

Abstract
NADPH oxidase of phagocytic cells transfers a single electron from intracellular NADPH to extracellular O2, producing superoxide , the precursor to several other reactive oxygen species. The finding that a genetic defect of the enzyme causes chronic granulomatous disease (CGD), characterized by recurrent severe bacterial infections, linked generation to destruction of potentially pathogenic micro-organisms. In this review, we focus on the consequences of the electrogenic functioning of NADPH oxidase. We show that enzyme activity depends on the possibilities for compensating charge movements. In resting neutrophils K+ conductance dominates, but upon activation the plasma membrane rapidly depolarizes beyond the opening threshold of voltage-gated H+ channels and H+ efflux becomes the major charge compensating factor. K+ release is likely to contribute to the killing of certain bacteria but complete elimination only occurs if production can proceed at full capacity. Finally, the reversed membrane potential of activated neutrophils inhibits Ca2+ entry, thereby preventing overloading the cells with Ca2+. Absence of this limiting mechanism in CGD cells may contribute to the pathogenesis of the disease.
Keywords: neutrophilic granulocytes, NADPH oxidase, bacterial killing, intracellular Ca2+ homeostasis, o2− production, chronic granulomatous disease
1. Introduction
The NADPH oxidase of phagocytic cells (in the new terminology NOX2) is a complex enzyme, the subunits of which are distributed between different compartments of the resting cells. In neutrophilic granulocytes, cytochrome b558 (consisting of gp91phox and p22phox) resides in the plasma membrane and in the membrane of the secretory vesicles, whereas p47phox, p67phox and Rac are cytosolic proteins. In human neutrophils, Rac2 is involved in the oxidase, but in other cell types Rac1 is the participating small GTPase. Upon activation of the phagocyte, p47phox, p67phox and Rac2 (or Rac1) translocate to the membrane where the functioning oxidase complex is formed. The site of oxidase activation depends on the nature of the stimulus: soluble stimulants induce activation of the enzyme in the plasma membrane whereas during phagocytosis this occurs in the membrane of the forming phagosome.
The active oxidase complex carries out the transfer of one electron from cytosolic NADPH to molecular oxygen, producing thereby superoxide anion in the extracellular or in the intraphagosomal space and H+ in the cytosol. The electrogenic functioning of the oxidase was suggested by Henderson et al. (1987) and later clearly demonstrated by recording of electron currents in activated granulocytes (Schrenzel et al. 1998). Detailed analysis of the current–voltage relationship revealed that the oxidase is able to transfer electrons against a steep potential gradient: electron current ceases only when the transmembrane potential reaches +200 mV (DeCoursey et al. 2003; Petheo & Demaurex 2005).
On the basis of the amplitude of the recorded electron current and the capacitance of the plasma membrane, Thomas DeCoursey (2003) calculated that activated neutrophils depolarize with a speed of 1.1 V s−1. Starting from the resting membrane potential of −60 mV, the critical value of +200 mV would thus be arrived at within 250 ms when—in the absence of any compensating ion movement— production would cease. In eosinophils, the duration of generation would be about 10 times shorter, limited to approximately 25 ms. Thus, sustained function of the NADPH oxidase and long-lasting production depend on the possibility and intensity of compensating ion movements. Sustained activity of the NADPH oxidase seems to be essential for optimal destruction of invading micro-organisms, as genetic defect of any oxidase subunit results in impaired production and development of serious infectious states in chronic granulomatous disease (CGD) patients. We suggested in an earlier review (Geiszt et al. 2001) that electrophysiological alterations may contribute to the pathogenesis of CGD.
2. Possibilities of charge compensation in phagocytes
H+ ions were proposed as charge compensation for -production very early (Henderson et al. 1987) and since then several groups have described and analysed in detail the electrogenic H+ transporting pathways of phagocytic cells (for review see DeCoursey 2003). Although the molecular identity of the H+ channels is still a topic of intensive debate (DeCoursey et al. 2002; Henderson & Meech 2002; Maturana et al. 2002; Touret & Grinstein 2002; Petheo et al. 2003), all the authors agree on the voltage dependency of the process. The threshold of H+ efflux is modified by several factors: activators such as arachidonic acid or phorbol miristate acetate (PMA)-treatment of the cell or an increase of the transmembrane pH gradient shift it to more negative voltages, whereas a shift to more positive voltages occurs in the presence of inhibitors such as Zn2+ or Cd2+. In any case, the threshold lies by tens of millivolts to the positive side of the prevailing resting membrane potential, thus depolarization of the cell membrane is required for opening of the voltage-gated proton channels (DeCoursey & Cherny 1993; Demaurex et al. 1993; Kapus et al. 1993). Compensation of electron transfer by H+ efflux prevents cytosolic acidification and at the same time provides protons for intraphagosomal dismutation of superoxide (figure 1).
