). TIM gene family members reside in syntenic chromosomal regions, 5q32.2 in human and 11B1.1 in mouse, that have been linked to both atopy and autoimmune diseases. In this review we will summarize our current understanding on the function of TIM family members in the modulation of immune responses, with special emphasis on their roles in the regulation of allo- and autoimmunity.
1. Inhibition of Th1 immune responses by TIM-3
TIM-3 is expressed only by differentiated Th1 cells, and was first identified by V. K. Kuchroo's laboratory, while using Th1 clones to immunize rats and generate monoclonal antibodies (mAb) capable of staining the surface of Th1 but not Th2 clones (Monney et al. 2002). TIM-3 protein is not expressed by naive T cells, and only minor amounts of TIM-3 can be detected after several rounds of antigenic stimulation in the presence of IL-4. In contrast, repetitive in vitro re-stimulations in the presence of Th1-driving conditions (IL-12 and anti-IL-4) leads to an abundance of Th1 cells expressing TIM-3 (Monney et al. 2002; Sanchez-Fueyo et al. 2003). Upregulation of TIM-3 in vivo also requires several rounds of cell division, and is closely associated with γIFN production, although not all γIFN-producing T cells are TIM-3 positive (Sabatos et al. 2003; Sanchez-Fueyo et al. 2003). TIM-3 is also expressed by differentiated type 1 CD8+T cells, but not other cell types (Monney et al. 2002). TIM-3 intracellular domain contains six tyrosine residues, including a tyrosine phosphorylation motif RSEENIY (Monney et al. 2002), and immunoprecipitation studies have shown that upon engagement TIM-3 has the ability to interact with members of the src family (A. J. Coyle, personal communication).
In order to assess the distribution of the putative TIM-3-ligand (L), TIM-3-Ig fusion proteins were constructed by X. X. Zheng from T. B. Strom's laboratory (Sabatos et al. 2003; Sanchez-Fueyo et al. 2003). Using TIM-3-Ig and flow cytometry, the putative TIM-3L is noted on the surface of CD4+CD25− T cells (both naive and memory), as well as on regulatory CD4+CD25+T cells. In addition, some binding can also be found on splenic CD11c+ and CD11b+ cell populations. Interestingly, in vitro stimulation of effector CD4+CD25− T cells leads to downregulation of TIM-3L, while regulatory CD4+CD25+T cells maintain similar levels of TIM-3L expression (Sabatos et al. 2003; Sanchez-Fueyo et al. 2003).
The functional role of TIM-3 in vivo has been mainly explored by using anti-TIM-3 monoclonal antibodies or TIM-3-Ig fusion proteins in a variety of Th1 driven murine models, in which blockade of TIM-3–TIM-3L interactions consistently induces increased Th1 responses. For instance, in a model in which diabetes is induced in NOD-scid mice through the adoptive transfer of T cell-enriched splenocytes from overtly diabetic donor NOD mice, treatment of recipient mice with a mAb specific for TIM-3 significantly hastens the onset of diabetes (Sanchez-Fueyo et al. 2003). Interestingly, similar results are obtained by using a TIM-3-Ig fusion protein, indicating that TIM-3 triggers a negative signal on islet destroying Th1 cells (Sanchez-Fueyo et al. 2003). Furthermore, administration of anti-TIM-3 mAb can aggravate Th1-dependent colitis after adoptive transfer of CD4+ CD45RBlow T cells into Scid mice (A. J. Coyle, personal communication), and increase the severity of experimental autoimmune encephalomyelitis (EAE) in immunized SJL mice (Monney et al. 2002; Sabatos et al. 2003). Surprisingly, in this model of EAE induction anti-TIM-3 mAb administration leads to massive activation and clonal expansion of macrophages (Monney et al. 2002; Sabatos et al. 2003), indicating that TIM-3 blockade may interfere with an interaction between Th1 cells and TIM-3L-expressing macrophages. An alternative model to account for TIM-3 biological effects involves its effect on CD4+CD25+T cells, which has been described in a peripheral tolerance transplantation model (Sanchez-Fueyo et al. 2003). In this model, long term survival and tolerance to MHC mismatched allografts is achieved through the administration of donor specific transfusion (DST) plus anti-CD154 (CD40L) co-stimulation blockade (Sanchez-Fueyo et al. 2002, 2003). These potent tolerizing effects are critically dependent on the presence of CD4+CD25+ regulatory T cells, which greatly augment their immunosuppressive properties in a donor-specific manner after recognizing donor alloantigens in the presence of CD40–CD154 co-stimulation blockade (Sanchez-Fueyo et al. 2003). Administration