

Abstract
The appropriate secretion of insulin from pancreatic β-cells is critically important to the maintenance of energy homeostasis. The β-cells must sense and respond suitably to postprandial increases of blood glucose, and perturbation of glucose-sensing in these cells can lead to hypoglycaemia or hyperglycaemias and ultimately diabetes. Here, we review β-cell glucose-sensing with a particular focus on the regulation of cellular excitability and exocytosis. We examine in turn: (i) the generation of metabolic signalling molecules; (ii) the regulation of β-cell membrane potential; and (iii) insulin granule dynamics and exocytosis. We further discuss the role of well known and putative candidate metabolic signals as regulators of insulin secretion.
Keywords: glucose, insulin secretion, metabolism, ion channels, membrane potential, exocytosis
1. Introduction
Beta-cells of the pancreatic islets of Langerhans act as glucose sensors, adjusting insulin output to the prevailing blood glucose level. Insulin is critically important for the promotion of glucose storage and the prevention of glycogen breakdown. Thus, the glucose-sensing mechanism of the β-cell is essential for maintenance of glucose homeostasis. This is exemplified by the role that the β-cell plays in the pathogenesis of type 2 diabetes mellitus, as a relative defect in the insulin response to glucose is at least as important as peripheral insulin resistance in the development of the disease (LeRoith 2002; Ashcroft & Rorsman 2004). Deterioration of β-cell function also contributes to the progression of type 2 diabetes (Kahn 2000). As well, several monogenic forms of diabetes are directly linked to defects in the glucose-sensing machinery of the β-cell (Porter & Barrett 2005) and type 2 diabetes has been linked to polymorphisms in the glucose signal transduction machinery (Ashcroft & Rorsman 2004). Thus, knowledge of the mechanisms central to β-cell glucose-sensing is critical toward understanding potential sites of dysfunction in type 2 diabetes, and potential targets for therapeutic intervention.
The glucose-sensing mechanism of pancreatic β-cells can be roughly divided into two components: (i) the proximal events of glucose entry and metabolism and (ii) the distal mechanism of insulin secretion, spanning from mitochondrial signal generation and initiation of electrical activity to the ultimate effectors of insulin granule exocytosis (figure 1). We believe this is appropriate since the metabolic signals and effectors controlling triggering, amplification, and indeed maintenance of secretion overlap. In the present review, we summarize in brief the proximal steps leading to generation of mitochondrial signals and review the effects of these signals on components of the distal glucose-sensing mechanism. Further, we present prospects for additional downstream effects of glucose by speculating on the actions of alternate candidate metabolic signals.
Figure 1.

Proximal and distal events in pancreatic β-cell glucose-sensing. Proximal events, including glucose entry, glycolysis and entry into mitochondrial metabolism, lead to the generation of mitochondrial signals. These signals regulate insulin secretion by controlling distal effectors including membrane excitability, Ca2+ signalling, insulin granule recruitment and exocytosis. This scheme differs somewhat from the popular concept of ‘triggering’ and ‘amplifying’ pathways, which overlap within the distal mechanism.
2. Proximal glucose-sensing and metabolic signal generation
(a) Glucose transport and glycolysis
There are three main characteristics of cytosolic glucose metabolism in the β-cell. First, glucose can equilibrate rapidly across the β-cell membrane due to the expression of the high capacity, low affinity glucose transporter-2 (Newgard & McGarry 1995). Second, after glucose has entered the β-cell, it is phosphorylated to glucose-6-phospate by the high KM glucokinase (GK, hexokinase IV), which constitutes the flux determining step for glycolysis (Iynedjian 1993; De Vos et al. 1995; Newgard & McGarry 1995; Matschinsky 1996) and is considered as the ‘glucose sensor’ in the pancreatic β-cell (Matschinsky 1996). The fact that mutations in the GK gene lead to impaired insulin secretion in maturity onset diabetes of the young type 2 patients further supports a role for GK as a glucose sensor (Porter & Barrett 2005). Third, once phosphorylated, glucose is metabolized by glycolysis to produce pyruvate, NADH and ATP.
The glycolytic product pyruvate is a substrate for the tricarboxylic acid (TCA) cycle in the mitochondria and is suggested to be an important modulator of insulin secretion (figure 2). Pyruvate, and its membrane permeable forms, fail to consistently stimulate insulin secretion (Sener et al. 1978; Mertz et al. 1996; Zawalich & Zawalich 1997) and inhibitors of pyruvate transport or the TCA cycle are not well correlated with glucose-stimulated insulin secretion (Dukes et al. 1994; Mertz et al. 1996). However, mitochondrial pyruvate metabolism is still likely important in glucose-sensing. Pancreatic β-cells express low levels of lactate dehydrogenase, the enzyme catalysing the conversion of pyruvate to lactate (Sekine et al. 1994; Schuit et al. 1997; Ishihara et al. 1999), and this prevents shunting of pyruvate away from the mitochondrial metabolic pathways (Zhao et al. 2001). Therefore, even though pyruvate provides a substrate to the TCA cycle for ATP production it is not the critical (or at least only) glycolytic factor that initiates insulin secretion. There must be other factor(s) derived from glycolysis, other than pyruvate, required for the generation of mitochondrial signals that lead to insulin secretion. One such candidate factor is NADH (Malaisse et al. 1979; Hedeskov et al. 1987; Pralong et al. 1990; Eto et al. 1999).
Figure 2.

Mitochondrial signal generation. NADH generated from pyruvate entry into the TCA cycle and NADH transported into the mitochondria by the malate–aspartate (Mal–Asp) and glycerol–phosphate (Gly–P) shuttles drive respiratory chain generation of a H+ gradient and hyperpolarization of the mitochondrial matrix. H+ gradient dissipation drives ATP production by ATP synthase. Matrix hyperpolarization promotes the entry of Ca2+ which further stimulates TCA cycle activity and export of ATP. The export of malate and citrate (also isocitrate, not shown) and subsequent conversion to pyruvate generates an increase in cytoplasmic NADPH. Also, exported citrate can be converted to acetyl-CoA and then malonyl-CoA which blocks the LC-CoA transporter carnitine palmitoyltransferase 1 (CPT1). This leads to an increase in cytoplasmic LC-CoA and potential downstream lipid-derived messengers such as diacylglycerol (DAG). Important signals for distal signalling are boxed in grey.
(b) Entry into mitochondrial metabolism
Both cytosolic pyruvate and NADH enter the mitochondria and are metabolized to produce ATP (figure 2). The glycolytic factor NADH enters the mitochondria via the glycerol–phosphate and the malate–aspartate shuttle systems (MacDonald 1982; Giroix et al. 1991; Sener & Malaisse 1992; Eto et al. 1999). These shuttles may play an important role in the regulation of insulin secretion (MacDonald 1990). In support of this, glycerol–phosphate shuttle and mitochondrial glycerol-phosphate dehydrogenase (mGPDH) activities are very high in β-cells relative to other mammalian cells (MacDonald 1990). Further support for the model has come from studies of mGPDH −/− mice (Eto et al. 1999). Neither absence of the glycerol–phosphate shuttle (in mGPDH −/− islets) nor suppression of the malate–aspartate shuttle alone (in wild-type islets) altered glucose-stimulated insulin secretion. However, blocking these two pathways together completely ablated the response to glucose, suggesting that both NADH shuttle systems are important for glucose-sensing.
(c) Generation of ATP
In addition to cytosolic NADH, NADH is produced from pyruvate in the mitochondria by the TCA cycle (figure 2). Both cytosolic and mitochondrial sources of NADH stimulate the electron transport chain to pump H+ ions out of the mitochondrial matrix, hyperpolarizing the inner mitochondrial membrane (Duchen et al. 1993; Maechler et al. 1997; Kennedy et al. 1998; Eto et al. 1999). The generation of ATP by ATP synthase is coupled to dissipation of the H+ gradient. Also, hyperpolarization of the mitochondrial inner membrane can stimulate the mitochondrial membrane potential-dependent Ca2+-uniporter to increase mitochondrial Ca2+ (Litsky & Pfeiffer 1997), augmenting Ca2+-dependent dehydrogenase activity and further increasing the production of NADH and ATP from the TCA cycle (Hansford 1991). A rise in matrix Ca2+ may also promote dissociation of an ATP synthase inhibitor, providing a positive feedback mechanism on ATP production (Moreno-Sanchez 1985; Territo et al. 2001). In addition, the increase in mitochondrial Ca2+ stimulates ATP transport into the cytosol, increasing cytosolic ATP concentration and the ATP/ADP ratio (Moreno-Sanchez 1985).
Mitochondrial oxidative metabolism has been estimated to produce 98% of β-cell ATP (Erecinska et al. 1992). The exact role of cytosolic or mitochondrial ATP production in the regulation of insulin secretion is controversial. Some investigators have suggested that cytosolic ATP production takes precedence in the regulation of ATP-sensitive channels (Mertz et al. 1996), such as occurring in cardiac muscle (Weiss & Lamp 1989). Support for the important role of mitochondrial ATP production in β-cells is that α-ketoisocaproic acid, which is entirely metabolized in mitochondria by the TCA cycle, can inactivate ATP-sensitive K+ (KATP) channels (Ashcroft & Ashcroft 1990) and stimulate insulin secretion (Best 1997). However, α-ketoisocaproic acid may block KATP channels by a direct mechanism (Branstrom et al. 1998a) and it has been argued that ATP production alone is insufficient for the physiological regulation of insulin secretion and the generation of other mitochondrial signals plays an important role in glucose-sensing (MacDonald et al. 2005). Glutamate may be this mitochondrial factor (Maechler & Wollheim 1999; Maechler et al. 2000; Hoy et al. 2002) although its role in insulin secretion is not currently favoured (MacDonald et al. 2005). Rather, evidence supports an important role for long-chain acyl-CoAs and the mitochondrial export of reducing equivalents (in the form of NADPH) as important distal signals. The generation of these signals, which is summarized below, is the subject of a recent excellent review (MacDonald et al. 2005).
(d) Long chain acyl CoA
Prentki, Corkey and co-workers suggested LC-CoA as a potential modulator of insulin secretion (Corkey et al. 1989; Prentki et al. 1992). This model (Deeney et al. 2000b) holds that glucose is in part metabolized to citrate, which is transported out of the mitochondria and converted to malonyl CoA (figure 2). Malonyl CoA then inhibits carnitine palmitoyltransferase 1 (CPT1), a key regulatory enzyme of fatty acid oxidation. Inhibition of CPT1 leads to an increase in cytosolic LC-CoA, the elevation of which could potentiate insulin secretion by direct acylation of regulatory proteins. Alternatively, LC-CoAs may be converted to other bioactive metabolites, for example diacylglycerol (DAG), which activate downstream effectors such as PKC. Correlative experiments have provided support for this model (Corkey et al. 1989; Chen et al. 1994), although it is still controversial since there is no alteration of insulin secretion after the link between glucose and lipid metabolism has been directly perturbed (Antinozzi et al. 1998; Mulder et al. 2001).
(e) NADPH
NADPH is another potential mitochondrial signalling molecule derived from glucose metabolism. Metabolizable insulin secretagogues increase the NADPH-to-NADP ratio in rodent islets (Ashcroft & Christie 1979; Hedeskov et al. 1987) and inhibition of NADPH formation reduces glucose-stimulated insulin secretion (Ammon & Steinke 1972; MacDonald et al. 1974). Recently, Newgard's group (Lu et al. 2002) found, by application of 13C NMR analysis of newly developed INS-1 derived cell lines, that recycling of pyruvate across the mitochondrial inner membrane (figure 2) correlates well with glucose-responsiveness. There are several pathways for pyruvate recycling and NADPH production. One key pathway is the pyruvate–malate shuttle system. The first step of this pathway involves a TCA cycle anaplerotic step where pyruvate is converted to oxaloacetate (OAA) via the pyruvate carboxylase (PC) reaction. This anaplerotic step is a key step in β-cells since 40–50% of all pyruvate in mitochondria enters the PC reaction (MacDonald 1993). While OAA is part of the TCA cycle, it can be converted to malate by mitochondrial malate dehydrogenase. Malate can participate in numerous pathways including the malate–aspartate shuttle (shuttles NADH into mitochondria) and the pyruvate–malate shuttle (shuttles NADPH out of the mitochondria). In the latter, malate is transported out of the mitochondria via a malate-Pi antiporter and then converted to pyruvate via malic enzyme resulting in the generation of CO2 and NADPH. Pyruvate can then be transported back into the mitochondria via a pyruvate-H+ symporter, and the cycle continues. Additionally, NADPH reducing equivalents can also be exported from the mitochondria as citrate and isocitrate (MacDonald et al. 2005).