Figure 1.
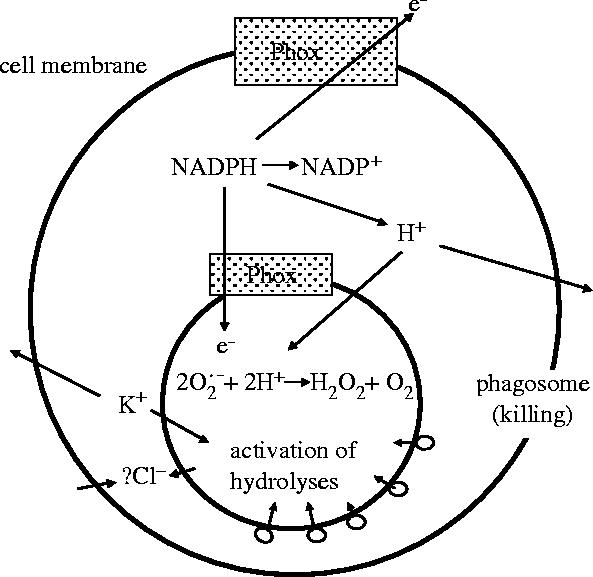
Possible mechanisms of charge compensation for electron transfer through the phagocytic NADPH oxidase (Phox).
Recently, attention has been directed to K+ as potential charge compensating ion and some experimental findings do support this suggestion. (i) The resting membrane potential of granulocytes is around −60 mV (Jankowski & Grinstein 1999; DeCoursey 2003), only approximately 10 mV more positive than the equilibrium potential for K+. The proximity of these two values indicates that under resting conditions, the dominating conductance of the plasma membrane is for K+. (ii) In the intraphagosomal pH, an initial alkalinization was observed, followed by later acidification, indicating a delay in H+ accumulation in the phagosome (Segal et al. 1981). The advantage of K+ accumulation in the phagosome could be the activation of cationic proteases by promoting their dissociation from the anionic proteoglycan matrix in which form they are stored in the neutrophilic granules (Reeves et al. 2002). In later studies, this ingenious suggestion was turned into an exclusive theory according to which in the presence of granule proteins and its derivatives do not contribute at all to the destruction of ingested micro-organisms (Reeves et al. 2003) and the entire killing activity of neutrophils depends only on K+ movements (Ahluwalia et al. 2004). In this view, the role of NADPH oxidase is restricted to providing the driving force for K+ movements.
A further possibility for charge compensation would be influx of anions into the cytoplasm (figure 1). The major mobile anion present in the extracellular fluids of the human body is Cl−. However, total Cl− content of phagosomes would only allow the compensation of less than 4% of electron transfer in activated neutrophils (DeCoursey 2004).
In view of these controversies on the species and role of compensating ion movements accompanying electron transfer via the NADPH oxidase, we decided to carry out a detailed quantitative analysis. The altered parameter was the rate of -generation that we decreased by increasing concentrations of the inhibitor diphenylene-iodonium (DPI). Changes in the plasma membrane potential, K+ release (on the basis of 86Rb efflux) and bacterial survival were followed in parallell and data were analysed in relation to each other (Rada et al. 2004).
3. Relation of -production to membrane depolarization and K+ release
Initiation of production results in a rapid change of the plasma membrane potential of neutrophilic granulocytes from the resting value of −60 mV up to +58 mV (Jankowski & Grinstein 1999). Identical stimulation does not induce any change in the membrane potential of CGD neutrophils, indicating that the depolarization is the result of electron transfer via the assembled oxidase complex and not of other signalling events (Seligmann & Gallin 1980; Geiszt et al. 1997).
Comparing the extent of membrane depolarization to the rate of production revealed a highly nonlinear relationship (figure 2a). Already 50% of the maximal depolarization was achieved at the lowest measurable rate (approx. 2–3% of the maximal rate) of generation. On the other hand, almost maximal depolarization occurred when the rate of production was around 20–25% of the maximal value. A remarkably similar relationship was observed when cells were stimulated by the chemoattractant formyl-methionyl-leucyl-phenylalanine (fMLP) or by the PKC activator PMA (figure 2a), although the absolute values of generation differed by a factor of four.
Figure 2.