of TIM-3-Ig together with DST plus anti-CD154 abrogates tolerance induction (Sanchez-Fueyo et al. 2002, 2003). Likewise, TIM-3 deficient mice cannot be rendered tolerant to islet allografts after DST plus anti-CD154 treatment (Sanchez-Fueyo et al. 2003), although wild-type CD4+CD25+T cells can perfectly neutralize the cytopathic effects of TIM-3 deficient effector T cells (Sanchez-Fueyo et al. 2003). In fact, TIM-3-Ig does not interfere with the suppressive effects of regulatory T cells but neutralizes the capacity of DST plus anti-CD154 to enhance CD4+CD25+T cell function (Sanchez-Fueyo et al. 2003). The mechanisms through which this phenomenon takes place have not been thoroughly determined. It is clear from several reports that the absence of CD40–CD154 co-stimulatory signals promotes peripheral tolerance and regulatory T cell activity (Jarvinen et al. 2003; Martin et al. 2003; Quezada et al. 2004). Thus, CD40 and TIM-3 could have opposing effects on antigen presenting cells serving as a checkpoint in the decision between tolerance and immunity. Alternatively, the Th1 cytokine environment promoted by TIM-3 blockade might interfere with the generation of powerfully suppressive regulatory T cells. The latter hypothesis is supported by data indicating that Th1 to Th2 immune deviation enhances the regulatory properties of CD4+CD25+T cells (our unpublished observations).
In short, TIM-3 functions to inhibit aggressive Th1 mediated auto- and allo-immune responses. These effects appear to be mediated, at least in part, by the regulation of the immunosuppressive potency of CD4+CD25+ regulatory T cells (figure 1). In addition, an inhibitory effect on Th1 cells through modulation of macrophage activation is also likely particularly in heavily Th1-biased inflammatory conditions.
Figure 1.

Potential mechanisms by which TIM-2 and TIM-3 modulate Th1–Th2 response. (a) TIM-3 and TIM-2 are reciprocally expressed by Th1 and Th2 cells, but TIM-3 is only expressed at the terminal stage of Th1 differentiation. Interaction between TIM-2 and its ligand delivers a negative signal to T cells and downregulates Th2 response (1). At the early stage of differentiation, Th1 cells don't express TIM-3 and expand (2). Terminally differentiated Th1 cells upregulate TIM-3. Interaction of TIM-3 with its ligand on antigen presenting cells and/or CD4+CD25+ regulatory T cells provides a negative signal that limits Th1 cells expansion (3). (b) Blocking the inhibitory interaction between TIM-2 and TIM-2L (either by using a TIM-2 fusion protein or an anti-sema4A mAb) allows the expansion of Th2 cells (1). IL-4, secreted by Th2 cells, reinforces the Th2 deviation by directly inhibiting the Th1 subset (2). Additionally, the blockade of the TIM-2/TIM-2L pathway might enhance CD4+CD25+ regulatory T cells expansion/function by providing a favourable Th2 cytokine milieu and/or by relieving TIM-2+ regulatory T cells from an inhibitory signal (3).
2. TIM-2 regulates Th2 responses
TIM-2 is structurally very similar to TIM-1 and is also expressed on CD4+T cells, but no orthologue for TIM-2 has been found in humans (McIntire et al. 2001; Kumanogoh et al. 2002). In mice, TIM-2, in contrast to TIM-3, is specifically upregulated in Th2 cells, and is downregulated in T cells cultured under Th1-inducing conditions (V. K. Kuchroo, unpublished observations). A ligand for TIM-2 has been identified as Sema4A, a protein mostly expressed by bone-marrow derived and splenic dendritic cells (Kumanogoh et al. 2002). Interestingly, Sema4A-dependent signal has been shown to be necessary for in vitro Th1 differentiation and T-bet expression, and Sema4A-deficient mice exhibit defective Th1 response (Kuchroo et al. 2003). In an EAE model, a blocking anti-Sema4A mAb is capable of inhibiting the production of Th1 cytokines by myelin-specific T cells if administered in the early phases (Kuchroo et al. 2003). In transplantation, the blockade of TIM-2/TIM-2L interaction using a TIM-2-Ig fusion protein synergizes with CD154 co-stimulation blockade to prevent allograft rejection, a phenomenon in which CD4+CD25+ regulatory T cells appear to be involved (our unpublished observations). Taken together these data indicate that, similarly to TIM-3 on Th1 cells, TIM-2 might provide an inhibitory signal on Th2 cells and downregulate Th2 responses. Hence blockade of TIM-2/TIM-2L interaction may relieve Th2 cells from this inhibition, skew the immune response toward a Th2 phenotype, and thereby favouring the induction/maintenance of peripheral tolerance (figure 1).