3. Distal sensing of metabolic signals: membrane excitability
Glucose depolarises the pancreatic β-cell membrane potential and initiates firing of action potentials (Dean & Matthews 1968, 1970a). It was recognized quite soon that membrane depolarization was dependent on metabolism of the sugar (Dean & Matthews 1970b). In the absence of stimulatory glucose (generally less than 5 mM), rodent β-cells are electrically silent, with a resting membrane potential of approximately −70 mV (Meissner & Schmelz 1974; Matthews & Sakamoto 1975) due to a high resting K+ conductance in these cells (Meissner et al. 1978). Reduction of the resting K+ conductance by stimulatory glucose leads to membrane depolarization and initiation of electrical activity characterized by slow wave depolarization with superimposed bursts of action potentials (Dean & Matthews 1970b; Pace & Price 1972; Meissner & Schmelz 1974; Matthews & Sakamoto 1975). As described below and outlined in figure 3, ATP-sensitive K+ (KATP) channels set the β-cell membrane potential and closure of these leads to depolarization. Membrane depolarization triggers action potential firing and opening of voltage-dependent Ca2+ channels (VDCCs), leading to Ca2+ influx which triggers exocytosis. Action potentials are terminated by the opening of voltage-dependent K+ channels which limit Ca2+ entry and thus insulin release.
Figure 3.
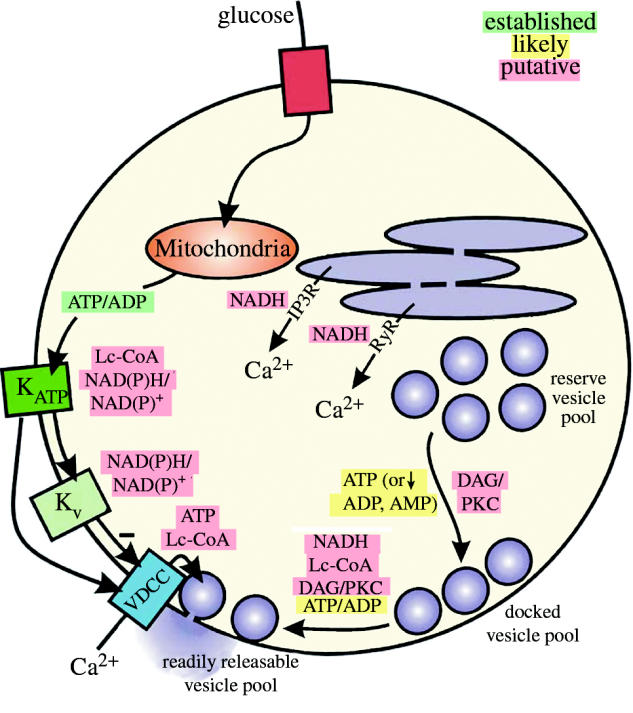
Stimulus–secretion coupling in the pancreatic β-cell. Glucose entry and mitochondrial metabolism increases the intracellular ATP-to-ADP ratio which leads to closure of KATP channels and membrane depolarization. This activates VDCCs, allowing influx of Ca2+ which triggers exocytosis of insulin granules. Kv channels also activate upon depolarization to mediate action potential repolarization, limiting Ca2+ entry and insulin secretion. Established and putative metabolic signalling molecules may regulate insulin secretion at a number of sites. These include the activity of ion channels, release of intracellular Ca2+ stores, mobilization and priming of secretory vesicles, and exocytosis. Here, we have shown the established (green), likely (yellow) and putative (red) regulatory interactions.
(a) ATP-sensitive K+ channels
As early as 1970, a role for ATP generation was implicated in the initiation of β-cell electrical activity based on the ability of 2,4-dinitrophenol (an uncoupler of oxidative phosphorylation) to prevent glucose-induced electrical responses in mouse islets (Dean & Matthews 1970b). Glucose was known to induce a reduction in rodent islet K+ permeability (Sehlin & Taljedal 1975; Henquin 1978). Ashcroft et al. (1984) and Cook & Hales (1984) demonstrated the existence of K+ channels in rat β-cells that were closed by glucose and ATP, respectively. Subsequently, in mouse β-cells these were shown to be the same channel (Rorsman & Trube 1985), closure of which precedes depolarization-induced Ca2+ influx (Arkhammar et al. 1987). ATP-sensitive K+ channels were first described in guinea pig cardiac muscle (Noma 1983) and are now known to be expressed in numerous tissues including smooth and skeletal muscle, neurons, peripheral axons and epithelial cells (Ashcroft & Ashcroft 1992). The β-cell KATP channel was the first cloned from insulinoma cells in 1995, along with its regulatory sulphonylurea receptor (SUR) subunit (Aguilar-Bryan et al. 1995; Inagaki et al. 1995; Sakura et al. 1995). There are currently two genes known for each of the KATP pore-forming subunits (Kir6.1 and 6.2) and the associated regulatory subunits (SUR1, 2), although diversity is further increased by alternative splicing of the SUR2 gene (Ashcroft & Gribble 1999). KATP channels are composed of four pore forming inward-rectifier K+ channel subunits (Kir6.2 in β-cells; two transmembrane domains) and four regulatory sulphonylurea receptor subunits (SUR1 in β-cells; total 17 transmembrane domains; Aguilar-Bryan & Bryan 1999; Ashcroft & Gribble 1999).
In the absence of glucose KATP channels have a high open probability and mediate an outward K+ flux that holds the membrane potential near the equilibrium potential for K+ (approximately −70 mV; Atwater et al. 1979; Dukes & Philipson 1996; Rorsman 1997). ATP can inhibit KATP channels at the Kir6.2 subunit in the absence of SUR1 (Tucker et al. 1997; John et al. 1998; Mikhailov et al. 1998) and point mutations within Kir6.2 can attenuate the effect of ATP (Tucker et al. 1997; Drain et al. 1998; Tucker et al. 1998; Koster et al. 1999). A similar effect is mediated by the anti-diabetic sulphonylurea drugs through an interaction with SUR1, a member of the ATP-binding cassette super-family (Aguilar-Bryan et al. 1995; Philipson & Steiner 1995; Ashfield et al. 1999), stimulating insulin secretion even in the absence of glucose. The SUR1 subunit also binds ATP (both with and without Mg) and MgADP via the presence of two cytoplasmic nucleotide binding domains (Ashcroft 2000). Binding of MgADP to SUR1 is thought to stabilize the channel in the open configuration, and when the ATP-to-ADP ratio increases, MgADP is displaced, and the Kir6.2 subunit becomes available for block by ATP (Ashcroft 2000). A number of excellent reviews focus with more detail on the function and role of KATP channels (Aguilar-Bryan & Bryan 1999; Ashcroft & Gribble 1999; Ashcroft 2000).
Evidence from rat hypothalamic neurons suggests that KATP channels may be regulated independently of ATP (Ainscow et al. 2002); however, the nature of this signal is unknown. One possibility is that KATP activity is modulated by additional metabolically regulated nucleotides. Vascular smooth muscle relaxation following hypoxic pulmonary vasoconstriction is characterized by a time-dependent increase in the NADH-to-NAD ratio and concomitant activation of KATP channels (Shigemori et al. 1996). High micromolar concentrations NAD(P) and NAD(P)H block KATP channels (Dunne et al. 1988; Harding et al. 1997), likely due to an interaction with the nucleotide inhibitory site of Kir6.2 when SUR1 is co-expressed (Dabrowski et al. 2003). It has been hypothesized that this interaction contributes to the resting level of channel inhibition (Dabrowski et al. 2003); however, a role for this in glucose-sensing is not clear. Since, the NAD(P)-to-NAD(P)H ratio is regulated by glucose it is possible that this plays a physiological role in the modulation of KATP during glucose-sensing.
In addition to regulation by nucleotides, KATP channels are modulated by lipid-derived signals that may play an important physiological role in relation to glucose-sensing. LC-CoA esters activate KATP in mouse (Branstrom et al. 1997) and human β-cells (Branstrom et al. 2004), and also activates the cloned channel (Branstrom et al. 1998b; Fox et al. 2003; Manning Fox et al. 2004). This effect, however, is expected to hyperpolarize the β-cell and thus not contribute to glucose-stimulated secretion per se, but may play an important role in the reduced secretory response observed in type 2 diabetes.
(b) Voltage-dependent calcium channels
The ability of pancreatic β-cells to respond to glucose has long been known to depend on extracellular Ca2+ (Curry et al. 1968; Hales & Milner 1968). Action potentials in rodent β-cells result largely from activation of a Ca2+ rather than a Na+ current (Dean & Matthews 1970a; Meissner & Schmelz 1974; Matthews & Sakamoto 1975). A limited role for Na+ currents in the rodent models was also suggested by the inability of tetrodotoxin to alter glucose-stimulated electrical activity and insulin secretion (Milner & Hales 1968; Meissner & Schmelz 1974), although Na+ currents may contribute to action potentials in human β-cells (Pressel & Misler 1990). The activation of VDCCs in mouse β-cells was first inferred from voltage noise analysis (Atwater et al. 1981). Subsequently, VDCC currents were measured from isolated mouse (Satin & Cook 1985; Rorsman & Trube 1986) and human (Kelly et al. 1991) β-cells. Depolarization of insulinoma and rat pancreatic β-cells leads to opening of L-type VDCCs (Keahey et al. 1989; Horvath et al. 1998; Ligon et al. 1998). A role for other VDCCs in insulin secreting cells (P/Q-type, N-type and T-type) is controversial, although R-type channels have a recently demonstrated role in later stages (or second phase) of insulin secretion (Jing et al. 2005). Antagonism of L-type channels is sufficient to inhibit insulin secretion in humans, rodents, dogs, and from insulinoma cells (Giugliano et al. 1980; Ohneda et al. 1983; Kanatsuna et al. 1985; Ohta et al. 1993). Therefore, L-type Ca2+ channels are considered the effectors of insulin secretion.
VDCC activity in mouse β-cells can be upregulated directly by glucose-derived signals (Smith et al. 1989), the nature of which remain elusive. One possibility is a direct regulation by ATP as L-type channel activity in rat vascular smooth muscle cells is increased by ATP, independent of channel phosphorylation (Yokoshiki et al. 1997). Application of DAG has also been shown to increase Ca2+ channel activity in insulinoma cells (Velasco & Petersen 1989) and palmitate, which presumably stimulates the accumulation of LC-CoAs, enhances Ca2+ channel activity in mouse β-cells (Olofsson et al. 2004). Finally, redox regulation of these channels cannot be ruled out as diphenylene iodonium, a blocker of NAD(P)H oxidase, antagonizes VDCCs in rat type 1 carotid body cells (Wyatt et al. 1994).
Release of intracellular Ca2+ stores is also thought to be involved in regulating insulin secretion. Intracellular Ca2+ stores may be released by the influx of extracellular Ca2+, called Ca2+-induced Ca2+ release (Graves & Hinkle 2003), or by other external signals in order to enhance or prolong insulin secretion rather than trigger it directly. Acetylcholine and cholecystokinin, for example, signal through inositol trisphosphate (IP3) to release internal Ca2+ stores and activate protein kinase c (PKC) (Lang 1999), thereby increasing insulin secretion. Interestingly, IP3-receptor mediated Ca2+ release is potentiated by NADH in a reconstituted lipid vesicle system (Kaplin et al. 1996), and in intact PC12 or cerebellar Purkinge cells exposed to hypoxia the metabolic generation of NADH is implicated in release of Ca2+ stores (Kaplin et al. 1996). Also, the skeletal muscle Ca2+ release channel RyR1 contains an oxidoreductase like domain that may bind adenine nucleotides and function as a redox sensor (Baker et al. 2002). NADH inhibited cardiac ryanodine receptor (RyR) activity but activated skeletal RyRs, likely through interaction with the RyR ATP binding site(s) (Zima et al. 2003). Thus, it is interesting to speculate that NAD(P)H (or the cellular redox state) may have an amplifying effect on Ca2+-induced Ca2+ release or second messenger mediated release of Ca2+ stores (Hidalgo et al. 2004).
