

Abstract
Periods of prolonged hypoxia are associated clinically with an increased incidence of dementia, the most common form of which is Alzheimer's disease. Here, we review recent studies aimed at providing a cellular basis for this association. Hypoxia promoted an enhanced secretory response of excitable cells via formation of a novel Ca2+ influx pathway associated with the formation of amyloid peptides of Alzheimer's disease. More strikingly, hypoxia potentiated Ca2+ influx specifically through L-type Ca2+ channels in three distinct cellular systems. This effect was post-transcriptional, and evidence suggests it occurred via increased formation of amyloid peptides which alter Ca2+ channel trafficking via a mechanism involving increased production of reactive oxygen species by mitochondria. This action of hypoxia is likely to contribute to dysregulation of Ca2+ homeostasis, which has been proposed as a mechanism of cell death in Alzheimer's disease. We suggest, therefore, that our data provide a cellular basis to account for the known increased incidence of Alzheimer's disease in patients who have suffered prolonged hypoxic episodes.
Keywords: hypoxia, calcium channel, Alzheimer's disease, reactive oxygen species
1. Hypoxia and Alzheimer's disease: the clinical evidence
Adaptation to conditions of chronic hypoxia is usually only seen in healthy individuals when they spend prolonged periods of time at high altitude. However, arterial O2 levels can be markedly reduced at sea level in patients suffering from a wide variety of cardiorespiratory diseases. Examples include congestive heart failure, chronic obstructive pulmonary disease and emphysema. Prolonged periods of hypoxia can lead to damage of higher brain functions such as memory and cognition and indeed, a wealth of clinical reports indicate clearly that in such individuals the likelihood of subsequently developing dementias—particularly Alzheimer's disease—is greatly increased (Incalzi et al. 1993; Tatemichi et al. 1994; Kokmen et al. 1996; Moroney et al. 1996). Our limited understanding of this important clinical link is that it appears to occur because hypoxia can promote selectively the expression of genes which encode key proteins associated with Alzheimer's disease (Kogure & Kato 1993; Koistinaho et al. 1996). However, the mechanisms underlying such effects, and their physiological consequences, have not, to date, been studied in depth and so our current awareness is poor. Nevertheless, the clear link between hypoxic/ischaemic episodes and increased incidence of Alzheimer's disease strongly suggests that one or more of these parameters are capable of precipitating this increasingly widespread disease.
2. Hypoxia and Alzheimer's disease: evidence for cellular mechanisms
Our previous studies, along with those of others, have shown that prolonged hypoxia causes dramatic changes in the functional expression of key proteins (particularly ion channels) in neuronally derived cells and cell lines (Wyatt et al. 1995; Taylor et al. 1999; Green & Peers 2001; Peers & Kemp 2001; Colebrooke et al. 2002) and also, more recently, in primary cultures of central neurons (Plant et al. 2002). Most importantly, we have shown that many of these effects of hypoxia involve formation of amyloid β peptides (AβPs) associated with Alzheimer's disease and can be mimicked by exposing cells to AβPs under normoxic conditions. Indeed, these dramatic effects of hypoxia appear to require formation of AβPs (Taylor et al. 1999; Green & Peers 2002; Green et al. 2002). Thus, of all the parameters required for normal cell function that can be disturbed during hypoxic/ischaemic episodes (e.g. lack of substrates, acidosis, accumulation of metabolic waste products) reduction in available O2 appears to be a crucial factor in the initiation of Alzheimer's disease.
3. Hypoxia and exocytosis
A wealth of evidence suggests that neuronal cell death in Alzheimer's disease arises from disruption of Ca2+ homeostasis. We first provided potential support for this in the actions of chronic hypoxia when examining exocytosis from individual PC12 cells, which release catecholamines in response to depolarizing stimuli that is dependent on voltage-gated Ca2+ entry. Exocytosis was monitored in real time, using single cell amperometry (Wightman et al. 1991; Finnegan et al. 1996). As shown in figure 1a, secretion (stimulated by exposure of cells to 50 mM K+) was entirely dependent on Ca2+ influx, since removal of Ca2+ from the extracellular solution fully and reversibly prevented exocytosis. This was the case in cells cultured normoxically and also in cells cultured under hypoxic conditions (figure 1a, upper traces). However, when Ca2+ entry via voltage-gated Ca2+ channels was prevented by application of 200 μM Cd2+, a non-selective Ca2+ channel blocker, a significant amount of exocytosis was evident in hypoxic (but not normoxic) cells. Thus, hypoxia induced a Cd2+-resistant Ca2+ influx pathway coupled to catecholamine secretion.
Figure 1.