3. TIM-1, adjuvant of T cell response
TI5M-1 was first described as KIM-1 (kidney injury molecule 1), an epithelial cell adhesion glycoprotein, which is upregulated in cell regeneration of proximal tubules in post-ischaemic kidney (Ichimura et al. 1998; Kuchroo et al. 2003). Rat KIM-1 gene exhibits a high homology of sequence with a human receptor for the hepatitis A virus (HAVcr1), and it is only recently that murine TIM-1 has been identified as the orthologue of both rat KIM-1 and human HAVcr1 (Kuchroo et al. 2003).
The possible involvement of TIM-1 in regulating Th1–Th2 cell differentiation stems from the work of McIntire and colleagues (Kuchroo et al. 2003), who identified a single locus on murine chromosome 11 that confers protection against the Th2-mediated development of airway hyper-reactivity in mice (AHR), an experimental model of asthma. This locus, named the T cell and airway-phenotype regulator (Tapr), is homologous to the human 5q32.2 locus, a region of the human genome linked with susceptibility to asthma and allergy (Kuchroo et al. 2003).
The TIM gene family has been shown to be located in this particular locus and among the different TIM proteins, the sequencing of TIM-1 showed major polymorphism between the mouse strains susceptible and resistant to AHR (Kuchroo et al. 2003). In addition to its linkage with AHR, TIM-1 was found to be transcribed during primary antigen stimulation in CD4+T cells (Kuchroo et al. 2003), a period of time that is crucial in influencing Th1–Th2 orientation. Together with the initial assumption that TIM-1 was selectively expressed by Th2-type T cells, these data led to the hypothesis that TIM-1 might be a key factor in regulating Th2 response. This hypothesis was further supported by several epidemiological studies that have documented an association between exposure to HAV and protection from atopy and asthma (Kuchroo et al. 2003). The common explanation to this association is that infection with viruses early in childhood promotes the development of Th1 cells, which in turn downregulate the development of Th2 cell response (Kuchroo et al. 2003). As an alternative, McIntire and colleagues propose that HAV may directly inhibit Th2 differentiation via its interaction with TIM-1/HAVcr1, and thereby reduce the likelihood of developing asthma (Kuchroo et al. 2003).
Umetsu's group provided the first mechanistic insights into the immunological function of TIM-1 by generating an agonist monoclonal antibody (mAb) specific for TIM-1 (clone 3B3; Kuchroo et al. 2003). This mAb exhibits remarkable co-stimulatory effect on T cells, greatly enhancing proliferation and cytokine production both in vitro and in vivo. In keeping with the identification of TIM-1 as an asthma-susceptibility gene, Umetsu and colleagues found that TIM-1 co-stimulation prevents the induction of respiratory tolerance and results in the development of AHR. They confirmed the expression of TIM-1 on non-polarized CD4+T cells early after activation and, interestingly, observed that co-stimulation by anti-TIM-1 increased production of both γINF and IL4. These data, collectively with the finding that TIM-1 is also significantly expressed on murine and human Th1 cell lines (Kuchroo et al. 2003), suggest that TIM-1 might rather function as an adjuvant of T cell responses, regardless of the Th1/Th2 orientation. In this respect, we have found that the same anti-TIM 1 clone is also able to stimulate the activation of allo-reactive cytopathic cells in a model of islet transplantation characterized by a Th1-dominant response (manuscript in preparation).
Thus far the expression of TIM-1 has been emphasized only on T cells and little attention has been paid to other cell types involved in the immune response. Using real-time quantitative PCR, we have been able to detect high expression of TIM-1 mRNA in antigen presenting cells, including different subset of dendritic cells (unpublished data). Whether the function of dendritic cells can be modulated by Tim-1, and whether dendritic cells can mediate the stimulatory signal provided by Tim-1 to T cells is still unresolved but it is tempting to speculate that TIM-1 could act by targeting both sides of the immunological synapse.
In summary, we are just at the very beginning of elucidating the role of TIM-1 in immunity. The data currently available seem to indicate that TIM-1 is probably not merely a Th2 regulator as initially hypothesized, but potentially a broader T cell costimulatory molecule that might be used as an effective adjuvant to enhance T cell immunity (figure 2).
Figure 2.