Dependence of membrane depolarization (a) and K+(86Rb) release (b) on the rate of production. This figure was originally published by Rada, B. K., Geiszt, M., Káldi, K., Timár, Cs., & Ligeti, E. 2004 Dual role of phagocytic NADPH oxidase in bacterial killing. Blood 104, 2947–2953. © the American Society of Hematology.
Similarly to other cells, depolarization induces K+ release also from granulocytes (Reeves et al. 2002). We measured K+ release in the presence of various concentrations of DPI. Relating the extent of K+ efflux to the rate of production (figure 2b) revealed a similar relationship as observed for the membrane potential change (figure 2a). Forty percent of the total K+ release occurred already at the lowest measurable rate (2% of the maximum) of production and a fourfold increase (from 25 to 100%) of generation was accompanied only by a rise of K+ efflux from 70 to 100%.
According to previous calculations, K+ efflux accounts for 6% of the total charge movement occurring in stimulated granulocytes (Reeves et al. 2002; DeCoursey 2004). Taking this value in consideration, we calculated for each experimental point of figure 2b the fraction of electron flow compensated by K+ efflux (figure 3a). At the lowest activity of the oxidase, approximately half of the electron transfer is compensated by K+, whereas this proportion rapidly decreases as the rate of production increases. This calculation is in good agreement with earlier measurements. In patch clamp experiments, carried out in activated eosinophils, the measured membrane potential showed linear relationship with the transmembrane pH gradient altered in a broad range by variations of the pH of the pipette and the bath solution (Bánfi et al. 1999; figure 3b). The correspondence of the membrane potential to the H+ equilibrium potential indicates that—in contrast to the resting state—in the fully activated state of the granulocytes, H+ conductance dominates over the conductance of any other ion.
Figure 3.

Dependence of the proportion of charge compensation occurring by K+ release on the rate of production (a) and correlation between membrane potential and transmembrane pH difference in activated eosinophilic granulocytes (b). In part b measurements were carried out either in the absence (open triangle) or in the presence (open square) of K+. Part (b) reproduced from The Journal of Experimental Medicine, 1999, 190, 183–194, by copyright permission of The Rockefeller University Press.
Finally, our experimental data allowed us to relate K+ release to membrane potential. As shown in figure 4, in the range where our techniques allowed us to carry out measurements, a linear relationship was obtained between these two parameters. Thus, in our experiments there was no indication of any change in the K+ conductance of the plasma membrane.
Figure 4.
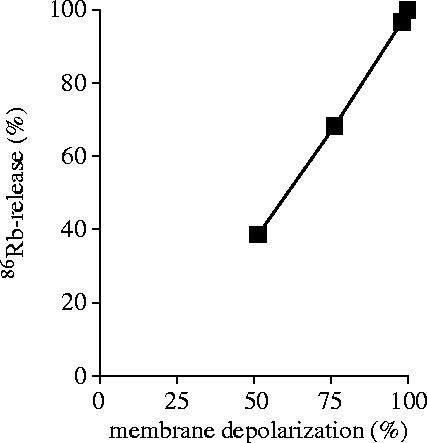
Relation of membrane depolarization and K+ release from PMA-activated neutrophils.
4. Relation of bacterial survival to production, membrane potential and K+ release
In order to investigate the functional role of the above parameters in the killing process, we carried out parallel titration with DPI of production induced by phagocytosed bacteria and of their survival. We obtained diverging results depending on whether Escherichia coli or Staphylococcus aureus were applied (figure 5). In case of E. coli, the killing process did not seem to depend on oxidase activity, as the survival rate increased only from 15 to 24% while production decreased from 100 to 2%. This result is in agreement with previous observations ascribing to non-oxidative processes the major role in killing of E. coli. In contrast, killing of S. aureus is strongly dependent on oxidative processes. Relating bacterial survival to the rate of production, we obtained a multiphase relationship. Decreasing the rate of generation from 100 to 20% resulted in a gradual, over tenfold increase of bacterial survival (from 7 to 89%) indicating a significant impairment of the killing capacity. It should be recalled that under these conditions there is almost no change either in the membrane potential or in K+ release (figure 2). Decrease of production below 20% was accompanied by a steeper increase of bacterial survival that became almost hyperbolic in the very low range: a decrease of production by only 2% (from 5 to 3% maximal capacity) increased bacterial survival from 130 to 195% of the original bacterial count. Under these conditions both membrane depolarization and K+ release changed significantly (figure 2).
Figure 5.