(a) Amperometric detection of secretion evoked in control (left traces) and chronically hypoxic (right) PC12 cells in response to 50 mM K+ (stimulus application began 10 s before beginning of traces). For the periods indicated by horizontal bars, cells were exposed to either Ca2+-free perfusate (upper traces), or to 200 μM Cd2+ (lower traces) in the presence of 2.5 mM Ca2+ (also in the continued presence of 50 mM K+) as indicated. Note that in chronically hypoxic cells, 200 μM Cd2+ failed to inhibit secretion completely. (b) Bar graph showing mean (+s.e.m. bars) exocytotic frequency recorded in cells previously exposed to 10% O2 for 21–26 h, in response 50 mM K+ in the presence of 200 μM Cd2+ alone (open bar), or following further blockade with Zn2+(10 mM), La3+(1 mM) Congo Red (10 mM) or 3D6 antibody (5 μg ml−1) as indicated. All blockers produced significant inhibition (p<0.04–0.001) of Cd2+-resistant release (n=8–12 recordings in each case).
To examine the Cd2+-resistant Ca2+ influx pathway pharmacologically, we examined secretion in the presence of Cd2+ but in the additional presence of other compounds. La3+ and Zn2+, known blockers of various Ca2+ influx pathways in other cells, suppressed secretion above that seen in the presence of Cd2+ alone (figure 1b). Secretion was also further suppressed by Congo Red; both Congo Red and Zn2+ have been shown previously to inhibit AβP-mediated Ca2+ fluxes (Lorenzo & Yankner 1994; Rhee et al. 1998), suggesting the surprising possibility that a 12–24 h period of hypoxia induced formation of Ca2+-permeable amyloid peptide channels which coupled closely to exocytosis. In further support of this, we found that exposure of cells to a monoclonal antibody (3D6) raised against the extracellularly located N′ terminus of amyloid peptides (anti-AβP1–5; Johnson-Wood et al. 1997) for 1 h at 37 °C also significantly suppressed the Cd2+-resistant component of the secretory response (figure 1b). These findings strongly suggested that hypoxia caused formation of Ca2+ permeable channels which were tightly coupled to exocytosis and are in some way associated with amyloid peptide formation.
4. Hypoxia and native Ca2+ channels
Clearly, the implied amyloid-related Ca2+ entry pathway required further study. We therefore used whole-cell patch clamp recordings to monitor Ca2+ influx directly, and we identified a marked increase in whole cell Ca2+ current density in cells cultured under hypoxic conditions (2.5% O2, 24 h), as compared with normoxic, control cells (figure 2a,b). Interestingly, this effect of hypoxia could be mimicked by exposure of cells to amyloid peptides, either Aβ(1–40) (figure 2c) or Aβ(25–35) (figure 2d). This was in accordance with the studies on exocytosis (figure 1). However, when cells were exposed to Cd2+ currents were virtually completely blocked (figure 2), suggesting that any Cd2+-resistant Ca2+ influx pathway was extremely small. Closer inspection revealed this to be the case: figure 2e,f illustrates mean Ca2+ currents recorded in the presence of Cd2+ in hypoxic (figure 2e) and Aβ(1–40) (figure 2f) treated cells. Note that this current could be fully blocked by the 3D6 monoclonal antibody. Note also that these tiny currents could not account for the marked increase in total whole-cell Ca2+ current, indicating that an additional effect of hypoxia (and amyloid) must have occurred.
Figure 2.

Augmentation of Ca2+ channel currents in PC12 cells by hypoxia and AβPs. Mean (+s.e.m. bars) Ca2+ channel current–voltage (I–V) relationships obtained from PC12 cells in the absence (solid circles) and presence (open circles) of 200 μM Cd2+. (a) I–V relationships obtained from 20 control (normoxically cultured) cells. (b) I–V relationships obtained from 28 cells maintained in 10% O2 for 24 h prior to recordings. (c) I–V relationships from nine cells exposed to 100 nM AβP1–40 for 24 h prior to recording (also shown are mean data from 14 cells exposed to 20 nM AβP1–40 for 24 h, in the absence of Cd2+; solid triangles). (d) I–V relationships from nine cells exposed to AβP25–35 for 24 h. (e) Mean I–V relationships taken from hypoxically cultured cells, recorded in the presence of 200 μM Cd2+ (solid circles). Open circles show data from identically treated cells, except that they were exposed to the 3D6 antibody (5 μg ml−1; 1 h). (f) As in (e), except that cells were exposed to amyloid petides (as in (c)) rather than hypoxia (n=8–15 cells in each case).
PC12 cells, like many excitable neuronal or neuroendocrine cells, possess multiple types of voltage-gated Ca2+ channels. Of these, we found that hypoxia appeared to selectively increase the L-type Ca2+ channels in these cells, as reflected in the increased sensitivity to 2 μM nifedipine (figure 3a,b). This was also the case in cells treated with amyloid peptides (figure 3c,d). It was noteworthy that the nifedipine-resistant current densities in each cell group were similar. Thus, hypoxia (and amyloid peptides) selectively augmented L-type Ca2+ currents.