(c) Voltage-dependent K+ channels
We have recently reviewed the role of voltage-dependent K+ channels in pancreatic β-cell function (MacDonald & Wheeler 2003). These channels open upon membrane depolarization to mediate repolarization during the action potential down-stroke. Kv channel α-subunits consist of six transmembrane domains, a voltage sensor (transmembrane domain 4), a pore forming loop and cytosolic C- and N-termini. Kv channels open in response to membrane depolarization and mediate outwardly rectifying K+ currents that act to repolarize action potentials, although different channel homologues exhibit different properties such as voltage-sensitivity and kinetics of activation and inactivation. Functional channels are a homo- or heterotetrameric complex of α-subunits of the same family (Kv1.X–4.X). Kv2.1 is the major β-cell Kv channel isoform (MacDonald et al. 2001, 2002). Cloned Kv channel subunits of the Kv5.X–11.X channel families do not mediate currents alone, but instead appear to modulate or inhibit other Kv channel families (Hugnot et al. 1996; Salinas et al. 1997a,b; Stocker & Kerschensteiner 1998). Besides regulatory α-subunits, Kv channel kinetics can be modulated by large N-terminal domains that mediate fast inactivation, or by the presence of cytoplasmic β-subunits (Kvβ1–3) that associate with the channel N-terminus (Rettig et al. 1994; Jing et al. 1999).
Numerous studies suggest that Kv channels, including the predominant β-cell channel Kv2.1, are regulated by mitochondrially derived signals (Michelakis et al. 2004). While the direct redox modulation of Kv α-subunits (particularly cysteine residues) may modify channel properties (Ruppersberg et al. 1991), the regulatory β-subunits in particular are proposed to act as sensors of intracellular redox potential (McCormack & McCormack 1994; Chouinard et al. 1995) and regulate channels dependent on NADPH-binding (Bahring et al. 2001; Peri et al. 2001). At least one study has demonstrated that mutation of the Kvβ (Kvβ1.1) oxidoreductase active site attenuated the ability of this subunit to confer fast inactivation to a Kv1 channel (Kv1.5; Bahring et al. 2001). Others have shown that mutation of regions putatively involved in NADPH co-factor binding alter the ability of β-subunits to promote channel surface expression (Peri et al. 2001; Campomanes et al. 2002). Regulatory β-subunits have also been implicated in the regulation of Kv channels in response to changes in [O2] (Coppock et al. 2001).
Recent evidence suggests the existence of membrane associated aldehyde oxidoreductase-like enzyme activity in rat islets (Laclau et al. 2001) and a number of oxidoreductase-like Kvβ subunits (Kvβ1, 2 and 3) are expressed in human and rat islets and INS-1 insulinoma cells (Chouinard et al. 2000). The ability of the aldehyde reductase antagonist diphenylhydantoin to prevent glucose-stimulated insulin secretion from rat islets supports a role for an NADPH-oxidoreductase activity in stimulus-secretion coupling (Kizer et al. 1970; Levin et al. 1972; Laclau et al. 2001). Indeed, Kv channel activity may be regulated by the NADPH-to-NADP ratio (Tipparaju et al. 2005), analogous to KATP channel regulation by the ATP-to-ADP ratio. We recently described a potent regulation of native Kv2.1 channels in rat β-cells whereby raising the cytoplasmic NADPH-to-NADP+ ratio caused currents to inactivate quickly and more completely and a leftward shift in the voltage-dependence of steady-state inactivation (MacDonald et al. 2003). Thus, more channels were already inactivated, and therefore unavailable to repolarize action potentials. This has important implications since the metabolic generation of NADPH may reduce the efficacy of Kv channels in repolarizing the β-cell and therefore contribute to β-cell electrical excitability in response to glucose, broadening action potentials and increasing calcium influx. This may in part account for the ability of glucose to modify β-cell electrical responses even when KATP channels are already closed by sulphonylureas (Henquin 1998; Best 2002).
(d) Additional electrogenic mechanisms
A number of additional factors may control β-cell electrical activity in addition to those mentioned above. These include other channels such as Cl− and non-selective cation channels, and electrogenic pumps such as the Na+/K+ ATPase. While the role of these have not been extensively characterized in pancreatic β-cells, their metabolic regulation may nonetheless yet prove to make important contributions to glucose-sensing. An insulinoma Cl− current has been shown to be regulated by ATP (Kinard & Satin 1995; Best et al. 1996). This may contribute to β-cell depolarization in response to glucose (Best 1997; Best et al. 2000). As well, while an early study suggested a limited role for glucose regulation of the Na+/K+ ATPase in β-cells (Lebrun et al. 1983), more recent work suggests that a glucose-stimulated reduction in the activity of this electrogenic pump, mediated in part by PKC, may contribute to membrane depolarization (Owada et al. 1999). Certainly, other potential contributors will be revealed as we gain a more complete understanding of the control of β-cell membrane potential.
4. Distal sensing of metabolic signals: insulin granule dynamics and exocytosis
Glucose-stimulated electrical activity in pancreatic β-cells serves the general purpose of activating VDCCs and delivering the Ca2+ stimulus to sites of insulin granule exocytosis. While the membrane potential is exquisitely controlled by glucose, the distribution and exocytotic competence of the insulin granules themselves must be equally well controlled to ensure an appropriate physiological response to glucose. Insulin granules, similar to secretory vesicles in numerous other cell types, exist within the cell in various functional pools (Rorsman & Renstrom 2003). Shown in figure 3, these include an intracellular reserve pool (90%), a morphologically docked pool (ca 10%), and a readily releasable pool (RRP) that is chemically ‘primed’ for release (0.3–2.2%; Rorsman et al. 2000; Bratanova-Tochkova et al. 2002; Olofsson et al. 2002).
The size of the RRP is a major determinant of the magnitude of the initial secretory response. While in the short-term, priming of granules already docked at the membrane may account for refilling of the RRP, ultimately granules must be mobilized from intracellular reserve pools. The initial (or first phase) exocytotic response can be replicated by any stimuli that increases intracellular Ca2+, causing the release of already docked and primed vesicles. Sustained (or second phase) secretion on the other hand, which is dependent on vesicle mobilization and priming, can only be elicited by metabolizable fuel secretagogues (Gembal et al. 1992; Henquin 2000). Thus, glucose-derived signals are important for amplifying and maintaining secretion by promoting the mobilization and priming of insulin granules from reserve pools.
(a) Mobilization of insulin granules
Granules within the cell undergo extensive movement (Somers et al. 1979; Pouli et al. 1998; Barg et al. 2002b; Varadi et al. 2002). Studies employing total internal reflection fluorescence microscopy demonstrated that second phase secretion results largely from the exocytosis of insulin granules newly recruited to the plasma membrane (Ohara-Imaizumi et al. 2002; Ohara-Imaizumi et al. 2004). In rat β-cells, glucose stimulates an increase in the number of insulin granules associated with the plasma membrane (Straub et al. 2004). Insulin granule movement can be stimulated by glucose (Pouli et al. 1998) or ATP (Varadi et al. 2002). Agents that raise cAMP also increase insulin granule movement (Lacy et al. 1975; Hisatomi et al. 1996) and PKC mobilizes catecholamine granules of chromaffin cells into the RRP (Gillis et al. 1996), although PKC may control secretion at a point between vesicle docking with the plasma membrane and exocytosis (Ammala et al. 1994).
Within the cytoplasm vesicle movement likely occurs along microtubules (Orci et al. 1972; Malaisse et al. 1974), mediated by the ATP-dependent motor activity of the conventional kinesin KIF5B (Varadi et al. 2002, 2003). Disruption of the microtubule network impairs granule movement and both the initial exocytotic response and refilling of the RRP (Ivarsson et al. 2004). Inhibitors of 5′-AMP activated kinase (AMPK) enhance insulin secretion (da Silva et al. 2003; Rutter et al. 2003) and a constitutively active AMPK mutant inhibited glucose stimulated granule movement (Tsuboi et al. 2003). AMPK is, therefore, suggested as a negative regulator of granule recruitment and is inhibited by an increased ATP-to-ADP ratio (and reduced AMP). Thus, metabolically generated ATP (or reciprocal changes in ADP and AMP) may be a crucial factor in recruitment of insulin granules from intracellular reserve pools to the plasma membrane.
Near the plasma membrane, insulin granules are transported along the cortical actin network in an ATP-dependent manner, likely by the motor protein myosin 5A (Varadi et al. 2005). By analogy to melanosomes (Hume et al. 2002; Wu et al. 2002), myosin 5A recruitment to the granule may be dependent on small Rab G-proteins such as Rab27a, which are also shown to be important for insulin secretion (Waselle et al. 2003). Actin remodelling stimulated by glucose (mediated by Cdc42 inhibition) is also suggested to be an important pre-requisite for granule recruitment and insulin secretion (Nevins & Thurmond 2003). Thus proteins dependent on GTP/GDP cycling may play an important role in recruitment of vesicles to the plasma membrane. There are conflicting reports as to the role of membrane depolarization and Ca2+ entry through VDCCs in regulating insulin granule traffic (Somers et al. 1979; Hisatomi et al. 1996; Niwa et al. 1998; Pouli et al. 1998). A role for IP3 mediated release of Ca2+ stores, which may be augmented by NADH (see above), and Ca2+-dependent phosphorylation of myosin light chain has been proposed (Iida et al. 1997; Niki 1999).
(b) Exocytosis of insulin granules
The machinery regulating exocytosis has been reviewed extensively (Burgoyne & Morgan 2003; Sollner 2003). A comparison of insulin versus neurotransmitter exocytosis is reviewed by Gerber & Sudhof (2002) and β-cell exocytosis has also been reviewed specifically (Bratanova-Tochkova et al. 2002). Fusion of an exocytotic vesicle with the plasma membrane is mediated by soluble NSF attachment protein receptor (SNARE) proteins. These include the plasma membrane proteins syntaxin and SNAP-25 and the vesicular protein VAMP (synaptobrevin). Synaptotagmin is the likely Ca2+ sensor for exocytosis (Meldolesi & Chieregatti 2004), and in β-cells has been variously reported to be represented by the synaptotagmin III, V, VII, VIII and IX (Mizuta et al. 1997; Brown et al. 2000; Gao et al. 2000; Gut et al. 2001; Iezzi et al. 2004). Various SNARE proteins interact with the VDCCs (Catterall 1999), and this mediates a tight coupling of Ca2+ entry and exocytosis of vesicles in the RRP (Barg et al. 2002a).
Prior to Ca2+-triggered and SNARE-mediated exocytosis, a docked insulin granule must first be primed for release. That is, certain chemical modifications that are not yet completely defined are required to render a granule competent for release. ATP is essential for granule priming prior to exocytosis (Eliasson et al. 1997). The role of ATP may, however, be limited to a permissive one and ADP seems to be a more dynamic regulator of exocytosis at this distal step (Olsen et al. 2003). The ultimate effector of the ADP effect on exocytosis is not entirely clear, but may involve PI4 kinase activation and downstream effects on the Ca2+-dependent activator protein for secretion (Olsen et al. 2003). Additionally, there is evidence that intra-vesicular acidification by a V-type H+ ATPase plays an important role in insulin granule priming (Barg et al. 2001; Renstrom et al. 2002). While vesicles that were already primed for release were capable of undergoing exocytosis, inhibition of granule acidification prevented refilling of the RRP and subsequent exocytosis (Barg et al. 2001). Thus, metabolically generated changes in the ATP-to-ATP ratio modulate RRP size by regulating steps between vesicle docking with the plasma membrane and the ultimate priming of the granule for release.
Lipid derived LC-CoA signals may also play an important role in regulating RRP size and exocytosis. Palmitoyl-CoA stimulated secretion from permeabilized insulinoma cells and Ca2+-dependent exocytosis in mouse β-cells (Deeney et al. 2000a). Also, palmitate was found to increase the initial exocytotic response of mouse β-cells (Olofsson et al. 2004); however, in this study the direct application of palmitoyl-CoA failed to enhance exocytosis. The role of LC-CoAs as direct regulator of the late stages of exocytosis, therefore, remains unclear, although it has been speculated that fatty acylation of exocytotic proteins mediates a LC-CoA effect on exocytosis (Corkey et al. 2000). Consistent with a potential role for acylation in the regulation of exocytosis, palmitoylation of SNAP-23 enhances membrane fusion (Pallavi & Nagaraj 2003) and the protein acylation inhibitor cerulenin attenuates time-dependent potentiation of secretion by glucose (Yamada et al. 2002).