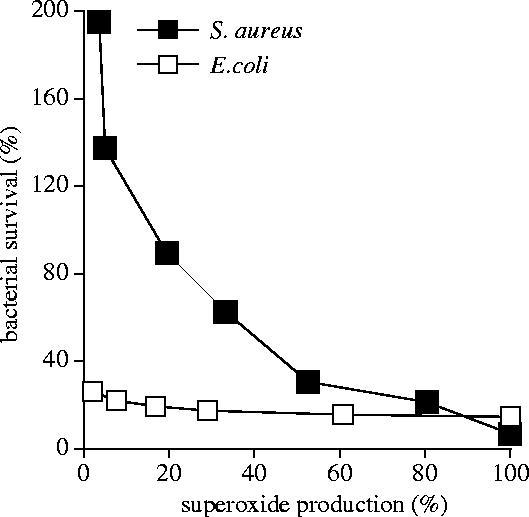
Relation of bacterial survival to the rate of generation. Note: bacteria divide during the time of the experiment, explaining why in the case of severely impaired killing capacity, there are more bacteria at the end than at the start. This figure was originally published by Rada, B. K., Geiszt, M., Káldi, K., Timár, Cs., & Ligeti, E. 2004 Dual role of phagocytic NADPH oxidase in bacterial killing. Blood 104, 2947–2953. © the American Society of Hematology.
To get more insight into the possible role of the electrophysiological changes in the killing process, we related our data on bacterial survival to changes of membrane potential (figure 6a) and K+ release (figure 6b) occurring under similar experimental conditions. Our quantitative analysis revealed for both electrophysiological parameters a clear linear relationship with bacterial survival (figure 6), suggesting that both membrane potential changes and K+ release may have a functional role in the killing process of neutrophilic granulocytes.
Figure 6.

Relation of bacterial survival to membrane depolarization (a) or K+ release (b).
Next, we tried to modulate membrane potential and production separately. Zn2+ was shown to inhibit electrogenic H+ pathway(s) in granulocytes (Kapus et al. 1992; DeCoursey et al. 2003) and increase both the depolarization and cytosolic acidification following stimulation of granulocytes. In presence of 10 μM ZnSO4, we detected clearly increased depolarization (figure 7a) and other authors demonstrated an increase of K+ release under similar conditions (Ahluwalia et al. 2004). The rate of generation was slightly lower in the presence of 10 μM ZnSO4 than in the presence of 5 nM DPI (figure 7a). However, bacterial survival showed different tendency in the presence of the two inhibitors: whereas 5 nM DPI increased bacterial survival approximately threefold (from 6.7 to 21%), in the presence of 10 μM ZnSO4 we observed slightly, but consistently lower survival of S. aureus than in the control, untreated samples (figure 7b). Thus, impaired production may be compensated by enhanced depolarization and K+ release.
Figure 7.

Comparison of the effect of 5 nM DPI and 10 μM ZnSO4 on membrane depolarization and generation (a) or bacterial survival (b).
5. Both production and K+ release contribute to destruction of micro-organisms
On the basis of our experimental and calculated data, we propose that both generation and K+ release initiated as charge compensation for electron transfer via the NADPH oxidase participate in the complex killing process of micro-organisms taking place in the isolated space of phagosomes. However, the significance of one or other process may vary depending on the exact conditions. In figure 8, we summarize the dominating events of different phases of bacterial killing.
Figure 8.
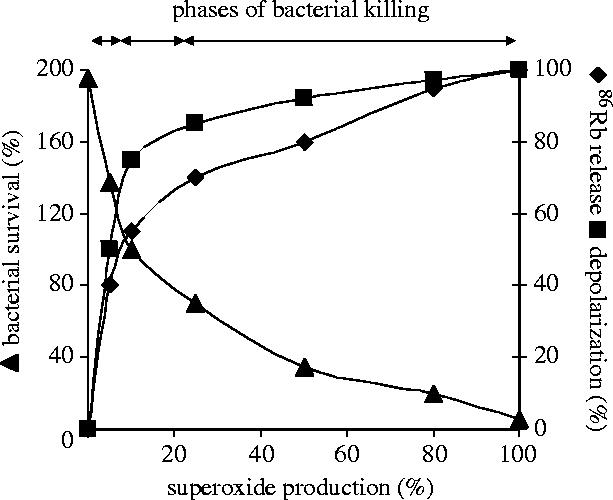
Summary of the changes of K+ release (filled diamond), membrane depolarization (filled square) and bacterial survival (filled triangle) in the function of the rate of generation.