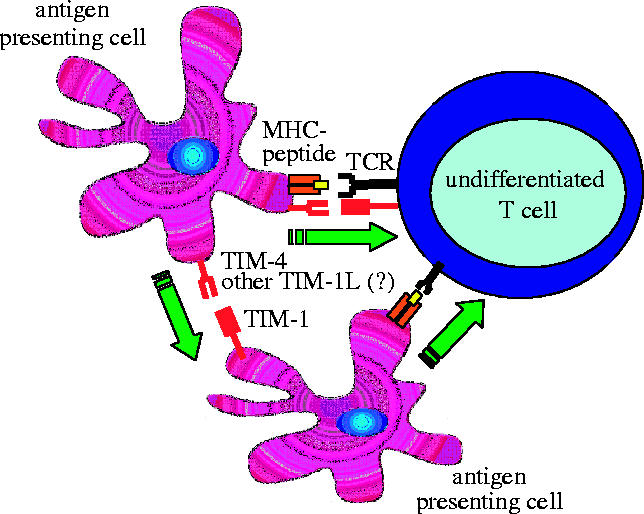
TIM-1-dependent costimulation of T cells. TIM-1 is upregulated on T cell early after activation and interacts with its ligand expressed on the stimulating antigen-presenting cell (APC). TIM-1/TIM-1L interaction provides a positive signal that costimulates T cell activation. Alternatively, TIM-1 expressed on APC might directly deliver a positive signal to the APC, which in turn amplifies T cell activation.
4. TIM-4 is a natural ligand for TIM-1
As compared to the other TIM-family members, TIM-4 lacks the signaling intracellular kinase phosphorylation motif (a putative signalling motif) and displays a RGD sequence in its IgV domain, a sequence found in many ligands that bind T cells integrins. Consistent with this structural feature, Meyers and colleagues recently reported that TIM-4 is not expressed by T cells but restricted to antigen-presenting cells with a particularly high level of expression on mature lymphoid dendritic cells (Meyers et al. 2005). While looking for a possible interaction between TIM-4 and other members of the TIM family, we find that a specific binding between a TIM-4 fusion protein and TIM-1-transfected cells line, suggesting that TIM-4 is the endogenous ligand of TIM-1 in antigen-presenting cells. Interestingly, in agreement with the effect of anti-TIM-1 mAb, we observed that the TIM-4/TIM-1 interaction delivers a positive signal and costimulates T cell proliferation. However, TIM-4 might not be the unique ligand of TIM-1 since Myers and colleagues also reported some staining of TIM-4− cells by a TIM-1-Ig (Meyers et al. 2005). In this regard, the precise interplay between different dendritic cell subsets (TIM-4+ and TIM-4− DC) and TIM-1+ T cells will have to be deciphered in order to assess the exact functional relevance of the TIM-4/TIM-1 interaction during the immune response (figure 2).
5. Concluding remarks
The studies on distribution and function of TIM proteins are still in their early phase in terms of understanding the exact role each of these molecules plays in modulating the immune response. Many additional studies will be required to obtain a detailed picture of the regulatory role these molecules play. For example, TIM-1 appears to be an activation molecule for many T cells but it is only expressed on T cells after they have been activated. In addition, it is not clear whether signalling via TIM-1 synergizes with TCR signal transduction. TIM-1 may be important in the activation of the DC, where it is easily detected, but the consequences of TIM-1 stimulation on DCs has not yet been carried out, and the physiological consequences of TIM-1 and TIM-4 interactions are still unknown. The idea that TIM-2 and TIM-3 molecules function by negatively regulating Th2 and Th1 immune responses is well supported by the findings presented in this review. These data show that TIM-2 and TIM-3-specific antagonistic antibodies or Ig blocking agents enhance Th2 and Th1 immune response, respectively. It will be important to demonstrate that agonistic antibodies and/or purified ligands also down regulate the Th1 and Th2 immune response. The production of knockout mice for each of the TIM proteins and their ligands will also help to definitively elucidate the complex interactions and functions these molecules carry out in helping to regulate the immune response.
Footnotes
One contribution of 16 to a Theme Issue ‘Immunoregulation: harnessing T cell biology for therapeutic benefit’.