The latter steps of insulin granule priming and filling of the RRP may also be regulated by lipid-derived signals further downstream of LC-CoA, namely the generation of diacylglycerol and activation of PKC. Recruitment of PKC isoforms to the plasma membrane is stimulated by glucose (Zaitsev et al. 1995), and the PKCϵ isoform in particular is recruited to insulin granules and appears to be important for exocytosis (Mendez et al. 2003). While PKC translocation likely results from elevated intracellular Ca2+ rather than a direct effect of lipid-derived signals (Deeney et al. 1996; Pinton et al. 2002), lipid-derived signals may still be important for activating or augmenting PKC activity since phorbol esters enhance insulin secretion even when intracellular Ca2+ is raised by direct depolarization with high K+ (Ravier et al. 1999). A recently identified highly Ca2+-sensitive pool (HCSP) of granules is increased in size by activation of PKC (Yang & Gillis 2004) and may provide a mechanism for increasing the release competence of granules in the absence of tight coupling to a particular VDCC. PKC activation may also stimulate Ca2+-independent but GTP dependent mechanisms of exocytosis in β-cells (Regazzi et al. 1989; Komatsu et al. 1998; Lee et al. 2003). The target(s) by which PKC mediates these effects is unknown. Although SNAP-25 is phosphorylated by PKC in a glucose-dependent manner, this does not correlate with an effect on insulin secretion (Gonelle-Gispert et al. 2002). It is also of interest to note that lipid-derived signals, such as DAG, can interact directly with proteins involved in exocytosis and vesicular cycling including small GTP-binding proteins and Munc13 (Kazanietz et al. 2000). Recently, Ivarsson et al. (2005) have provided evidence suggesting that NADPH may regulate exocytosis directly through the glutaredoxin and thioredoxin proteins.
5. Conclusions and future perspective
Glucose-sensing in the pancreatic β-cell is of critical importance for maintaining energy homeostasis. Despite this central role, we still do not completely understand how the numerous signals generated by mitochondria regulate the important mediators of cellular excitability and exocytosis. The role of ATP, or perhaps more appropriately the ATP-to-ADP ratio, and its effect on KATP channels is generally well understood. However, KATP inhibition alone cannot account for the complex secretory response of these cells. Perhaps just as important are numerous other metabolically regulated targets, including other ion channels and electrogenic ion pumps, and certainly effects on insulin granule dynamics and exocytosis. Future study of the alternate mitochondrial signals and distal mechanisms with which they interact will be crucial towards a more complete understanding of β-cell glucose-sensing and eventually the development of new and better treatments for type 2 diabetes.
Acknowledgments
This work was supported by a grant to PR from the Wellcome Trust. JWJ and PEM were supported by fellowships from the Canadian Institutes of Health Research. PEM currently holds the European Foundation for the Study of Diabetes/AstraZeneca Fellowship in Islet Biology and PR is a Royal Society-Wolfson Research Fellow.
Glossary
- AMPK
5′-AMP kinase
- CPT1
carnitine palmitoyltransferase 1
- DAG
diacylglycerol
- GK
glucokinase
- IP3
inositol trisposphate
- KATP
ATP-sensitive K+
- Kv
voltage-dependent K+
- LC-CoA
long-chain acyl-CoA
- LDH
lactate dehydrogenase
- mGPDH
mitochondrial glycerol-phosphate dehydrogenase
- OAA
oxaloacetate
- PC
pyruvate carboxylase
- PKC
protein kinase C
- RRP
readily releasable pool
- RyR
ryanodine receptor
- SNARE
soluble NSF attachment protein receptor
- SUR
sulphonylurea receptor
- TCA
tricarboxylic acid
- VDCC
voltage-dependent Ca2+ channel
Footnotes
One contribution of 18 to a Theme Issue ‘Reactive oxygen species in health and disease’.
References
- Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. 10.1210/er.20.2.101 [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ainscow E.K, Mirshamsi S, Tang T, Ashford M.L, Rutter G.A. Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: evidence for ATP-independent control of ATP-sensitive K+ channels. J. Physiol. 2002;544:429–445. doi: 10.1113/jphysiol.2002.022434. 10.1113/jphysiol.2002.022434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammala C, Eliasson L, Bokvist K, Berggren P.O, Honkanen R.E, Sjoholm A, Rorsman P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic β cells. Proc. Natl Acad. Sci. USA. 1994;91:4343–4347. doi: 10.1073/pnas.91.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammon H.P, Steinke J. 6-Amnionicotinamide (6-AN) as a diabetogenic agent. In vitro and in vivo studies in the rat. Diabetes. 1972;21:143–148. doi: 10.2337/diab.21.3.143. [DOI] [PubMed] [Google Scholar]
- Antinozzi P.A, Segall L, Prentki M, McGarry J.D, Newgard C.B. Molecular or pharmacologic perturbation of the link between glucose and lipid metabolism is without effect on glucose-stimulated insulin secretion. A re-evaluation of the long-chain acyl-CoA hypothesis. J. Biol. Chem. 1998;273:16 146–16 154. doi: 10.1074/jbc.273.26.16146. 10.1074/jbc.273.26.16146 [DOI] [PubMed] [Google Scholar]
- Arkhammar P, Nilsson T, Rorsman P, Berggren P.O. Inhibition of ATP-regulated K+ channels precedes depolarization-induced increase in cytoplasmic free Ca2+ concentration in pancreatic β-cells. J. Biol. Chem. 1987;262:5448–5454. [PubMed] [Google Scholar]
- Ashcroft S.J. The β-cell KATP channel. J. Membr. Biol. 2000;176:187–206. doi: 10.1007/s00232001095. 10.1007/s002320001095 [DOI] [PubMed] [Google Scholar]
- Ashcroft S.J, Ashcroft F.M. Properties and functions of ATP-sensitive K-channels. Cell Signal. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. 10.1016/0898-6568(90)90048-F [DOI] [PubMed] [Google Scholar]
- Ashcroft S.J, Ashcroft F.M. The sulfonylurea receptor. Biochim. Biophys. Acta. 1992;1175:45–59. doi: 10.1016/0167-4889(92)90008-y. 10.1016/0167-4889(92)90008-Y [DOI] [PubMed] [Google Scholar]
- Ashcroft S.J, Christie M.R. Effects of glucose on the cytosolic ratio of reduced/oxidized nicotinamide-adenine dinucleotide phosphate in rat islets of Langerhans. Biochem. J. 1979;184:697–700. doi: 10.1042/bj1840697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F.M, Gribble F.M. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. 10.1007/s001250051247 [DOI] [PubMed] [Google Scholar]
- Ashcroft F.M, Rorsman P. Molecular defects in insulin secretion in type-2 diabetes. Rev. Endocr. Metab. Disord. 2004;5:135–142. doi: 10.1023/B:REMD.0000021435.87776.a7. 10.1023/B:REMD.0000021435.87776.a7 [DOI] [PubMed] [Google Scholar]
- Ashcroft F.M, Harrison D.E, Ashcroft S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. 10.1038/312446a0 [DOI] [PubMed] [Google Scholar]
- Ashfield R, Gribble F.M, Ashcroft S.J, Ashcroft F.M. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- Atwater I, Rojas E, Scott A. Simultaneous measurements of insulin release and electrical activity from single microdissected mouse Islets of Langerhans. J. Physiol. 1979;291:57P. [PubMed] [Google Scholar]
- Atwater I, Dawson C.M, Eddlestone G.T, Rojas E. Voltage noise measurements across the pancreatic beta-cell membrane: calcium channel characteristics. J. Physiol. 1981;314:195–212. doi: 10.1113/jphysiol.1981.sp013701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahring R, et al. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvβ subunits. J. Biol. Chem. 2001;276:22 923–22 929. doi: 10.1074/jbc.M100483200. 10.1074/jbc.M100483200 [DOI] [PubMed] [Google Scholar]
- Baker M.L, Serysheva I.I, Sencer S, Wu Y, Ludtke S.J, Jiang W, Hamilton S.L, Chiu W. The skeletal muscle Ca2+ release channel has an oxidoreductase-like domain. Proc. Natl Acad. Sci. USA. 2002;99:12 155–12 160. doi: 10.1073/pnas.182058899. 10.1073/pnas.182058899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg S, Huang P, Eliasson L, Nelson D.J, Obermuller S, Rorsman P, Thevenod F, Renstrom E. Priming of insulin granules for exocytosis by granular Cl− uptake and acidification. J. Cell Sci. 2001;114:2145–2154. doi: 10.1242/jcs.114.11.2145. [DOI] [PubMed] [Google Scholar]
- Barg S, Eliasson L, Renstrom E, Rorsman P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse β-cells. Diabetes. 2002a;51(Suppl. 1):S74–S82. doi: 10.2337/diabetes.51.2007.s74. [DOI] [PubMed] [Google Scholar]
- Barg S, Olofsson C.S, Schriever-Abeln J, Wendt A, Gebre-Medhin S, Renstrom E, Rorsman P. Delay between fusion pore opening and peptide release from large dense-core vesicles in neuroendocrine cells. Neuron. 2002b;33:287–299. doi: 10.1016/s0896-6273(02)00563-9. 10.1016/S0896-6273(02)00563-9 [DOI] [PubMed] [Google Scholar]
- Best L. Glucose and alpha-ketoisocaproate induce transient inward currents in rat pancreatic beta cells. Diabetologia. 1997;40:1–6. doi: 10.1007/s001250050635. 10.1007/s001250050635 [DOI] [PubMed] [Google Scholar]
- Best L. Evidence that glucose-induced electrical activity in rat pancreatic β-cells does not require KATP channel inhibition. J. Membr. Biol. 2002;185:193–200. doi: 10.1007/s00232-001-0114-1. 10.1007/s00232-001-0114-1 [DOI] [PubMed] [Google Scholar]
- Best L, Sheader E.A, Brown P.D. A volume-activated anion conductance in insulin-secreting cells. Pflug. Arch. 1996;431:363–370. doi: 10.1007/BF02207273. 10.1007/BF02207273 [DOI] [PubMed] [Google Scholar]
- Best L, Brown P.D, Sheader E.A, Yates A.P. Selective inhibition of glucose-stimulated β-cell activity by an anion channel inhibitor. J. Membr. Biol. 2000;177:169–175. doi: 10.1007/s002320001110. 10.1007/s002320001110 [DOI] [PubMed] [Google Scholar]
- Branstrom R, Corkey B.E, Berggren P.O, Larsson O. Evidence for a unique long chain acyl-CoA ester binding site on the ATP-regulated potassium channel in mouse pancreatic beta cells. J. Biol. Chem. 1997;272:17 390–17 394. doi: 10.1074/jbc.272.28.17390. 10.1074/jbc.272.28.17390 [DOI] [PubMed] [Google Scholar]
- Branstrom R, Efendic S, Berggren P.O, Larsson O. Direct inhibition of the pancreatic β-cell ATP-regulated potassium channel by alpha-ketoisocaproate. J. Biol. Chem. 1998a;273:14 113–14 118. doi: 10.1074/jbc.273.23.14113. [DOI] [PubMed] [Google Scholar]
- Branstrom R, Leibiger I.B, Leibiger B, Corkey B.E, Berggren P.O, Larsson O. Long chain coenzyme A esters activate the pore-forming subunit (Kir6.2) of the ATP-regulated potassium channel. J. Biol. Chem. 1998b;273:31 395–31 400. doi: 10.1074/jbc.273.47.31395. 10.1074/jbc.273.47.31395 [DOI] [PubMed] [Google Scholar]
- Branstrom R, et al. Long-chain CoA esters activate human pancreatic beta-cell KATP channels: potential role in Type 2 diabetes. Diabetologia. 2004;47:277–283. doi: 10.1007/s00125-003-1299-x. [DOI] [PubMed] [Google Scholar]
- Bratanova-Tochkova T.K, et al. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes. 2002;51(Suppl. 1):S83–S90. doi: 10.2337/diabetes.51.2007.s83. [DOI] [PubMed] [Google Scholar]