In the first phase, at very low rate of production, K+ is the major charge compensating cation. Approximately half of the charge moved via the NADPH oxidase is compensated for by K+ movement and the majority of K+ efflux to the extracellular or intraphagosomal space occurs in this phase. However, the K+ conductance of the membrane does not allow full charge compensation to take place, hence the rapid depolarization observed when the rate of production is below 5–10% of its maximal capacity. Several observations suggest that the electrophysiological changes taking place in this phase contribute to the killing process: (i) bacterial survival decreases rapidly in this initial phase of NADPH oxidase activation. (ii) A linear relationship was revealed between bacterial survival and the extent of depolarization or K+ efflux. (iii) A decrease of production could be compensated by an increase in depolarization and K+ release.
In an intermediate phase, as the rate of production increases, depolarization of the plasma membrane attains the threshold of the voltage-gated H+ channels and H+ efflux gradually takes over as charge compensation for electron transfer. Parallel to this, contribution of K+ ions to charge compensation decreases.
Finally, approximately above 20% of maximal capacity of the NADPH oxidase, a stable membrane potential is achieved. Charge compensation occurs mainly by H+ efflux whereas K+ release is minimal. Nevertheless, there is a monotonous and significant decrease in bacterial survival proportional to the increase of the rate of production. We ascribe this increase in the killing capacity to the accumulation of anions, H+ ions and the derivatives formed thereof. Thus, while our experiments indicate that K+ release is likely to provide detectable contribution to destruction of certain micro-organisms, it should be noted that profound and almost complete elimination took place only when NADPH oxidase was allowed to work at full capacity.
In our experiments, the activity of the NADPH oxidase was limited by varying concentrations of the inhibitor DPI. At the moment, no endogenous inhibitor of the oxidase is known. However, substrate supply could limit the activity of this enzyme in vivo. The Km of the phagocytic oxidase for oxygen was reported to be in the range of 5–10 μM (Cross & Segal 2004). In human extracellular fluids, this concentration of oxygen corresponds to a partial pressure of 4–8 mmHg. There are thus places and conditions in the human body, such as poorly perfused regions or the interior of abscesses, when the partial pressure of oxygen is below the Km of the oxidase, allowing only low rate of production. According to recent patch clamp measurements, also the intracellular NADPH concentration may be limiting the enzyme activity (Petheo & Demaurex 2005). Under these conditions, the role of electrophysiological processes could become critical in the battle between pathogenic invaders and antimicrobial defense of the body.
Taken together, we propose that production of on the one hand, and the release of compensating cations on the other hand, should not be regarded as alternative mechanisms, but rather as complementary processes both contributing significantly to the final outcome.
6. Activity of NADPH oxidase alters Ca2+ homeostasis of granulocytes
Activation of the chemotactic, chemokine and purinergic receptors in the plasma membrane of neutrophilic granulocytes initiates a rapid Ca2+ signal. The first phase of this Ca2+ signal is due to Ca2+ release from intracellular stores, whereas the sustained phase can be ascribed to delayed entry of Ca2+ via store-dependent (or capacitative) Ca2+ channels (Scharff & Foder 1993). Opening of the capacitative Ca2+ channels is the consequence of depletion of intracellular Ca2+ stores and the ion flux through these channels follows the prevailing electrochemical gradient of Ca2+ across the plasma membrane (Geiszt et al. 1997).
As detailed above, the membrane potential of resting or activated neutrophils may differ by as much as 120 mV, thus the driving force for Ca2+ entry is significantly lower during active production than in rest. In fact, we have shown in an earlier study (Geiszt et al. 1997) that Ca2+ entry via the capacitative Ca2+ channels is completely blocked during production. This inhibition was independent of the type of stimulus used to activate the NADPH oxidase (fMLP or the PKC activator PMA), could not be mimicked by producing by the xanthine/xanthine oxidase system, but could be partially relieved by the K+ ionophore valinomycin that allowed increased charge compensation and thus decreased depolarization. In genetically modified neutrophilic cell-lines (differentiated PLB-985 cells with different levels of expression of the NADPH oxidase), clear correlation was found between the intensity of production, extent of plasma membrane depolarization and inhibition of Ca2+ entry through opened capacitative channels (Rada et al. 2003). Thus, we concluded that inhibition of Ca2+ influx during production is entirely due to a decrease in the driving force. Using several techniques, we observed almost complete inhibition of Ca2+ entry related to NADPH oxidase activity. In view of the extremely high concentration gradient for Ca2+ ions across the plasma membrane of resting neutrophils ([Ca2+]ic around 100 nM), one may be surprised that depolarization up to a maximum of +60 mV would be sufficient to block Ca2+ entry completely. However, recent investigations have shown that in the subplasmalemmal space of activated neutrophils, [Ca2+] may reach and possibly exceed 50 μM (Davies & Hallett 1998; Dewitt & Hallett 2002), substantiating our findings nicely.