Figure 3.

Nifedipine sensitivity of Ca2+ channel currents in PC12 cells. Mean (+s.e.m. bars) Ca2+ channel current–voltage (I–V) relationships obtained from PC12 cells before (filled circles) and during (open circles) bath application of 2 μM nifedipine. Cells were either cultured normoxically (a, n=10 cells), exposed to chronic hypoxia for 24 h (b, n=8), exposed to 100 nM AβP1–40 for 24 h (c, n=9), or to 100 nM AβP25–35 for 24 h (d, n=9).
While such effects were interesting, they were somewhat removed from the question of development of Alzheimer's disease, since they were performed in a clonal cell line. Thus, to address a more pertinent cell type, we examined this phenomenon in cerebellar granule neurons (CGNs). Figure 4a indicates that, as in PC12 cells, Ca2+ currents recorded in CGNs were augmented by a period of hypoxia, and this effect was not seen when the L-type component of the Ca2+ current was blocked by nifedipine (figure 4b). At the same time, immunoreactivity for amyloid β peptide was markedly enhanced in these cells (figure 4c), an effect which was fully blocked by inhibition of either β- or γ-secretase, the two enzymes required for cleavage of Aβ from its precursor protein, APP (figure 4c). This was a primary indication that the effects of hypoxia required Aβ formation. Amyloid immunoreactivity was examined using the monoclonal antibody, 3D6 (Johnson-Wood et al. 1997).
Figure 4.

Hypoxia augments voltage-gated Ca2+ channels in cerebellar granule neurons. (a) Mean (+s.e.m. bars) current density versus voltage relationships obtained from neurons cultured under normoxic (open squares; n=13 cells) or chronically hypoxic (solid squares; n=17 cells) conditions. (b) Mean (+s.e.m. bars) current density versus voltage relationships obtained from neurons cultured under normoxic (open squares; n=12 cells) and chronically hypoxic (solid squares; n=14 cells) conditions, and recorded in the presence throughout of 2 μM nimodipine. (c) Immunofluorescent images of clusters of cerebellar granule cell bodies (together with bright field images, to the right of each) cultured either normoxically (left) or under chronically hypoxic conditions (right) in the absence of secretase inhibitors (top row) or in the presence of the γ-secretase inhibitor γ-IV (3 μM; middle row) or the β-secretase inhibitor, BS-I (30 nM; bottom row). Scale bar shown in the bottom of the figure indicates 10 μm and applies to all panels.
5. Hypoxia and recombinant Ca2+ channels
This phenomenon of hypoxia-induced increases in functional L-type Ca2+ channel expression was also faithfully reproduced in a recombinant expression system. Thus, using a HEK-293 cell line stably expressing the major subunit of the human L-type Ca2+ channel (α1C) in the absence of auxiliary subunits, whole-cell patch clamp recordings showed current augmentation by either hypoxia (figure 5a) or by AβPs (figure 5b). Furthermore, the effect of hypoxia could be almost fully inhibited by co-incubation with inhibitors of either β- or γ-secretases (figure 5c), again indicating that the effects of hypoxia required amyloid formation. This finding suggested that hypoxia did not augment current amplitudes via transcriptional regulation of the channel protein, since such transcription was under the control of the promoter regions of the plasmid introduced artificially into the HEK-293 cells. We reasoned, therefore, that increased functional expression might be attributed to a post-transcriptional event such as altered trafficking. Initial studies using double immunohistochemistry suggested that amyloid peptides and the α1C subunit co-localized, particularly under hypoxic conditions (Scragg et al. 2005). To probe further the possible association of these two proteins, we performed immunoprecipitation and western blot experiments. Figure 6a shows blots taken from homogenates precipitated using the 3D6 (anti-amyloid) antibody then probed with the anti-α1C antibody. In control cells (left), the signal was extremely weak. However, a clear band of appropriate molecular weight was detected in hypoxic cells (right). These results (representative of six paired experiments) were supported in experiments where homogenates were precipitated with the anti-α1C antibody then probed using the 3D6 antibody (figure 6b). In this case, amyloid peptides were detected in both control (figure 6b, left) and hypoxic (right) cell samples, proof of physical interaction under both conditions, but band intensity was consistently (n=6 experiments) greater in hypoxic samples.
Figure 5.