References
- Anderson G.P, Coyle A.J. TH2 and ‘TH2-like’ cells in allergy and asthma: pharmacological perspectives. Trends. Pharmacol. Sci. 1994;15:324–332. doi: 10.1016/0165-6147(94)90027-2. doi:10.1016/0165-6147(94)90027-2 [DOI] [PubMed] [Google Scholar]
- Ichimura T, Bonventre J.V, Bailly V, Wei H, Hession C.A, Cate R.L, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J. Biol. Chem. 1998;273:4135–4142. doi: 10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- Jarvinen L.Z, Blazar B.R, Adeyi O.A, Strom T.B, Noelle R.J. CD154 on the surface of CD4+CD25+ regulatory T cells contributes to skin transplant tolerance. Transplantation. 2003;76:1375–1379. doi: 10.1097/01.TP.0000093462.16309.73. doi:10.1097/01.TP.0000093462.16309.73 [DOI] [PubMed] [Google Scholar]
- Kuchroo V.K, Umetsu D.T, DeKruyff R.H, Freeman G.J. The TIM gene family: emerging roles in immunity and disease. Nat. Rev. Immunol. 2003;3:454–462. doi: 10.1038/nri1111. doi:10.1038/nri1111 [DOI] [PubMed] [Google Scholar]
- Kumanogoh A, et al. Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature. 2002;419:629–633. doi: 10.1038/nature01037. doi:10.1038/nature01037 [DOI] [PubMed] [Google Scholar]
- Kundig T.M, Shahinian A, Kawai K, Mittrucker H.W, Sebzda E, Bachmann M.F, Mak T.W, Ohashi P.S. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity. 1996;5:41–52. doi: 10.1016/s1074-7613(00)80308-8. doi:10.1016/S1074-7613(00)80308-8 [DOI] [PubMed] [Google Scholar]
- Li X.C, Zand M.S, Li Y, Zheng X.X, Strom T.B. On histocompatibility barriers, Th1 to Th2 immune deviation, and the nature of the allograft responses. J. Immunol. 1998;161:2241–2247. [PMC free article] [PubMed] [Google Scholar]
- Locksley R.M, Killeen N, Lenardo M.J. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. doi:10.1016/S0092-8674(01)00237-9 [DOI] [PubMed] [Google Scholar]
- Martin E, O'Sullivan B, Low P, Thomas R. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin-10. Immunity. 2003;18:155–167. doi: 10.1016/s1074-7613(02)00503-4. doi:10.1016/S1074-7613(02)00503-4 [DOI] [PubMed] [Google Scholar]
- McIntire J.J, Umetsu S.E, Akbari O, Potter M, Kuchroo V.K, Barsh G.S, Freeman G.J, Umetsu D.T, DeKruyff R.H. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat. Immunol. 2001;2:1109–1116. doi: 10.1038/ni739. doi:10.1038/ni739 [DOI] [PubMed] [Google Scholar]
- Meyers J.H, et al. TIM-4 is the ligand for TIM-1, and the TIM-1–TIM-4 interaction regulates T cell proliferation. Nat. Immunol. 2005;6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- Monney L, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. doi:10.1038/415536a [DOI] [PubMed] [Google Scholar]
- Quezada S.A, Jarvinen L.Z, Lind E.F, Noelle R.J. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. doi:10.1146/annurev.immunol.22.012703.104533 [DOI] [PubMed] [Google Scholar]
- Refaeli Y, Van Parijs L, London C.A, Tschopp J, Abbas A.K. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. doi:10.1016/S1074-7613(00)80566-X [DOI] [PubMed] [Google Scholar]
- Romagnani S. Lymphokine production by human T cells in disease states. Annu. Rev. Immunol. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. doi:10.1146/annurev.iy.12.040194.001303 [DOI] [PubMed] [Google Scholar]
- Sabatos C.A, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. doi:10.1038/ni988 [DOI] [PubMed] [Google Scholar]
- Salomon B, Bluestone J.A. Complexities of CD28/B7:CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. doi:10.1146/annurev.immunol.19.1.225 [DOI] [PubMed] [Google Scholar]
- Sanchez-Fueyo A, Weber M, Domenig C, Strom T.B, Zheng X.X. Tracking the immunoregulatory mechanisms active during allograft tolerance. J. Immunol. 2002;168:2274–2281. doi: 10.4049/jimmunol.168.5.2274. [DOI] [PubMed] [Google Scholar]
- Sanchez-Fueyo A, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. doi:10.1038/ni987 [DOI] [PubMed] [Google Scholar]
- Strom T.B, Roy-Chaudhury P, Manfro R, Zheng X.X, Nickerson P.W, Wood K, Bushell A. The Th1/Th2 paradigm and the allograft response. Curr. Opin. Immunol. 1996;8:688–693. doi: 10.1016/s0952-7915(96)80087-2. doi:10.1016/S0952-7915(96)80087-2 [DOI] [PubMed] [Google Scholar]