In our view, if production is able to interfere with Ca2+ entry so dramatically, there should be significant differences in the Ca2+ homeostasis of healthy and CGD neutrophils. However, the only previous investigation, using a Ca2+-specific dye of high chelating capacity, did not find any difference in the fMLP-induced Ca2+ signal of CGD cells (Lew et al. 1984). Using a more sensitive dye, we obtained clear difference in the fMLP-induced Ca2+ signal of healthy and NADPH oxidase deficient PLB-985 cells (figure 9a). We did not see any difference in the ATP-induced Ca2+ signal (ATP does not activate the NADPH oxidase) or if fMLP-stimulation was carried out in Ca2+-free solutions, indicating that it was really Ca2+ entry that was impaired during production. The difference of the Ca2+ signal observed in genetically modified myeloid cells could be mimicked in healthy neutrophils by inhibition of the oxidase by DPI (figure 9b). Acceleration of Ca2+ influx and hyperactivation of CGD neutrophils has also been reported by other groups (Tintinger et al. 2001).
Figure 9.

Alteration of the Ca2+ signal in cells defective in generation. The fMLP-induced Ca2+ signal was compared in differentiated PLB-985 myeloid cells containing an intact NADPH oxidase or no functioning enzyme (marked CGD) (a) and in peripheral blood neutrophils inhibited by 5 μM DPI (fMLP+DPI) or investigated in the absence of inhibitor (fMLP only) (b). This figure was originally published by Rada, B. K., Geiszt, M., van Bruggen, R., Német, K., Roos, D., & Ligeti, E. 2003 Clin. Exp. Immunol. 132, 53–60. © the British Society of Immunology.
As discussed recently in an Editorial Review (Hallett 2003), inhibition of Ca2+ entry into activated neutrophils may represent a self-regulatory and self-protecting mechanism. Activation of plasma membrane receptors induces a Ca2+ signal that is necessary for diverse functions, such as efficient activation of the NADPH oxidase or exocytosis of the various granule populations (Scharff & Foder 1993). However, inititation of production leads very rapidly to significant depolarization, stopping further Ca2+ supply and protecting thereby the cell from an excess Ca2+ load. This mechanism resembles the coupled activation of voltage-gated Ca2+ channels and Ca2+-dependent K+ channels in other cell types. In excitable cells such as smooth muscle or chromaffin cells, depolarization opens voltage-gated Ca2+ channels, but Ca2+ entry via these channels initiates the opening of Ca2+-dependent K+ channels, the ensuing K+ efflux then leads to hyperpolarization and closing of the voltage-gated Ca2+ channels, limiting thereby the Ca2+ load during each activation event (Orio et al. 2002). Lack of the self-protecting mechanism in neutrophils expressing a defective NADPH oxidase could contribute to the pathogenesis of CGD. In neutrophils, an elevation of [Ca2+]ic was shown to inhibit locomotion (Laffalian & Hallett 1995), activate phospholipase A2 (Tintinger et al. 2001), induce the de novo synthesis of cytokines, such as the chemotactic agent IL-8 (Kuhns et al. 1998), and—contrarily to many other cell types— to retard apoptosis (Whyte et al. 1993). In fact, in CGD cells, both spontaneous and induced apoptosis were shown to be significantly slower than in healthy neutrophils (Kasahara et al. 1997). Thus, all these changes may result in accumulation and immobilization of granulocytes with prolonged lifetime at certain sites, and may contribute to the formation of granulomas typical for the CGD patients.
7. Concluding remarks
Data and experimental results summarized in this review were obtained in phagocytic cells, mostly in neutrophilic and eosinophilic granulocytes expressing the NADPH oxidase NOX2. As detailed in other reviews of this issue, several homologues of the phagocytic NADPH oxidase have been discovered in the recent years. They all carry out transmembrane electron transport, thus their function is electrogenic as well and depends on possibilities of charge compensation. The problems and ideas communicated in this review have relevance for the entire NOX family and will surely be investigated in various NOX-expressing cell types in the future.
Acknowledgements
Experimental work carried out in the authors' laboratory was financially supported by grants from the Hungarian Research Fund (OTKA T037755, Ts040865, Ts049851, M036310) and the Hungarian Ministry of Health (ETT 041/2003). The corresponding author also acknowledges the inspiring ambience of the conference supported by the Tschira Foundation.