- Brown H, et al. Synaptotagmin III isoform is compartmentalized in pancreatic β-cells and has a functional role in exocytosis. Diabetes. 2000;49:383–391. doi: 10.2337/diabetes.49.3.383. [DOI] [PubMed] [Google Scholar]
- Burgoyne R.D, Morgan A. Secretory granule exocytosis. Physiol. Rev. 2003;83:581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- Campomanes C.R, Carroll K.I, Manganas L.N, Hershberger M.E, Gong B, Antonucci D.E, Rhodes K.J, Trimmer J.S. Kv β subunit oxidoreductase activity and Kv1 potassium channel trafficking. J. Biol. Chem. 2002;277:8298–8305. doi: 10.1074/jbc.M110276200. 10.1074/jbc.M110276200 [DOI] [PubMed] [Google Scholar]
- Catterall W.A. Interactions of presynaptic Ca2+ channels and SNARE proteins in neurotransmitter release. Ann. NY Acad. Sci. 1999;868:144–159. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- Chen S, Ogawa A, Ohneda M, Unger R.H, Foster D.W, McGarry J.D. More direct evidence for a malonyl-CoA-carnitine palmitoyltransferase I interaction as a key event in pancreatic β-cell signaling. Diabetes. 1994;43:878–883. doi: 10.2337/diab.43.7.878. [DOI] [PubMed] [Google Scholar]
- Chouinard S.W, Wilson G.F, Schlimgen A.K, Ganetzky B. A potassium channel beta subunit related to the aldo-keto reductase superfamily is encoded by the Drosophila hyperkinetic locus. Proc. Natl Acad. Sci. USA. 1995;92:6763–6767. doi: 10.1073/pnas.92.15.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouinard S.W, Lu F, Ganetzky B, MacDonald M.J. Evidence for voltage-gated potassium channel beta-subunits with oxidoreductase motifs in human and rodent pancreatic β-cells. Receptor Channel. 2000;7:237–243. [PubMed] [Google Scholar]
- Cook D.L, Hales C.N. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311:271–273. doi: 10.1038/311271a0. 10.1038/311271a0 [DOI] [PubMed] [Google Scholar]
- Coppock E.A, Martens J.R, Tamkun M.M. Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:L1–12. doi: 10.1152/ajplung.2001.281.1.L1. [DOI] [PubMed] [Google Scholar]
- Corkey B.E, Glennon M.C, Chen K.S, Deeney J.T, Matschinsky F.M, Prentki M. A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic β-cells. J. Biol. Chem. 1989;264:21 608–21 612. [PubMed] [Google Scholar]
- Corkey B.E, Deeney J.T, Yaney G.C, Tornheim K, Prentki M. The role of long-chain fatty acyl-CoA esters in β-cell signal transduction. J. Nutr. 2000;130:299S–304S. doi: 10.1093/jn/130.2.299S. [DOI] [PubMed] [Google Scholar]
- Curry D.L, Bennett L.L, Grodsky G.M. Requirement for calcium ion in insulin secretion by the perfused rat pancreas. Am. J. Physiol. 1968;214:174–178. doi: 10.1152/ajplegacy.1968.214.1.174. [DOI] [PubMed] [Google Scholar]
- Dabrowski M, Trapp S, Ashcroft F.M. Pyridine nucleotide regulation of the KATP channel Kir6.2/SUR1 expressed in Xenopus oocytes. J. Physiol. 2003;550:357–363. doi: 10.1113/jphysiol.2003.041715. 10.1113/jphysiol.2003.041715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva X, Leclerc I, Varadi A, Tsuboi T, Moule S.K, Rutter G.A. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem. J. 2003;371:761–774. doi: 10.1042/BJ20021812. 10.1042/BJ20021812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P.M, Matthews E.K. Electrical activity in pancreatic islet cells. Nature. 1968;219:389–390. doi: 10.1038/219389a0. [DOI] [PubMed] [Google Scholar]
- Dean P.M, Matthews E.K. Electrical activity in pancreatic islet cells: effect of ions. J. Physiol. 1970a;210:265–275. doi: 10.1113/jphysiol.1970.sp009208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P.M, Matthews E.K. Glucose-induced electrical activity in pancreatic islet cells. J. Physiol. 1970b;210:255–264. doi: 10.1113/jphysiol.1970.sp009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeney J.T, Cunningham B.A, Chheda S, Bokvist K, Juntti-Berggren L, Lam K, Korchak H.M, Corkey B.E, Berggren P.O. Reversible Ca2+-dependent translocation of protein kinase C and glucose-induced insulin release. J. Biol. Chem. 1996;271:18 154–18 160. doi: 10.1074/jbc.271.30.18154. 10.1074/jbc.271.18.10623 [DOI] [PubMed] [Google Scholar]
- Deeney J.T, Gromada J, Hoy M, Olsen H.L, Rhodes C.J, Prentki M, Berggren P.O, Corkey B.E. Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI β-cells) J. Biol. Chem. 2000a;275:9363–9368. doi: 10.1074/jbc.275.13.9363. 10.1074/jbc.275.13.9363 [DOI] [PubMed] [Google Scholar]
- Deeney J.T, Prentki M, Corkey B.E. Metabolic control of β-cell function. Semin. Cell Dev. Biol. 2000b;11:267–275. doi: 10.1006/scdb.2000.0175. 10.1006/scdb.2000.0175 [DOI] [PubMed] [Google Scholar]
- De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, Schuit F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Invest. 1995;96:2489–2495. doi: 10.1172/JCI118308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P, Li L, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl Acad. Sci. USA. 1998;95:13 953–13 958. doi: 10.1073/pnas.95.23.13953. 10.1073/pnas.95.23.13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen M.R, Smith P.A, Ashcroft F.M. Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic beta-cells. Biochem. J. 1993;294:35–42. doi: 10.1042/bj2940035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes I.D, Philipson L.H. K+ channels: generating excitement in pancreatic β-cells. Diabetes. 1996;45:845–853. doi: 10.2337/diab.45.7.845. [DOI] [PubMed] [Google Scholar]
- Dukes I.D, McIntyre M.S, Mertz R.J, Philipson L.H, Roe M.W, Spencer B, Worley J.F., III Dependence on NADH produced during glycolysis for β-cell glucose signaling. J. Biol. Chem. 1994;269:10 979–10 982. [PubMed] [Google Scholar]
- Dunne M.J, Findlay I, Petersen O.H. Effects of pyridine nucleotides on the gating of ATP-sensitive potassium channels in insulin-secreting cells. J. Membr. Biol. 1988;102:205–216. doi: 10.1007/BF01925714. 10.1007/BF01925714 [DOI] [PubMed] [Google Scholar]
- Eliasson L, Renstrom E, Ding W.G, Proks P, Rorsman P. Rapid ATP-dependent priming of secretory granules precedes Ca2+-induced exocytosis in mouse pancreatic B-cells. J. Physiol. 1997;503:399–412. doi: 10.1111/j.1469-7793.1997.399bh.x. 10.1111/j.1469-7793.1997.399bh.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Bryla J, Michalik M, Meglasson M.D, Nelson D. Energy metabolism in islets of Langerhans. Biochim. Biophys. Acta. 1992;1101:273–295. doi: 10.1016/0005-2728(92)90084-f. [DOI] [PubMed] [Google Scholar]
- Eto K, et al. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999;283:981–985. doi: 10.1126/science.283.5404.981. 10.1126/science.283.5404.981 [DOI] [PubMed] [Google Scholar]
- Fox J.E, Magga J, Giles W.R, Light P.E. Acyl coenzyme A esters differentially activate cardiac and β-cell adenosine triphosphate-sensitive potassium channels in a side-chain length-specific manner. Metabolism. 2003;52:1313–1319. doi: 10.1016/s0026-0495(03)00199-9. 10.1016/S0026-0495(03)00199-9 [DOI] [PubMed] [Google Scholar]
- Gao Z, Reavey-Cantwell J, Young R.A, Jegier P, Wolf B.A. Synaptotagmin III/VII isoforms mediate Ca2+-induced insulin secretion in pancreatic islet β-cells. J. Biol. Chem. 2000;275:36 079–36 085. doi: 10.1074/jbc.M004284200. 10.1074/jbc.M004284200 [DOI] [PubMed] [Google Scholar]
- Gembal M, Gilon P, Henquin J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Invest. 1992;89:1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber S.H, Sudhof T.C. Molecular determinants of regulated exocytosis. Diabetes. 2002;51(Suppl. 1):S3–S11. doi: 10.2337/diabetes.51.2007.s3. [DOI] [PubMed] [Google Scholar]
- Gillis K.D, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. 10.1016/S0896-6273(00)80147-6 [DOI] [PubMed] [Google Scholar]
- Giroix M.H, Rasschaert J, Bailbe D, Leclercq-Meyer V, Sener A, Portha B, Malaisse W.J. Impairment of glycerol phosphate shuttle in islets from rats with diabetes induced by neonatal streptozocin. Diabetes. 1991;40:227–232. doi: 10.2337/diab.40.2.227. [DOI] [PubMed] [Google Scholar]
- Giugliano D, Torella R, Cacciapuoti F, Gentile S, Verza M, Varricchio M. Impairment of insulin secretion in man by nifedipine. Eur. J. Clin. Pharmacol. 1980;18:395–398. doi: 10.1007/BF00636791. 10.1007/BF00636791 [DOI] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Costa M, Takahashi M, Sadoul K, Halban P. Phosphorylation of SNAP-25 on serine-187 is induced by secretagogues in insulin-secreting cells, but is not correlated with insulin secretion. Biochem. J. 2002;368:223–232. doi: 10.1042/BJ20020896. 10.1042/BJ20020896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves T.K, Hinkle P.M. Ca2+-induced Ca2+ release in the pancreatic β-cell: direct evidence of endoplasmic reticulum Ca2+ release. Endocrinology. 2003;144:3565–3574. doi: 10.1210/en.2002-0104. 10.1210/en.2002-0104 [DOI] [PubMed] [Google Scholar]
- Gut A, Kiraly C.E, Fukuda M, Mikoshiba K, Wollheim C.B, Lang J. Expression and localisation of synaptotagmin isoforms in endocrine β-cells: their function in insulin exocytosis. J. Cell Sci. 2001;114:1709–1716. doi: 10.1242/jcs.114.9.1709. [DOI] [PubMed] [Google Scholar]
- Hales C.N, Milner R.D. Cations and the secretion of insulin from rabbit pancreas in vitro. J. Physiol. 1968;199:177–187. doi: 10.1113/jphysiol.1968.sp008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford R.G. Dehydrogenase activation by Ca2+ in cells and tissues. J. Bioenerg. Biomembr. 1991;23:823–854. doi: 10.1007/BF00786004. 10.1007/BF00786004 [DOI] [PubMed] [Google Scholar]
- Harding E.A, Kane C, James R.F, London N.J, Dunne M.J. Modulation of three types of potassium selective channels by NAD and other pyridine nucleotides in human pancreatic beta-cells. NAD and K+ channels in human beta-cells. Adv. Exp. Med. Biol. 1997;426:43–50. doi: 10.1007/978-1-4899-1819-2_6. [DOI] [PubMed] [Google Scholar]
- Hedeskov C.J, Capito K, Thams P. Cytosolic ratios of free [NADPH]/[NADP+] and [NADH]/[NAD+] in mouse pancreatic islets, and nutrient-induced insulin secretion. Biochem. J. 1987;241:161–167. doi: 10.1042/bj2410161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin J.C. d-glucose inhibits potassium efflux from pancreatic islet cells. Nature. 1978;271:271–273. doi: 10.1038/271271a0. 10.1038/271271a0 [DOI] [PubMed] [Google Scholar]
- Henquin J.C. A minimum of fuel is necessary for tolbutamide to mimic the effects of glucose on electrical activity in pancreatic β-cells. Endocrinology. 1998;139:993–998. doi: 10.1210/endo.139.3.5783. 10.1210/en.139.3.993 [DOI] [PubMed] [Google Scholar]
- Henquin J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Bull R, Behrens M.I, Donoso P. Redox regulation of RyR-mediated Ca2+ release in muscle and neurons. Biol. Res. 2004;37:539–552. doi: 10.4067/s0716-97602004000400007. [DOI] [PubMed] [Google Scholar]
- Hisatomi M, Hidaka H, Niki I. Ca2+/calmodulin and cyclic 3,5′ adenosine monophosphate control movement of secretory granules through protein phosphorylation/dephosphorylation in the pancreatic β-cell. Endocrinology. 1996;137:4644–4649. doi: 10.1210/endo.137.11.8895328. 10.1210/en.137.11.4644 [DOI] [PubMed] [Google Scholar]