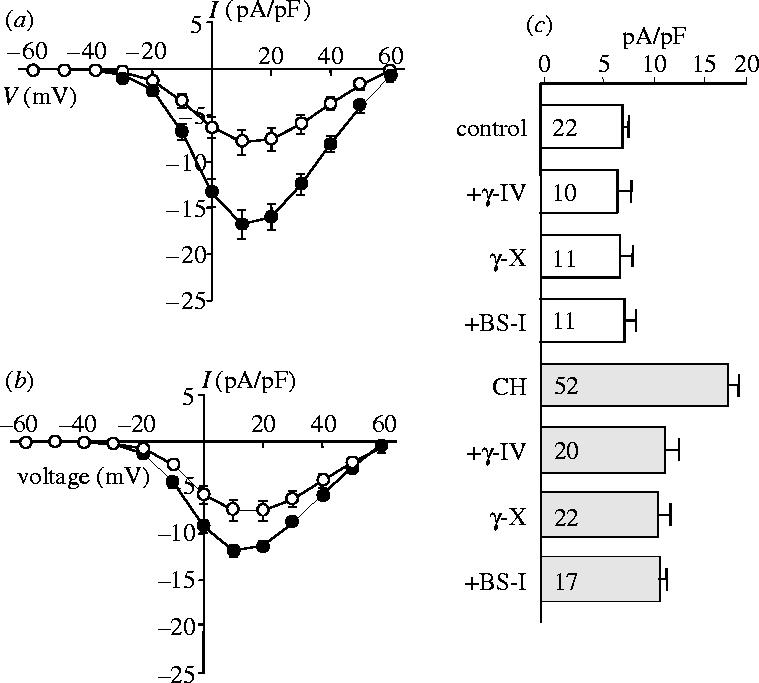
Hypoxia enhances Ca2+ channel currents in HEK-293 cells stably expressing the human cardiac L-type Ca2+ channel α1C subunit. (a) Mean (+s.e.m. bars) current density versus voltage relationships obtained from control cells (open symbols, n=12) and from cells cultured in a hypoxic atmosphere of 2.5% O2 for 24 h prior to recording (solid symbols, n=15). (b) Mean current density (+s.e.m. bars) versus voltage relationships obtained from control cells (open symbols, n=9) and from cells pretreated for 24 h with 20 nM AβP1–40 (solid symbols circles, n=18). (c) Bar graph showing mean (+s.e.m.) current density (determined at a test potential of +20 mV) in control cells (open bars) and in hypoxically cultured (CH) cells (shaded bars) exposed to various secretase inhibitors (the β-secretase inhibitor, BS-I (30 nM), the γ-secretase inhibitor, γ-IV (2.5 μM) and the γ-secretase inhibitor, γ-X (100 nM), as indicated). Values in bars indicate n numbers.
Figure 6.
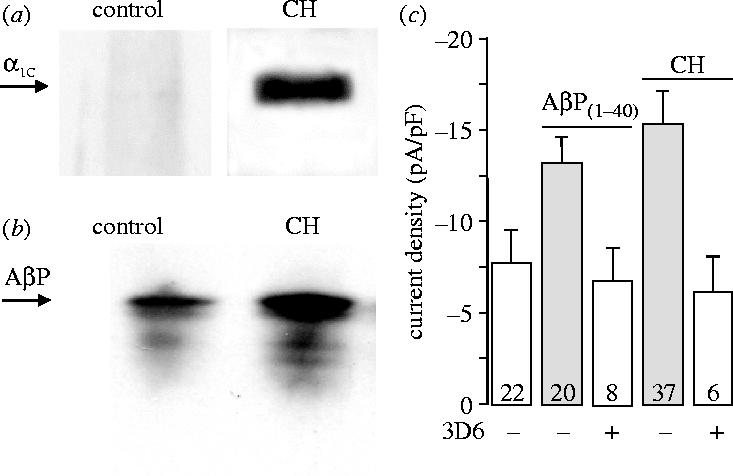
Evidence for co-localization of amyloid peptide and Ca2+ channels. (a) Western blots probed with the anti-α1C antibody, following immunoprecipitation of cell homogentates using the 3D6 monoclonal anti-amyloid antibody. Note marked immunodetection in hypoxic but not normoxic cells. Representative of six experiments. (b) Detection of AβPs in samples of control and hypoxic cell homogenates, immunoprecipitated using the anti-α1C antibody as indicated. Blots were probed using the 3D6 antibody. Representative of six experiments. (c) Bar graph plotting mean (+s.e.m.) current density observed at a test potential of +20 mV in control cells, cells exposed to 20 nM AβP1–40 for 24 h, and cells exposed to chronic hypoxia (as indicated) either with (open bars) or without (shaded bars) subsequent exposure to the anti-amyloid peptide monoclonal antibody 3D6 (5 μg ml−1). Values in bars indicate n numbers.