Footnotes
One contribution of 18 to a Theme Issue ‘Reactive oxygen species in health and disease’.
References
- Ahluwalia J, Tinker A, Clapp L.H, Duchen M.R, Abramov A.Y, Pope S, Nobles M, Segal A.W. The large-conductance Ca2+-activated K+ channel is essential for innate immunity. Nature. 2004;427:853–858. doi: 10.1038/nature02356. 10.1038/nature02356 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bánfi B, Schrenzel J, Nüsse O, Lew D.P, Ligeti E, Krause K.H, Demaurex N. A novel H+ conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J. Exp. Med. 1999;190:183–194. doi: 10.1084/jem.190.2.183. 10.1084/jem.190.2.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross A.R, Segal A.W. The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies E.V, Hallett M.B. High micromolar Ca2+ beneath the plasma membrane in stimulated neutrophils. Biochem. Biophys. Res. Commun. 1998;248:679–683. doi: 10.1006/bbrc.1998.9031. 10.1006/bbrc.1998.9031 [DOI] [PubMed] [Google Scholar]
- DeCoursey T.E. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- DeCoursey T.E. During the respiratory burst do phagocytes need proton channels or potassium channels, or both? Science STKE. 2004:pe21. doi: 10.1126/stke.2332004pe21. 10.1126/stke.2332004pe21 [DOI] [PubMed] [Google Scholar]
- DeCoursey T.E, Cherny V.V. Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys. J. 1993;65:1590–1598. doi: 10.1016/S0006-3495(93)81198-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey T.R, Morgan D, Cherny V.V. The gp91phox component of NADPH oxidase is not a voltage-gated proton channel. J. Gen. Physiol. 2002;120:773–779. doi: 10.1085/jgp.20028704. 10.1085/jgp.20028704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey T.E, Morgan D, Cherny V.V. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 2003;422:531–534. doi: 10.1038/nature01523. 10.1038/nature01523 [DOI] [PubMed] [Google Scholar]
- Demaurex N, Grinstein S, Jaconi M, Schlegel W, Lew D.P, Krause K.H. Proton currents in human granulocytes: regulation by membrane potential and intracellular pH. J. Physiol. 1993;466:329–344. [PMC free article] [PubMed] [Google Scholar]
- Dewitt S, Hallett M.B. Cytosolic free Ca2+ changes and calpain activation are required for β2 integrin-accelerated phagocytosis by human neutrophils. J. Cell Biol. 2002;159:181–189. doi: 10.1083/jcb.200206089. 10.1083/jcb.200206089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiszt M, Kapus A, Nemet K, Farkas L, Ligeti E. Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J. Biol. Chem. 1997;272:26 471–26 478. doi: 10.1074/jbc.272.42.26471. 10.1074/jbc.272.42.26471 [DOI] [PubMed] [Google Scholar]
- Geiszt M, Kapus A, Ligeti E. Chronic granulomatous disease: more than the lack of superoxide? J. Leukoc. Biol. 2001;69:191–196. [PubMed] [Google Scholar]
- Hallett M.B. Holding back neutrophil aggression; the oxidase has potential. Clin. Exp. Immunol. 2003;132:181–184. doi: 10.1046/j.1365-2249.2003.02158.x. 10.1046/j.1365-2249.2003.02158.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson L.M, Meech R.W. Proton conduction through gp91phox. J. Gen. Physiol. 2002;120:759–765. doi: 10.1085/jgp.20028708. 10.1085/jgp.20028708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson L.M, Chappell J.B, Jones O.T. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem. J. 1987;246:325–329. doi: 10.1042/bj2460325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski A, Grinstein S. A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. J. Biol. Chem. 1999;274:26 098–26 104. doi: 10.1074/jbc.274.37.26098. 10.1074/jbc.274.37.26098 [DOI] [PubMed] [Google Scholar]
- Kapus A, Szászi K, Ligeti E. Phorbol 12-myristate 13-acetate activates an electrogenic H+-conducting pathway in the membrane of neutrophils. Biochem. J. 1992;281:697–701. doi: 10.1042/bj2810697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapus A, Romanek R, Qu A.Y, Rotstein O.D, Grinstein S. A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J. Gen. Physiol. 1993;102:729–760. doi: 10.1085/jgp.102.4.729. 10.1085/jgp.102.4.729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H, Miyawaki T, Taniguchi N. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood. 1997;89:1748–1753. [PubMed] [Google Scholar]