- Horvath A, Szabadkai G, Varnai P, Aranyi T, Wollheim C.B, Spat A, Enyedi P. Voltage dependent calcium channels in adrenal glomerulosa cells and in insulin producing cells. Cell Calcium. 1998;23:33–42. doi: 10.1016/s0143-4160(98)90072-0. 10.1016/S0143-4160(98)90072-0 [DOI] [PubMed] [Google Scholar]
- Hoy M, Maechler P, Efanov A.M, Wollheim C.B, Berggren P.O, Gromada J. Increase in cellular glutamate levels stimulates exocytosis in pancreatic β-cells. FEBS Lett. 2002;531:199–203. doi: 10.1016/s0014-5793(02)03500-7. 10.1016/S0014-5793(02)03500-7 [DOI] [PubMed] [Google Scholar]
- Hugnot J.P, Salinas M, Lesage F, Guillemare E, de Weille J, Heurteaux C, Mattei M.G, Lazdunski M. Kv8.1, a new neuronal potassium channel subunit with specific inhibitory properties towards Shab and Shaw channels. EMBO J. 1996;15:3322–3331. [PMC free article] [PubMed] [Google Scholar]
- Hume A.N, Collinson L.M, Hopkins C.R, Strom M, Barral D.C, Bossi G, Griffiths G.M, Seabra M.C. The leaden gene product is required with Rab27a to recruit myosin Va to melanosomes in melanocytes. Traffic. 2002;3:193–202. doi: 10.1034/j.1600-0854.2002.030305.x. 10.1034/j.1600-0854.2002.030305.x [DOI] [PubMed] [Google Scholar]
- Iezzi M, Kouri G, Fukuda M, Wollheim C.B. Synaptotagmin V and IX isoforms control Ca2+-dependent insulin exocytosis. J. Cell Sci. 2004;117:3119–3127. doi: 10.1242/jcs.01179. 10.1242/jcs.01179 [DOI] [PubMed] [Google Scholar]
- Iida Y, Senda T, Matsukawa Y, Onoda K, Miyazaki J.I, Sakaguchi H, Nimura Y, Hidaka H, Niki I. Myosin light-chain phosphorylation controls insulin secretion at a proximal step in the secretory cascade. Am. J. Physiol. 1997;273:E782–E789. doi: 10.1152/ajpendo.1997.273.4.E782. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement J.P, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Wang H, Drewes L.R, Wollheim C.B. Overexpression of monocarboxylate transporter and lactate dehydrogenase alters insulin secretory responses to pyruvate and lactate in beta cells. J. Clin. Invest. 1999;104:1621–1629. doi: 10.1172/JCI7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivarsson R, Obermuller S, Rutter G.A, Galvanovskis J, Renstrom E. Temperature-sensitive random insulin granule diffusion is a prerequisite for recruiting granules for release. Traffic. 2004;5:750–762. doi: 10.1111/j.1600-0854.2004.00216.x. 10.1111/j.1600-0854.2004.00216.x [DOI] [PubMed] [Google Scholar]
- Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, Renstrom E, Schuit F.C. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes. 2005;54:2132–2142. doi: 10.2337/diabetes.54.7.2132. [DOI] [PubMed] [Google Scholar]
- Iynedjian P.B. Mammalian glucokinase and its gene. Biochem. J. 1993;293:1–13. doi: 10.1042/bj2930001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing J, Chikvashvili D, Singer-Lahat D, Thornhill W.B, Reuveny E, Lotan I. Fast inactivation of a brain K+ channel composed of Kv1.1 and Kvβ1.1 subunits modulated by G protein βγ subunits. EMBO J. 1999;18:1245–1256. doi: 10.1093/emboj/18.5.1245. 10.1093/emboj/18.5.1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X, et al. Cav2.3 calcium channels control second-phase insulin release. J. Clin. Invest. 2005;115:146–154. doi: 10.1172/JCI22518. 10.1172/JCI200522518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S.A, Monck J.R, Weiss J.N, Ribalet B. The sulphonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells (HEK 293) J. Physiol. 1998;510:333–345. doi: 10.1111/j.1469-7793.1998.333bk.x. 10.1111/j.1469-7793.1998.333bk.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S.E. The importance of the β-cell in the pathogenesis of type 2 diabetes mellitus. Am. J. Med. 2000;108(Suppl. 6a):2S–8S. doi: 10.1016/s0002-9343(00)00336-3. 10.1016/S0002-9343(00)00336-3 [DOI] [PubMed] [Google Scholar]
- Kanatsuna T, et al. Effects of nifedipine on insulin secretion and glucose metabolism in rats and in hypertensive type 2 (non-insulin dependent) diabetics. Arzneimittelforschung. 1985;35:514–517. [PubMed] [Google Scholar]
- Kaplin A.I, Snyder S.H, Linden D.J. Reduced nicotinamide adenine dinucleotide-selective stimulation of inositol 1,4,5-trisphosphate receptors mediates hypoxic mobilization of calcium. J. Neurosci. 1996;16:2002–2011. doi: 10.1523/JNEUROSCI.16-06-02002.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanietz M.G, Caloca M.J, Eroles P, Fujii T, Garcia-Bermejo M.L, Reilly M, Wang H. Pharmacology of the receptors for the phorbol ester tumor promoters: multiple receptors with different biochemical properties. Biochem. Pharmacol. 2000;60:1417–1424. doi: 10.1016/s0006-2952(00)00470-6. 10.1016/S0006-2952(00)00470-6 [DOI] [PubMed] [Google Scholar]
- Keahey H.H, Rajan A.S, Boyd A.E, III, Kunze D.L. Characterization of voltage-dependent Ca2+ channels in β-cell line. Diabetes. 1989;38:188–193. doi: 10.2337/diab.38.2.188. [DOI] [PubMed] [Google Scholar]
- Kelly R.P, Sutton R, Ashcroft F.M. Voltage-activated calcium and potassium currents in human pancreatic β-cells. J. Physiol. 1991;443:175–192. doi: 10.1113/jphysiol.1991.sp018829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy E.D, Maechler P, Wollheim C.B. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells. Diabetes. 1998;47:374–380. doi: 10.2337/diabetes.47.3.374. [DOI] [PubMed] [Google Scholar]
- Kinard T.A, Satin L.S. An ATP-sensitive Cl− channel current that is activated by cell swelling, cAMP, and glyburide in insulin-secreting cells. Diabetes. 1995;44:1461–1466. doi: 10.2337/diab.44.12.1461. [DOI] [PubMed] [Google Scholar]
- Kizer J.S, Vargas-Gordon M, Brendel K, Bressler R. The in vitro inhibition of insulin secretion by diphenylhydantoin. J. Clin. Invest. 1970;49:1942–1948. doi: 10.1172/JCI106413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Noda M, Sharp G.W. Nutrient augmentation of Ca2+-dependent and Ca2+-independent pathways in stimulus-coupling to insulin secretion can be distinguished by their guanosine triphosphate requirements: studies on rat pancreatic islets. Endocrinology. 1998;139:1172–1183. doi: 10.1210/endo.139.3.5859. 10.1210/en.139.3.1172 [DOI] [PubMed] [Google Scholar]
- Koster J.C, Sha Q, Shyng S, Nichols C.G. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. 10.1111/j.1469-7793.1999.019ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laclau M, Lu F, MacDonald M.J. Enzymes in pancreatic islets that use NADP(H) as a cofactor including evidence for a plasma membrane aldehyde reductase. Mol. Cell Biochem. 2001;225:151–160. doi: 10.1023/a:1012238709063. 10.1023/A:1012238709063 [DOI] [PubMed] [Google Scholar]
- Lacy P.E, Finke E.H, Codilla R.C. Cinemicrographic studies on beta granule movement in monolayer culture of islet cells. Lab. Invest. 1975;33:570–576. [PubMed] [Google Scholar]
- Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur. J. Biochem. 1999;259:3–17. doi: 10.1046/j.1432-1327.1999.00043.x. 10.1046/j.1432-1327.1999.00043.x [DOI] [PubMed] [Google Scholar]
- Lebrun P, Malaisse W.J, Herchuelz A. Na+–K+ pump activity and the glucose-stimulated Ca2+-sensitive K+ permeability in the pancreatic B-cell. J. Membr. Biol. 1983;74:67–73. doi: 10.1007/BF01870596. 10.1007/BF01870596 [DOI] [PubMed] [Google Scholar]
- Lee I.S, Hur E.M, Suh B.C, Kim M.H, Koh D.S, Rhee I.J, Ha H, Kim K.T. Protein kinase A- and C-induced insulin release from Ca2+-insensitive pools. Cell Signal. 2003;15:529–537. doi: 10.1016/s0898-6568(02)00137-7. 10.1016/S0898-6568(02)00137-7 [DOI] [PubMed] [Google Scholar]
- LeRoith D. Beta-cell dysfunction and insulin resistance in type 2 diabetes: role of metabolic and genetic abnormalities. Am. J. Med. 2002;113(Suppl. 6A):3S–11S. doi: 10.1016/s0002-9343(02)01276-7. 10.1016/S0002-9343(02)01276-7 [DOI] [PubMed] [Google Scholar]
- Levin S.R, Grodsky G.M, Hagura R, Smith D. Comparison of the inhibitory effects of diphenylhydantoin and diazoxide upon insulin secretion from the isolated perfused pancreas. Diabetes. 1972;21:856–862. doi: 10.2337/diab.21.8.856. [DOI] [PubMed] [Google Scholar]
- Ligon B, Boyd A.E, III, Dunlap K. Class A calcium channel variants in pancreatic islets and their role in insulin secretion. J. Biol. Chem. 1998;273:13 905–13 911. doi: 10.1074/jbc.273.22.13905. 10.1074/jbc.273.22.13905 [DOI] [PubMed] [Google Scholar]
- Litsky M.L, Pfeiffer D.R. Regulation of the mitochondrial Ca2+ uniporter by external adenine nucleotides: the uniporter behaves like a gated channel which is regulated by nucleotides and divalent cations. Biochemistry. 1997;36:7071–7080. doi: 10.1021/bi970180y. 10.1021/bi970180y [DOI] [PubMed] [Google Scholar]
- Lu D, Mulder H, Zhao P, Burgess S.C, Jensen M.V, Kamzolova S, Newgard C.B, Sherry A.D. 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS) Proc. Natl Acad. Sci. USA. 2002;99:2708–2713. doi: 10.1073/pnas.052005699. 10.1073/pnas.052005699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M.J. Evidence for the malate aspartate shuttle in pancreatic islets. Arch. Biochem. Biophys. 1982;213:643–649. doi: 10.1016/0003-9861(82)90594-x. 10.1016/0003-9861(82)90594-X [DOI] [PubMed] [Google Scholar]
- MacDonald M.J. Elusive proximal signals of β-cells for insulin secretion. Diabetes. 1990;39:1461–1466. doi: 10.2337/diab.39.12.1461. [DOI] [PubMed] [Google Scholar]
- MacDonald M.J. Estimates of glycolysis, pyruvate (de)carboxylation, pentose phosphate pathway, and methyl succinate metabolism in incapacitated pancreatic islets. Arch. Biochem. Biophys. 1993;305:205–214. doi: 10.1006/abbi.1993.1413. 10.1006/abbi.1993.1413 [DOI] [PubMed] [Google Scholar]
- MacDonald P.E, Wheeler M.B. Voltage-dependent K+ channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia. 2003;46:1046–1062. doi: 10.1007/s00125-003-1159-8. 10.1007/s00125-003-1159-8 [DOI] [PubMed] [Google Scholar]
- MacDonald M.J, Ammon H.P, Patel T, Steinke J. Failure of 6-aminonicotinamide to inhibit the potentiating effect of leucine and arginine on glucose-induced insulin release in vitro. Diabetologia. 1974;10:761–765. doi: 10.1007/BF01219538. 10.1007/BF01219538 [DOI] [PubMed] [Google Scholar]
- MacDonald P.E, Ha X.F, Wang J, Smukler S.R, Sun A.M, Gaisano H.Y, Salapatek A.M, Backx P.H, Wheeler M.B. Members of the Kv1 and Kv2 voltage-dependent K+ channel families regulate insulin secretion. Mol. Endocrinol. 2001;15:1423–1435. doi: 10.1210/mend.15.8.0685. 10.1210/me.15.8.1423 [DOI] [PubMed] [Google Scholar]
- MacDonald P.E, et al. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic β-cells enhances glucose-dependent insulin secretion. J. Biol. Chem. 2002;277:44 938–44 945. doi: 10.1074/jbc.M205532200. 10.1074/jbc.M205532200 [DOI] [PubMed] [Google Scholar]
- MacDonald P.E, Salapatek A.M, Wheeler M.B. Temperature and redox state dependence of native Kv2.1 currents in rat pancreatic β-cells. J. Physiol. 2003;546:647–653. doi: 10.1113/jphysiol.2002.035709. 10.1113/jphysiol.2002.035709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M.J, Fahien L.A, Brown L.J, Hasan N.M, Buss J.D, Kendrick M.A. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2005;288:E1–15. doi: 10.1152/ajpendo.00218.2004. 10.1152/ajpendo.00218.2004 [DOI] [PubMed] [Google Scholar]