Figure 6c plots mean current densities (taken at the membrane potential at which currents were maximal in amplitude) in control cells, and those cultured under hypoxic conditions or in the presence of amyloid peptides. Also shown are data obtained from hypoxic or amyloid-treated cells following a 1 h period in which cells were also exposed to 5 μg ml−1 of the 3D6 monoclonal antibody. The antibody fully reversed current augmentation under either experimental condition. This effect was comparable to the effects of autoantibodies present in the plasma of patients with Lambert–Eaton myasthenic syndrome (Vincent et al. 1989), who present with muscular weakness due to suppression of acetylcholine (ACh) release from motor nerve terminals. ACh release is suppressed because autoantibodies bind to, and cross-link, presynaptic Ca2+ channels (Vincent et al. 1989; Peers et al. 1993). This cross-linking in turn triggers endocytotic downregulation of channels. With this in mind, an attractive (though still speculative) explanation for the present studies is that 3D6 antibodies also trigger downregulation of α1C subunits via cross-linking of adjacent amyloid peptides which our co-immunoprecipitation experiments (figure 6a,b) suggest are tightly coupled.
6. The involvement of reactive oxygen species in hypoxic current augmentation
A growing body of evidence suggests that hypoxia can, seemingly paradoxically, cause an increase in the generation of reactive oxygen species (ROS; e.g. Duranteau et al. 1998; Chandel & Schumacker 2000; Waypa & Schumacker 2005). The ROS appear to be derived specifically from mitochondria. To examine whether ROS were involved in hypoxia-induced increases of Ca2+ channel currents, we examined the effects of antioxidants. Results are exemplified by the action of ascorbic acid (figure 7a), which fully prevented the augmentation of currents by hypoxia, while leaving control (normoxic) currents unaffected (not shown). Furthermore, this inhibitory effect of ascorbic acid was mimicked by co-culturing cells under hypoxic conditions in the additional presence of rotenone, an inhibitor of complex I of the mitochondrial electron transport chain. Thus, again, hypoxic augmentation of currents was fully blocked (figure 7b). These findings are in accordance with the suggestion of Schumacker and colleagues (e.g. Chandel & Schumacker 2000; Waypa & Schumacker 2005) that hypoxia increases ROS production from mitochondria.
Figure 7.

Involvement of mitochondrial ROS in hypoxic augmentation of Ca2+ channels. (a) Mean (+s.e.m.) current density–voltage relationships obtained in HEK-293 cells stably expressing L-type Ca2+ channel α1C subunits. Cells were cultured under normoxic conditions (open circles), hypoxic conditions (solid circles) or hypoxic conditions in the additional presence of 200 μM ascorbic acid (asc., squares). Data were obtained between 7 and 12 cells for each experimental condition. (b) Mean (+s.e.m.) current density–voltage relationships obtained in HEK-293 cells stably expressing L-type Ca2+ channel α1C subunits. Cells were cultured under normoxic conditions (open circles), hypoxic conditions (solid circles) or hypoxic conditions in the additional presence of 1 μM rotenone (squares). Data were obtained between 8 and 30 cells for each experimental condition.
7. Conclusions
The data presented herein provide a plausible cellular basis for the known association of prolonged hypoxia and dementia, as documented in several clinical studies (Incalzi et al. 1993; Tatemichi et al. 1994; Kokmen et al. 1996; Moroney et al. 1996). In PC12 cells, we noted two distinct effects of hypoxia—the induction of a Cd2+-resistant Ca2+ influx pathway which was tightly coupled to exocytosis, and the selective upregulation of L-type Ca2+ channels. Both effects involve amyloid β peptide formation. Indeed, where tested, the effects of hypoxia require amyloid formation, since inhibitors of either β- or γ-secretases (the enzymes required for cleavage of amyloid peptide from its precursor; Mattson 1997; Vassar & Citron 2000; LaFerla 2002) prevented effects of hypoxia.
The induction of an amyloid-associated influx pathway coupled to exocytosis has not been explored further in detail, but is reminiscent of amyloid-mediated ion channel activity previously reported (Arispe et al. 1993, 1996; Pollard et al. 1993). These previous studies have suggested that small aggregates of amyloid peptides can form membrane-spanning, Ca2+ permeable ion channels, thereby contributing to the dysregulation of Ca2+ homeostasis believed to be involved in neuronal cell death in Alzheimer's disease (LaFerla 2002). Further studies are require to explore this possibility but it is important to note that hypoxic induction of this influx pathway is distinct from the effect of hypoxia to upregulate native L-type Ca2+ channels. The distinction is emphasized by our findings that the influx pathway coupled to secretion was unaffected by inhibitors of the transcriptional regulator NFκ-B, whereas the enhancement of L-type Ca2+ channels was not (Green & Peers 2002).