- Kuhns D.B, Young H.A, Gallin E.K, Gallin J.I. Ca2+-dependent production and release of IL-8 in human neutrophils. J. Immunol. 1998;161:4332–4339. [PubMed] [Google Scholar]
- Laffalian I, Hallett M.B. Does cytosolic free Ca2+ signal neutrophil chemotaxis? J. Cell Sci. 1995;108:3199–3205. doi: 10.1242/jcs.108.10.3199. [DOI] [PubMed] [Google Scholar]
- Lew P.D, Wollheim C, Seger R.A, Pozzan T. Cytosolic free calcium changes induced by chemotactic peptide in neutrophils from patients with chronic granulomatous disease. Blood. 1984;63:231–233. [PubMed] [Google Scholar]
- Maturana A, Krause K.H, Demaurex N. NOX family NADPH oxidases: do they have built-in proton channels? J. Gen. Physiol. 2002;120:781–786. doi: 10.1085/jgp.20028713. 10.1085/jgp.20028713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P, Rojas P, Ferreira G, Latorre R. New disguises for an old channel: MaxiK channel β-subunits. News Physiol. Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- Petheo G.L, Demaurex N. Voltage- and NADPH-dependence of electron currents generated by the phagocytic NADPH oxidase. Biochem. J. 2005 doi: 10.1042/BJ20041889. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petheo G.L, Maturana A, Spät A, Demaurex N. Interactions between electron and proton currents in excised patches from human eosinophils. J. Gen. Physiol. 2003;122:713–726. doi: 10.1085/jgp.200308891. 10.1085/jgp.200308891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada B.K, Geiszt M, van Bruggen R, Német K, Roos D, Ligeti E. Calcium signaling is altered in myeloid cells with a deficiency in NADPH oxidase activity. Clin. Exp. Immun. 2003;132:53–60. doi: 10.1046/j.1365-2249.2003.02138.x. 10.1046/j.1365-2249.2003.02138.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada B.K, Geiszt M, Káldi K, Timár Cs, Ligeti E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 2004;104:2947–2953. doi: 10.1182/blood-2004-03-1005. 10.1182/blood-2004-03-1005 [DOI] [PubMed] [Google Scholar]
- Reeves E.P, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. 10.1038/416291a [DOI] [PubMed] [Google Scholar]
- Reeves E.P, Nagl M, Godovac-Zimmermann J, Segal A.W. Reassessment of the microbicidal activity of reactive oxygen species and hypochlorous acid with reference to the phagocytic vacuole of the neutrophil granulocyte. J. Med. Microbiol. 2003;52:643–651. doi: 10.1099/jmm.0.05181-0. 10.1099/jmm.0.05181-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharff O, Foder B. Regulation of cytosolic calcium in blood cells. Physiol. Rev. 1993;73:547–582. doi: 10.1152/physrev.1993.73.3.547. [DOI] [PubMed] [Google Scholar]
- Schrenzel J, Serrander L, Banfi B, Nusse O, Fouyouzi R, Lew D.P, Demaurex N, Krause K.H. Electron currents generated by the human phagocyte NADPH oxidase. Nature. 1998;392:734–737. doi: 10.1038/33725. 10.1038/33725 [DOI] [PubMed] [Google Scholar]
- Segal A.W, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290:406–409. doi: 10.1038/290406a0. 10.1038/290406a0 [DOI] [PubMed] [Google Scholar]
- Seligmann B.E, Gallin J.I. Use of lipophilic probes of membrane potential to assess human neutrophil activation. Abnormality in chronic granulomatous disease. J. Clin. Invest. 1980;66:493–503. doi: 10.1172/JCI109880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintinger G.R, Theron A.J, Steel H.C, Anderson R. Accelerated calcium influx and hyperactivation of neutrophils in chronic granulomatous disease. Clin. Exp. Immunol. 2001;123:254–263. doi: 10.1046/j.1365-2249.2001.01447.x. 10.1046/j.1365-2249.2001.01447.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touret N, Grinstein S. Voltage-gated proton ‘channels’: a spectator's viewpoint. J. Gen. Physiol. 2002;120:767–771. doi: 10.1085/jgp.20028706. 10.1085/jgp.20028706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte M.K, Hardwick S.J, Meagher L.C, Savill J.S, Haslett C. Transient elevations of cytosolic free calcium retard subsequent apoptosis in neutrophils in vitro. J. Clin. Invest. 1993;92:446–455. doi: 10.1172/JCI116587. [DOI] [PMC free article] [PubMed] [Google Scholar]