- Maechler P, Wollheim C.B. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402:685–689. doi: 10.1038/45280. 10.1038/45280 [DOI] [PubMed] [Google Scholar]
- Maechler P, Kennedy E.D, Pozzan T, Wollheim C.B. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic β-cells. EMBO J. 1997;16:3833–3841. doi: 10.1093/emboj/16.13.3833. 10.1093/emboj/16.13.3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P, Antinozzi P.A, Wollheim C.B. Modulation of glutamate generation in mitochondria affects hormone secretion in INS-1E beta cells. IUBMB Life. 2000;50:27–31. doi: 10.1080/15216540050176557. [DOI] [PubMed] [Google Scholar]
- Malaisse W.J, van Obberghen E, Devis G, Somers G, Ravazzola M. Dynamics of insulin release and microtubular–microfilamentous system. V. A model for the phasic release of insulin. Eur. J. Clin. Invest. 1974;4:313–318. doi: 10.1111/j.1365-2362.1974.tb00409.x. [DOI] [PubMed] [Google Scholar]
- Malaisse W.J, Sener A, Herchuelz A, Hutton J.C. Insulin release: the fuel hypothesis. Metabolism. 1979;28:373–386. doi: 10.1016/0026-0495(79)90111-2. 10.1016/0026-0495(79)90111-2 [DOI] [PubMed] [Google Scholar]
- Manning Fox J.E, Nichols C.G, Light P.E. Activation of adenosine triphosphate-sensitive potassium channels by acyl coenzyme A esters involves multiple phosphatidylinositol 4,5-bisphosphate-interacting residues. Mol. Endocrinol. 2004;18:679–686. doi: 10.1210/me.2003-0431. 10.1210/me.2003-0431 [DOI] [PubMed] [Google Scholar]
- Matschinsky F.M. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45:223–241. doi: 10.2337/diab.45.2.223. [DOI] [PubMed] [Google Scholar]
- Matthews E.K, Sakamoto Y. Electrical characteristics of pancreatic islet cells. J. Physiol. 1975;246:421–437. doi: 10.1113/jphysiol.1975.sp010897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack T, McCormack K. Shaker K+ channel β subunits belong to an NAD(P)H-dependent oxidoreductase superfamily. Cell. 1994;79:1133–1135. doi: 10.1016/0092-8674(94)90004-3. 10.1016/0092-8674(94)90004-3 [DOI] [PubMed] [Google Scholar]
- Meissner H.P, Schmelz H. Membrane potential of beta-cells in pancreatic islets. Pflug. Arch. 1974;351:195–206. doi: 10.1007/BF00586918. 10.1007/BF00586918 [DOI] [PubMed] [Google Scholar]
- Meissner H.P, Henquin J.C, Preissler M. Potassium dependence of the membrane potential of pancreatic B-cells. FEBS Lett. 1978;94:87–89. doi: 10.1016/0014-5793(78)80912-0. 10.1016/0014-5793(78)80912-0 [DOI] [PubMed] [Google Scholar]
- Meldolesi J, Chieregatti E. Fusion has found its calcium sensor. Nat. Cell Biol. 2004;6:476–478. doi: 10.1038/ncb0604-476. 10.1038/ncb0604-476 [DOI] [PubMed] [Google Scholar]
- Mendez C.F, Leibiger I.B, Leibiger B, Hoy M, Gromada J, Berggren P.O, Bertorello A.M. Rapid association of protein kinase C-epsilon with insulin granules is essential for insulin exocytosis. J. Biol. Chem. 2003;278:44 753–44 757. doi: 10.1074/jbc.M308664200. 10.1074/jbc.M308664200 [DOI] [PubMed] [Google Scholar]
- Mertz R.J, Worley J.F, Spencer B, Johnson J.H, Dukes I.D. Activation of stimulus-secretion coupling in pancreatic β-cells by specific products of glucose metabolism. Evidence for privileged signaling by glycolysis. J. Biol. Chem. 1996;271:4838–4845. doi: 10.1074/jbc.271.9.4838. 10.1074/jbc.271.9.4838 [DOI] [PubMed] [Google Scholar]
- Michelakis E.D, Thebaud B, Weir E.K, Archer S.L. Hypoxic pulmonary vasoconstriction: redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J. Mol. Cell. Cardiol. 2004;37:1119–1136. doi: 10.1016/j.yjmcc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Mikhailov M.V, Proks P, Ashcroft F.M, Ashcroft S.J. Expression of functionally active ATP-sensitive K-channels in insect cells using baculovirus. FEBS Lett. 1998;429:390–394. doi: 10.1016/s0014-5793(98)00640-1. 10.1016/S0014-5793(98)00640-1 [DOI] [PubMed] [Google Scholar]
- Milner R.D, Hales C.N. Cations and the secretion of insulin. Biochim. Biophys. Acta. 1968;150:165–167. doi: 10.1016/0005-2736(68)90022-9. [DOI] [PubMed] [Google Scholar]
- Mizuta M, Kurose T, Miki T, Shoji-Kasai Y, Takahashi M, Seino S, Matsukura S. Localization and functional role of synaptotagmin III in insulin secretory vesicles in pancreatic β-cells. Diabetes. 1997;46:2002–2006. doi: 10.2337/diab.46.12.2002. [DOI] [PubMed] [Google Scholar]
- Moreno-Sanchez R. Regulation of oxidative phosphorylation in mitochondria by external free Ca2+ concentrations. J. Biol. Chem. 1985;260:4028–4034. [PubMed] [Google Scholar]
- Mulder H, Lu D, Finley J, An J, Cohen J, Antinozzi P.A, McGarry J.D, Newgard C.B. Overexpression of a modified human malonyl-CoA decarboxylase blocks the glucose-induced increase in malonyl-CoA level but has no impact on insulin secretion in INS-1-derived (832/13) β-cells. J. Biol. Chem. 2001;276:6479–6484. doi: 10.1074/jbc.M010364200. 10.1074/jbc.M010364200 [DOI] [PubMed] [Google Scholar]
- Nevins A.K, Thurmond D.C. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am. J. Physiol. Cell Physiol. 2003;285:C698–C710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- Newgard C.B, McGarry J.D. Metabolic coupling factors in pancreatic β-cell signal transduction. Annu. Rev. Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. 10.1146/annurev.bi.64.070195.003353 [DOI] [PubMed] [Google Scholar]
- Niki I. Ca2+ signaling and the insulin secretory cascade in the pancreatic beta-cell. Jpn J. Pharmacol. 1999;80:191–197. doi: 10.1254/jjp.80.191. 10.1254/jjp.80.191 [DOI] [PubMed] [Google Scholar]
- Niwa T, Matsukawa Y, Senda T, Nimura Y, Hidaka H, Niki I. Acetylcholine activates intracellular movement of insulin granules in pancreatic β-cells via inositol trisphosphate-dependent mobilization of intracellular Ca2+ Diabetes. 1998;47:1699–1706. doi: 10.2337/diabetes.47.11.1699. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. 10.1038/305147a0 [DOI] [PubMed] [Google Scholar]
- Ohara-Imaizumi M, Nakamichi Y, Tanaka T, Ishida H, Nagamatsu S. Imaging exocytosis of single insulin secretory granules with evanescent wave microscopy: distinct behavior of granule motion in biphasic insulin release. J. Biol. Chem. 2002;277:3805–3808. doi: 10.1074/jbc.C100712200. 10.1074/jbc.C100712200 [DOI] [PubMed] [Google Scholar]
- Ohara-Imaizumi M, Nishiwaki C, Kikuta T, Nagai S, Nakamichi Y, Nagamatsu S. TIRF imaging of docking and fusion of single insulin granule motion in primary rat pancreatic β-cells: different behaviour of granule motion between normal and Goto-Kakizaki diabetic rat β-cells. Biochem. J. 2004;381:13–18. doi: 10.1042/BJ20040434. 10.1042/BJ20040434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohneda A, Kobayashi T, Nihei J. Effects of Ca antagonists, nifedipine, niludipine and verapamil, on endocrine function of the pancreas. Tohoku J. Exp. Med. 1983;140:153–159. doi: 10.1620/tjem.140.153. [DOI] [PubMed] [Google Scholar]
- Ohta M, Nelson J, Nelson D, Meglasson M.D, Erecinska M. Effect of Ca2+ channel blockers on energy level and stimulated insulin secretion in isolated rat islets of Langerhans. J. Pharmacol. Exp. Ther. 1993;264:35–40. [PubMed] [Google Scholar]
- Olofsson C.S, Gopel S.O, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic β-cells. Pflug. Arch. 2002;444:43–51. doi: 10.1007/s00424-002-0781-5. 10.1007/s00424-002-0781-5 [DOI] [PubMed] [Google Scholar]
- Olofsson C.S, Salehi A, Holm C, Rorsman P. Palmitate increases L-type Ca2+ currents and the size of the readily releasable granule pool in mouse pancreatic β-cells. J. Physiol. 2004;557:935–948. doi: 10.1113/jphysiol.2004.066258. 10.1113/jphysiol.2004.066258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen H.L, et al. Phosphatidylinositol 4-kinase serves as a metabolic sensor and regulates priming of secretory granules in pancreatic β cells. Proc. Natl Acad. Sci. USA. 2003;100:5187–5192. doi: 10.1073/pnas.0931282100. 10.1073/pnas.0931282100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Gabbay K.H, Malaisse W.J. Pancreatic beta-cell web: its possible role in insulin secretion. Science. 1972;175:1128–1130. doi: 10.1126/science.175.4026.1128. [DOI] [PubMed] [Google Scholar]
- Owada S, Larsson O, Arkhammar P, Katz A.I, Chibalin A.V, Berggren P.O, Bertorello A.M. Glucose decreases Na+, K+-ATPase activity in pancreatic β-cells. An effect mediated via Ca2+-independent phospholipase A2 and protein kinase C-dependent phosphorylation of the α-subunit. J. Biol. Chem. 1999;274:2000–2008. doi: 10.1074/jbc.274.4.2000. 10.1074/jbc.274.4.2000 [DOI] [PubMed] [Google Scholar]
- Pace C.S, Price S. Electrical responses of pancreatic islet cells to secretory stimuli. Biochem. Biophys. Res. Commun. 1972;46:1557–1563. doi: 10.1016/0006-291x(72)90785-1. 10.1016/0006-291X(72)90785-1 [DOI] [PubMed] [Google Scholar]
- Pallavi B, Nagaraj R. Palmitoylated peptides from the cysteine-rich domain of SNAP-23 cause membrane fusion depending on peptide length, position of cysteines, and extent of palmitoylation. J. Biol. Chem. 2003;278:12 737–12 744. doi: 10.1074/jbc.M208598200. 10.1074/jbc.M208598200 [DOI] [PubMed] [Google Scholar]
- Peri R, Wible B.A, Brown A.M. Mutations in the Kvβ2 binding site for NADPH and their effects on Kv1.4. J. Biol. Chem. 2001;276:738–741. doi: 10.1074/jbc.M008445200. 10.1074/jbc.M008445200 [DOI] [PubMed] [Google Scholar]
- Philipson L.H, Steiner D.F. Pas de deux or more: the sulfonylurea receptor and K+ channels. Science. 1995;268:372–373. doi: 10.1126/science.7716539. [DOI] [PubMed] [Google Scholar]
- Pinton P, Tsuboi T, Ainscow E.K, Pozzan T, Rizzuto R, Rutter G.A. Dynamics of glucose-induced membrane recruitment of protein kinase C β II in living pancreatic islet β-cells. J. Biol. Chem. 2002;277:37 702–37 710. doi: 10.1074/jbc.M204478200. 10.1074/jbc.M204478200 [DOI] [PubMed] [Google Scholar]
- Porter J.R, Barrett T.G. Monogenic syndromes of abnormal glucose homeostasis: clinical review and relevance to the understanding of the pathology of insulin resistance and β-cell failure. J. Med. Genet. 2005 doi: 10.1136/jmg.2005.030791. 10.1136/jmg.2005.030791 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouli A.E, Emmanouilidou E, Zhao C, Wasmeier C, Hutton J.C, Rutter G.A. Secretory-granule dynamics visualized in vivo with a phogrin-green fluorescent protein chimaera. Biochem. J. 1998;333:193–199. doi: 10.1042/bj3330193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralong W.F, Bartley C, Wollheim C.B. Single islet beta-cell stimulation by nutrients: relationship between pyridine nucleotides, cytosolic Ca2+ and secretion. EMBO J. 1990;9:53–60. doi: 10.1002/j.1460-2075.1990.tb08079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Vischer S, Glennon M.C, Regazzi R, Deeney J.T, Corkey B.E. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J. Biol. Chem. 1992;267:5802–5810. [PubMed] [Google Scholar]
- Pressel D.M, Misler S. Sodium channels contribute to action potential generation in canine and human pancreatic islet B cells. J. Membr. Biol. 1990;116:273–280. doi: 10.1007/BF01868466. 10.1007/BF01868466 [DOI] [PubMed] [Google Scholar]