The most striking effect of hypoxia was the enhancement of L-type Ca2+ channels, a phenomenon which was observed not only in PC12 cells, but also in central neurons and a recombinant expression system. This in itself suggests the effect is not tissue specific and so is likely to be a widespread effect. Most importantly, in all cell types this effect of hypoxia required amyloid formation. It is this requirement which convinces us that Ca2+ channel upregulation provides a cellular basis for the clinical association of hypoxia with dementia (see above). L-type Ca2+ channels serve a variety of important functions (Lipscombe et al. 2000; Striessnig et al. 2004; Wang et al. 2004), and so their selective targeting by amyloid peptides is likely to have profound (and deleterious) effects on cell activity and survival.
The fact that we could reproduce the effects of hypoxia on L-type Ca2+ channels in a recombinant expression system allowed us to draw immediate conclusions. First, channel upregulation was unlikely to occur via increased channel gene transcription, since the gene was part of a foreign plasmid incorporated into these cells which contained no known hypoxia-sensitive regions. Second, channel upregulation did not require the presence of auxiliary subunits. Normally L-type (and other) Ca2+ channels are protein complexes, with auxiliary subunits regulating normal channel activity (Dolphin 1995; Yamaguchi et al. 1998). Our recombinant expression system lacked auxiliary subunits, yet amyloid mediated hypoxic enhancement of currents was striking.
Looking for post-transcriptional mechanisms, we explored the possibility that amyloid peptides might act as chaperone molecules, altering the trafficking of Ca2+ channels. Evidence to support this idea came from a striking increase in the co-localization of the two proteins, as determined by double-labelling immunocytochemistry (Scragg et al. 2005). Furthermore, the two proteins co-immunoprecipitated (figure 6), suggesting they physically interact (although how direct this is, we cannot currently determine). We propose that this association is also present at the plasma membrane, since exposure of hypoxic (or amyloid-treated) cells to the 3D6 antibody raised against amyloid peptide suppressed any enhancement of current amplitude.
Importantly, we also provided evidence that the effects of hypoxia require formation of ROS. This is a contentious issue, since some groups believe ROS to increase during hypoxia (Waypa & Schumacker 2005), while others believe ROS levels to decrease in hypoxia (Michelakis et al. 2004), even when studied in the same tissue. The latter opinion is the more instinctive, since ROS are derived from O2, yet compelling evidence is accumulating for the former. Our findings are consistent with the idea that increased production of ROS is required for the observed effects of hypoxia on Ca2+ channels. Thus, not only do antioxidants prevent any effects of hypoxia, but exposure of cells to H2O2 mimics the effects of hypoxia (Green et al. 2002). Furthermore, hypoxic ROS generation appears to be from mitochondria (figure 7), in accordance with other studies (Waypa & Schumacker 2005).
A key fundamental question is how hypoxia triggers increased amyloidogenic processing (i.e. the formation of amyloid peptides from their precursor protein, APP). We are currently exploring the alternative processing pathways of APP, and our recent findings suggest that increased amyloid formation might arise due to increased expression of presenilin-1, the major component of γ-secretase, essential for amyloid formation (Smith et al. 2004). Clearly there is much further work to be done, but our data to date provide a plausible cellular basis to account for the known increased incidence of Alzheimer's disease in patients who have suffered prolonged hypoxic episodes.
Acknowledgments
This work was supported by the Medical Research Council, The Wellcome Trust and the British Heart Foundation and the Heart and Stroke Foundation of Ontario.
Footnotes
One contribution of 18 to a Theme Issue ‘Reactive oxygen species in health and disease’.
References
- Arispe N, Rojas E, Pollard H.B. Alzheimer-disease amyloid beta-protein forms calcium channels in bilayer-membranes—blockade by tromethamine and aluminum. Proc. Natl Acad. Sci. USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, Pollard H.B, Rojas E. Zn2+ interaction with Alzheimer amyloid-beta protein calcium channels. Proc. Natl Acad. Sci. USA. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. 10.1073/pnas.93.4.1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N.S, Schumacker P.T. Cellular oxygen sensing by mitochondria: old questions, new insight. J. Appl. Physiol. 2000;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. 10.1063/1.1303764 [DOI] [PubMed] [Google Scholar]
- Colebrooke R.L, Smith I.F, Kemp P.J, Peers C. Chronic hypoxia remodels voltage-gated Ca2+ entry in a human airway chemoreceptor cell line. Neurosci. Lett. 2002;318:69–72. doi: 10.1016/s0304-3940(01)02479-x. 10.1016/S0304-3940(01)02479-X [DOI] [PubMed] [Google Scholar]
- Dolphin A.C. The G. L. Brown Prize Lecture. Voltage-dependent calcium channels and their modulation by neurotransmitters and G proteins. Exp. Physiol. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- Duranteau J, Chandel N.S, Kulisz A, Shao Z, Schumacker P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998;273:11 619–11 624. doi: 10.1074/jbc.273.19.11619. 10.1074/jbc.273.19.11619 [DOI] [PubMed] [Google Scholar]
- Finnegan J.M, Pihel K, Cahill P.S, Huang L, Zerby S.E, Ewing A.G, Kennedy R.T, Wightman R.M. Vesicular quantal size measured by amperometry at chromaffin, mast, pheochromocytoma, and pancreatic beta-cells. J. Neurochem. 1996;66:1914–1923. doi: 10.1046/j.1471-4159.1996.66051914.x. [DOI] [PubMed] [Google Scholar]
- Green K.N, Peers C. Amyloid beta peptides mediate hypoxic augmentation of Ca2+ channels. J. Neurochem. 2001;77:953–956. doi: 10.1046/j.1471-4159.2001.00338.x. 10.1046/j.1471-4159.2001.00338.x [DOI] [PubMed] [Google Scholar]
- Green K.N, Peers C. Divergent pathways account for two distinct effects of amyloid β peptides on exocytosis and Ca2+ currents: involvement of ROS and NFκB. J. Neurochem. 2002;81:1043–1051. doi: 10.1046/j.1471-4159.2002.00907.x. 10.1046/j.1471-4159.2002.00907.x [DOI] [PubMed] [Google Scholar]
- Green K.N, Boyle J.P, Peers C. Hypoxia potentiates exocytosis and Ca2+ channels in PC12 cells via increased amyloid β peptide formation and ROS generation. J. Physiol. 2002;541:1013–1023. doi: 10.1113/jphysiol.2002.017582. 10.1113/jphysiol.2002.017582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incalzi R.A, Gemma A, Marra C, Muzzolon R, Capparella O, Carbonin P. Chronic obstructive pulmonary disease. An original model of cognitive decline. Am. Rev. Respir. Dis. 1993;148:418–424. doi: 10.1164/ajrccm/148.2.418. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, et al. Amyloid precursor protein processing and AB42 deposition in a transgenic mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. 10.1073/pnas.94.4.1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure K, Kato H. Altered gene expression in cerebral ischemia. Stroke. 1993;24:2121–2127. doi: 10.1161/01.str.24.12.2121. [DOI] [PubMed] [Google Scholar]
- Koistinaho J, Pyykonen I, Keinanen R, Hokfelt T. Expression of beta-amyloid precursor protein mRNAs following transient focal ischaemia. Neuroreport. 1996;7:2727–2731. doi: 10.1097/00001756-199611040-00064. [DOI] [PubMed] [Google Scholar]
- Kokmen E, Whisnant J.P, O'Fallon W.M, Chu C.P, Beard C.M. Dementia after ischemic stroke: a population-based study in Rochester, Minnesota (1960–1984) Neurology. 1996;46:154–159. doi: 10.1212/wnl.46.1.154. [DOI] [PubMed] [Google Scholar]
- LaFerla F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. 10.1038/nrn960 [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Helton T.D, Xu W. L-type calcium channels: the low down. J. Neurophysiol. 2000;92:2633–2641. doi: 10.1152/jn.00486.2004. 10.1152/jn.00486.2004 [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner B.A. B-amyloid neurotoxicity requires fibril formation and is inhibited by Congo Red. Proc. Natl Acad. Sci. USA. 1994;91:12 243–12 247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M.P. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Michelakis E.D, Thebaud B, Weir E.K, Archer S.L. Hypoxic pulmonary vasoconstriction: redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J. Mol. Cell. Cardiol. 2004;37:1119–1136. doi: 10.1016/j.yjmcc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Moroney J.T, Bagiella E, Desmond D.W, Paik M.C, Stern Y, Tatemichi T.K. Risk factors for incident dementia after stroke. Role of hypoxic and ischemic disorders. Stroke. 1996;27:1283–1289. doi: 10.1161/01.str.27.8.1283. [DOI] [PubMed] [Google Scholar]