- Ravier M.A, Gilon P, Henquin J.C. Oscillations of insulin secretion can be triggered by imposed oscillations of cytoplasmic Ca2+ or metabolism in normal mouse islets. Diabetes. 1999;48:2374–2382. doi: 10.2337/diabetes.48.12.2374. [DOI] [PubMed] [Google Scholar]
- Regazzi R, Li G, Ullrich S, Jaggi C, Wollheim C.B. Different requirements for protein kinase C activation and Ca2+-independent insulin secretion in response to guanine nucleotides. Endogenously generated diacylglycerol requires elevated Ca2+ for kinase C insertion into membranes. J. Biol. Chem. 1989;264:9939–9944. [PubMed] [Google Scholar]
- Renstrom E, Ivarsson R, Shears S.B. Inositol 3,4,5,6-tetrakisphosphate inhibits insulin granule acidification and fusogenic potential. J. Biol. Chem. 2002;277:26 717–26 720. doi: 10.1074/jbc.C200314200. 10.1074/jbc.C200314200 [DOI] [PubMed] [Google Scholar]
- Rettig J, Heinemann S.H, Wunder F, Lorra C, Parcej D.N, Dolly J.O, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. 10.1038/369289a0 [DOI] [PubMed] [Google Scholar]
- Rorsman P. The pancreatic beta-cell as a fuel sensor: an electrophysiologist's viewpoint. Diabetologia. 1997;40:487–495. doi: 10.1007/s001250050706. 10.1007/s001250050706 [DOI] [PubMed] [Google Scholar]
- Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. 10.1007/s00125-003-1153-1 [DOI] [PubMed] [Google Scholar]
- Rorsman P, Trube G. Glucose dependent K+-channels in pancreatic beta-cells are regulated by intracellular ATP. Pflug. Arch. 1985;405:305–309. doi: 10.1007/BF00595682. 10.1007/BF00595682 [DOI] [PubMed] [Google Scholar]
- Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic beta-cells under voltage-clamp conditions. J. Physiol. 1986;374:531–550. doi: 10.1113/jphysiol.1986.sp016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Eliasson L, Renstrom E, Gromada J, Barg S, Gopel S. The cell physiology of biphasic insulin secretion. News Physiol. Sci. 2000;15:72–77. doi: 10.1152/physiologyonline.2000.15.2.72. [DOI] [PubMed] [Google Scholar]
- Ruppersberg J.P, Stocker M, Pongs O, Heinemann S.H, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IKA channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. 10.1038/352711a0 [DOI] [PubMed] [Google Scholar]
- Rutter G.A, da Silva Xavier G, Leclerc I. Roles of 5′-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem. J. 2003;375:1–16. doi: 10.1042/BJ20030048. 10.1042/BJ20030048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Ammala C, Smith P.A, Gribble F.M, Ashcroft F.M. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. 10.1016/0014-5793(95)01369-5 [DOI] [PubMed] [Google Scholar]
- Salinas M, de Weille J, Guillemare E, Lazdunski M, Hugnot J.P. Modes of regulation of shab K+ channel activity by the Kv8.1 subunit. J. Biol. Chem. 1997a;272:8774–8780. doi: 10.1074/jbc.272.13.8774. 10.1074/jbc.272.39.24371 [DOI] [PubMed] [Google Scholar]
- Salinas M, Duprat F, Heurteaux C, Hugnot J.P, Lazdunski M. New modulatory alpha subunits for mammalian Shab K+ channels. J. Biol. Chem. 1997b;272:24 371–24 379. doi: 10.1074/jbc.272.39.24371. 10.1074/jbc.272.39.24371 [DOI] [PubMed] [Google Scholar]
- Satin L.S, Cook D.L. Voltage-gated Ca2+ current in pancreatic B-cells. Pflug. Arch. 1985;404:385–387. doi: 10.1007/BF00585354. 10.1007/BF00585354 [DOI] [PubMed] [Google Scholar]
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, Prentki M. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in β cells. J. Biol. Chem. 1997;272:18 572–18 579. doi: 10.1074/jbc.272.30.18572. 10.1074/jbc.272.30.18572 [DOI] [PubMed] [Google Scholar]
- Sehlin J, Taljedal I.B. Glucose-induced decrease in Rb+ permeability in pancreatic beta cells. Nature. 1975;253:635–636. doi: 10.1038/253635a0. 10.1038/253635a0 [DOI] [PubMed] [Google Scholar]
- Sekine N, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic β-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994;269:4895–4902. [PubMed] [Google Scholar]
- Sener A, Malaisse W.J. Hexose metabolism in pancreatic islets. Ca2+-dependent activation of the glycerol phosphate shuttle by nutrient secretagogues. J. Biol. Chem. 1992;267:13 251–13 256. [PubMed] [Google Scholar]
- Sener A, Kawazu S, Hutton J.C, Boschero A.C, Devis G, Somers G, Herchuelz A, Malaisse W.J. The stimulus-secretion coupling of glucose-induced insulin release. Effect of exogenous pyruvate on islet function. Biochem. J. 1978;176:217–232. doi: 10.1042/bj1760217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemori K, Ishizaki T, Matsukawa S, Sakai A, Nakai T, Miyabo S. Adenine nucleotides via activation of ATP-sensitive K+ channels modulate hypoxic response in rat pulmonary artery. Am. J. Physiol. 1996;270:L803–L809. doi: 10.1152/ajplung.1996.270.5.L803. [DOI] [PubMed] [Google Scholar]
- Smith P.A, Rorsman P, Ashcroft F.M. Modulation of dihydropyridine-sensitive Ca2+ channels by glucose metabolism in mouse pancreatic beta-cells. Nature. 1989;342:550–553. doi: 10.1038/342550a0. 10.1038/342550a0 [DOI] [PubMed] [Google Scholar]
- Sollner T.H. Regulated exocytosis and SNARE function. Mol. Membr. Biol. 2003;20:209–220. doi: 10.1080/0968768031000104953. 10.1080/0968768031000104953 [DOI] [PubMed] [Google Scholar]
- Somers G, Blondel B, Orci L, Malaisse W.J. Motile events in pancreatic endocrine cells. Endocrinology. 1979;104:255–264. doi: 10.1210/endo-104-1-255. [DOI] [PubMed] [Google Scholar]
- Stocker M, Kerschensteiner D. Cloning and tissue distribution of two new potassium channel α-subunits from rat brain. Biochem. Biophys. Res. Commun. 1998;248:927–934. doi: 10.1006/bbrc.1998.9072. 10.1006/bbrc.1998.9072 [DOI] [PubMed] [Google Scholar]
- Straub S.G, Shanmugam G, Sharp G.W. Stimulation of insulin release by glucose is associated with an increase in the number of docked granules in the β-cells of rat pancreatic islets. Diabetes. 2004;53:3179–3183. doi: 10.2337/diabetes.53.12.3179. [DOI] [PubMed] [Google Scholar]
- Territo P.R, French S.A, Dunleavy M.C, Evans F.J, Balaban R.S. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, AND light scattering. J. Biol. Chem. 2001;276:2586–2599. doi: 10.1074/jbc.M002923200. 10.1074/jbc.M002923200 [DOI] [PubMed] [Google Scholar]
- Tipparaju S.M, Saxena N, Liu S.Q, Kumar R, Bhatnagar A. Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am. J. Physiol. Cell Physiol. 2005;288:C366–C376. doi: 10.1152/ajpcell.00354.2004. 10.1152/ajpcell.00354.2004 [DOI] [PubMed] [Google Scholar]
- Tsuboi T, da Silva Xavier G, Leclerc I, Rutter G.A. 5′-AMP-activated protein kinase controls insulin-containing secretory vesicle dynamics. J. Biol. Chem. 2003;278:52 042–52 051. doi: 10.1074/jbc.M307800200. 10.1074/jbc.M307800200 [DOI] [PubMed] [Google Scholar]
- Tucker S.J, Gribble F.M, Zhao C, Trapp S, Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. 10.1038/387179a0 [DOI] [PubMed] [Google Scholar]
- Tucker S.J, Gribble F.M, Proks P, Trapp S, Ryder T.J, Haug T, Reimann F, Ashcroft F.M. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. 10.1093/emboj/17.12.3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Ainscow E.K, Allan V.J, Rutter G.A. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic β-cells. J. Cell Sci. 2002;115:4177–4189. doi: 10.1242/jcs.00083. 10.1242/jcs.00083 [DOI] [PubMed] [Google Scholar]
- Varadi A, Tsuboi T, Johnson-Cadwell L.I, Allan V.J, Rutter G.A. Kinesin I and cytoplasmic dynein orchestrate glucose-stimulated insulin-containing vesicle movements in clonal MIN6 β-cells. Biochem. Biophys. Res. Commun. 2003;311:272–282. doi: 10.1016/j.bbrc.2003.09.208. 10.1016/j.bbrc.2003.09.208 [DOI] [PubMed] [Google Scholar]
- Varadi A, Tsuboi T, Rutter G.A. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 β-cells. Mol. Biol. Cell. 2005;16:2670–2680. doi: 10.1091/mbc.E04-11-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco J.M, Petersen O.H. The effect of a cell-permeable diacylglycerol analogue on single Ca2+ (Ba2+) channel currents in the insulin-secreting cell line RINm5F. Q. J. Exp. Physiol. 1989;74:367–370. doi: 10.1113/expphysiol.1989.sp003280. [DOI] [PubMed] [Google Scholar]
- Waselle L, Coppola T, Fukuda M, Iezzi M, El Amraoui A, Petit C, Regazzi R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol. Biol. Cell. 2003;14:4103–4113. doi: 10.1091/mbc.E03-01-0022. 10.1091/mbc.E03-01-0022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J.N, Lamp S.T. Cardiac ATP-sensitive K+ channels. Evidence for preferential regulation by glycolysis. J. Gen. Physiol. 1989;94:911–935. doi: 10.1085/jgp.94.5.911. 10.1085/jgp.94.5.911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wang F, Rao K, Sellers J.R, Hammer J.A., III Rab27a is an essential component of melanosome receptor for myosin Va. Mol. Biol. Cell. 2002;13:1735–1749. doi: 10.1091/mbc.01-12-0595. 10.1091/mbc.01-12-0595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt C.N, Weir E.K, Peers C. Diphenylene iodonium blocks K+ and Ca2+ currents in type I cells isolated from the neonatal rat carotid body. Neurosci. Lett. 1994;172:63–66. doi: 10.1016/0304-3940(94)90663-7. 10.1016/0304-3940(94)90663-7 [DOI] [PubMed] [Google Scholar]
- Yamada S, Komatsu M, Sato Y, Yamauchi K, Kojima I, Aizawa T, Hashizume K. Time-dependent stimulation of insulin exocytosis by 3′,5′-cyclic adenosine monophosphate in the rat islet β-cell. Endocrinology. 2002;143:4203–4209. doi: 10.1210/en.2002-220368. 10.1210/en.2002-220368 [DOI] [PubMed] [Google Scholar]
- Yang Y, Gillis K.D. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J. Gen. Physiol. 2004;124:641–651. doi: 10.1085/jgp.200409081. 10.1085/jgp.200409081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoshiki H, Katsube Y, Sperelakis N. Regulation of Ca2+ channel currents by intracellular ATP in smooth muscle cells of rat mesenteric artery. Am. J. Physiol. 1997;272:H814–H819. doi: 10.1152/ajpheart.1997.272.2.H814. [DOI] [PubMed] [Google Scholar]
- Zaitsev S.V, Efendic S, Arkhammar P, Bertorello A.M, Berggren P.O. Dissociation between changes in cytoplasmic free Ca2+ concentration and insulin secretion as evidenced from measurements in mouse single pancreatic islets. Proc. Natl Acad. Sci. USA. 1995;92:9712–9716. doi: 10.1073/pnas.92.21.9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawalich W.S, Zawalich K.C. Influence of pyruvic acid methyl ester on rat pancreatic islets. Effects on insulin secretion, phosphoinositide hydrolysis, and sensitization of the beta cell. J. Biol. Chem. 1997;272:3527–3531. doi: 10.1074/jbc.272.6.3527. 10.1074/jbc.272.6.3527 [DOI] [PubMed] [Google Scholar]
- Zhao C, Wilson M.C, Schuit F, Halestrap A.P, Rutter G.A. Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes. 2001;50:361–366. doi: 10.2337/diabetes.50.2.361. [DOI] [PubMed] [Google Scholar]
- Zima A.V, Copello J.A, Blatter L.A. Differential modulation of cardiac and skeletal muscle ryanodine receptors by NADH. FEBS Lett. 2003;547:32–36. doi: 10.1016/s0014-5793(03)00664-1. 10.1016/S0014-5793(03)00664-1 [DOI] [PubMed] [Google Scholar]