- Peers C, Kemp P.J. Acute oxygen sensing: diverse but convergent mechanisms in airway and arterial chemoreceptors. Respir. Res. 2001;2:145–149. doi: 10.1186/rr51. 10.1186/rr51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C, Johnston I, Lang B, Wray D. Cross-linking of presynaptic calcium channels: a mechanism of action for Lambert–Eaton myasthenic syndrome antibodies at the mouse neuromuscular junction. Neurosci. Lett. 1993;153:45–48. doi: 10.1016/0304-3940(93)90073-t. 10.1016/0304-3940(93)90073-T [DOI] [PubMed] [Google Scholar]
- Plant L.D, Kemp P.J, Peers C, Henderson Z, Pearson H.A. Hypoxic depolarization of central neurones by specific inhibition of TASK-1. Stroke. 2002;33:2324–2328. doi: 10.1161/01.str.0000027440.68031.b0. 10.1161/01.STR.0000027440.68031.B0 [DOI] [PubMed] [Google Scholar]
- Pollard H.B, Rojas E, Arispe N. A new hypothesis for the mechanism of amyloid toxicity, based on the calcium-channel activity of amyloid-beta protein (AβP) in phospholipid-bilayer membranes. Ann. NY Acad. Sci. 1993;695:165–168. doi: 10.1111/j.1749-6632.1993.tb23046.x. [DOI] [PubMed] [Google Scholar]
- Rhee S.K, Quist A.P, Lal R. Amyloid B protein (1–42) forms calcium permeable, Zn2+-sensitive channel. J. Biol. Chem. 1998;273:13 379–13 382. doi: 10.1074/jbc.273.22.13379. 10.1074/jbc.273.22.13379 [DOI] [PubMed] [Google Scholar]
- Scragg J.L, Fearon I.M, Boyle J.P, Ball S.G, Varadi G, Peers C. Alzheimer's amyloid peptides mediate hypoxic up-regulation of L-type Ca2+ channels. FASEB J. 2005;19:150–152. doi: 10.1096/fj.04-2659fje. [DOI] [PubMed] [Google Scholar]
- Smith I.F, Boyle J.P, Green K.N, Pearson H.A, Peers C. Hypoxic remodeling of Ca2+ mobilization in type I cortical astrocytes: involvement of ROS and pro-amyloidogenic APP processing. J. Neurochem. 2004;88:869–877. doi: 10.1046/j.1471-4159.2003.02212.x. [DOI] [PubMed] [Google Scholar]
- Striessnig J, et al. L-type Ca2+ channels in Ca2+ channelopathies. Biochem. Biophys. Res. Commun. 2004;322:1341–1346. doi: 10.1016/j.bbrc.2004.08.039. 10.1016/j.bbrc.2004.08.039 [DOI] [PubMed] [Google Scholar]
- Tatemichi T.K, Paik M, Bagiella E, Desmond D.W, Stern Y, Sano M, Hauser W.A, Mayeux R. Risk of dementia after stroke in a hospitalized cohort: results of a longitudinal study. Neurology. 1994;44:1885–1891. doi: 10.1212/wnl.44.10.1885. [DOI] [PubMed] [Google Scholar]
- Taylor S.C, Batten T.F, Peers C. Hypoxic enhancement of quantal catecholamine secretion. Evidence for the involvement of amyloid beta-peptides. J. Biol. Chem. 1999;274:31 217–31 222. doi: 10.1074/jbc.274.44.31217. 10.1074/jbc.274.44.31217 [DOI] [PubMed] [Google Scholar]
- Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. 10.1016/S0896-6273(00)00051-9 [DOI] [PubMed] [Google Scholar]
- Vincent A, Lang B, Newsom-Davis J. Autoimmunity to the voltage-gated calcium channel underlies the Lambert–Eaton myasthenic syndrome, a paraneoplastic disorder. Trends Neurosci. 1989;12:496–502. doi: 10.1016/0166-2236(89)90109-4. 10.1016/0166-2236(89)90109-4 [DOI] [PubMed] [Google Scholar]
- Wang M.C, Dolphin A, Kitmitto A. L-type voltage-gated calcium channels: understanding function through structure. FEBS Lett. 2004;564:245–250. doi: 10.1016/S0014-5793(04)00253-4. 10.1016/S0014-5793(04)00253-4 [DOI] [PubMed] [Google Scholar]
- Waypa G.B, Schumacker P.T. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J. Appl. Physiol. 2005;98:404–414. doi: 10.1152/japplphysiol.00722.2004. 10.1152/japplphysiol.00722.2004 [DOI] [PubMed] [Google Scholar]
- Wightman R.M, Jankowski J.A, Kennedy R.T, Kawagoe K.T, Schroeder T.J, Leszczyszyn D.J, Near J.A, Diliberto E.J, Jr, Viveros O.H. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc. Natl Acad. Sci. USA. 1991;88:10 754–10 758. doi: 10.1073/pnas.88.23.10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt C.N, Wright C, Bee D, Peers C. O2− sensitive K+ currents in carotid-body chemoreceptor cells from normoxic and chronically hypoxic rats and their roles in hypoxic chemotransduction. Proc. Natl Acad. Sci. USA. 1995;92:295–299. doi: 10.1073/pnas.92.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hara M, Strobeck M, Fukasawa K, Schwartz A, Varadi G. Multiple modulation pathways of calcium channel activity by a beta subunit. J. Biol. Chem. 1998;273:19 348–19 356. doi: 10.1074/jbc.273.30.19348. 10.1074/jbc.273.30.19348 [DOI] [PubMed] [Google Scholar]