

Abstract
Amyloid beta peptide (Aβ) accumulates in the CNS in Alzheimer's disease. Both the full peptide (1–42) or the 25–35 fragment are toxic to neurons in culture. We have used fluorescence imaging technology to explore the mechanism of neurotoxicity in mixed asytrocyte/neuronal cultures prepared from rat or mouse cortex or hippocampus, and have found that Aβ acts preferentially on astrocytes but causes neuronal death. Aβ causes sporadic transient increases in [Ca2+]c in astrocytes, associated with a calcium dependent increased generation of reactive oxygen species (ROS) and glutathione depletion. This caused a slow dissipation of mitochondrial potential on which abrupt calcium dependent transient depolarizations were superimposed. The mitochondrial depolarization was reversed by mitochondrial substrates glutamate, pyruvate or methyl succinate, and by NADPH oxidase (NOX) inhibitors, suggesting that it reflects oxidative damage to metabolic pathways upstream of mitochondrial complex I. The Aβ induced increase in ROS and the mitochondrial depolarization were absent in cells cultured from transgenic mice lacking the NOX component, gp91phox. Neuronal death after 24 h of Aβ exposure was dramatically reduced both by NOX inhibitors and in gp91phox knockout mice. Thus, by raising [Ca2+]c in astrocytes, Aβ activates NOX, generating oxidative stress that is transmitted to neurons, causing neuronal death.
Keywords: amyloid, Alzheimer, NADPH oxidase, intracellular calcium, glutathione
1. Introduction
The accumulation of amyloid beta (Aβ) peptides in the neurofibrillary plaques of Alzheimer's disease (AD) is one of the diagnostic features of the disease, and Aβ peptides are strongly associated with the neuronal death and cognitive deficit associated with this appalling illness (Näslund et al. 2000). While there remains substantial controversy about the specific role played by Aβ in AD pathology, it is clear that the accumulation of the peptide alone is sufficient to cause dementia. Indeed, all of the known mutations which associate with familial AD lie within genes that are involved in processing Aβ—genes for amyloid precursor protein (APP) or for the presenilins, enzymes involved in the processing of APP—all of which result in Aβ accumulation (Tanzi et al. 1996). It is also well established experimentally that Aβ itself is neurotoxic, and the mechanism of that neurotoxicity is clearly of profound interest.
We have recently attempted to explore the mechanisms of Aβ neurotoxicity in a culture model in which neurons grow together with astrocytes, and we here review some of our recent findings and also present some new observations. In the context of this special issue, there is substantial evidence that places oxidative stress centre stage in AD and that associates Aβ neurotoxicity specifically with oxidative stress, although as with almost all studies in this field, the mechanisms remain controversial (see for example, Canevari et al. 2004 for a recent review).
2. Aβ and intracellular calcium signals
We have applied Aβ to primary cultures of neurons and astrocytes prepared from either the hippocampus or cortex of neonatal rats. Using fluorescent imaging techniques and using the calcium indicator fura-2 to measure intracellular calcium ([Ca2+]c), we have found that both the full peptide Aβ 1–42 and the short neurotoxic peptide fragment Aβ 25–35 caused sporadic [Ca2+]c signals in astrocytes after a delay which is usually of the order of 5–10 min (figure 1). The non-toxic reverse peptide Aβ 35–25 was routinely used as a control in all experiments we report here and was invariably without effect. Remarkably, intracellular calcium signals were confined to the astrocytes while adjacent neurons remained silent. The [Ca2+]c signals were modest in amplitude—for example, only very small signals were apparent when using the low calcium indicator fura-ff, suggesting that signals do not rise above ∼1μM, and signals could continue for many hours. The [Ca2+]c signals were highly variable in duration and frequency, but typically consisted of reversible transient increases in [Ca2+]c with durations of a few minutes (see figure 1). These signals were entirely dependent on extracellular calcium, were apparently independent of intracellular calcium stores and seem to be attributable to calcium influx through an Aβ mediated channel in the astrocyte membrane. Thus, a screen of channel inhibitors failed to have any major impact on the response and so we think it most likely that Aβ itself forms calcium permeant channels in the astrocyte membrane, a pathogenic mechanism proposed by (Arispe et al. 1993; Bhatia et al. 2000; Abramov et al. 2003).
Figure 1.

Aβ causes transient calcium signals, transient mitochondrial depolarizations, and a slow progressive loss of mitochondrial potential in mouse astrocytes. A culture of hippocampal cortical astrocytes was co-loaded with fura-2 (AM ester, 5 μM for 20 min) and with rhodamine 123 (100 μM for 15 min followed by washing). Application of Aβ 1–42 (2 μM) as indicated initiated transient calcium responses and also transient mitochondrial depolarizations which were superimposed on a slow progressive loss of mitochondrial potential. (a) Signals from many cells are superimposed. (b) Signals from just two cells are extracted so that individual cellular responses can be seen more clearly. These cellular responses in mouse astrocytes were not notably different from those we have described in cells from the rat (n=67 cells).
In fact, it was established in the early 1990s that Aβ peptides can form channels in artificial planar lipid membranes. These are cationic channels showing a selectivity Cs+>Li+>Ca2+=K+ and there are substantial data suggesting that these channels may mediate calcium influx into cells in response to Aβ. The channels show sporadic activity and seem capable of generating a number of different conductance states (Arispe et al. 1993). The peptide inserts into the membranes of lipid vesicles, where it can be visualized through immunofluorescence (Rhee et al. 1998; Lin et al. 2001) and where it forms a calcium permeable channel. The channel is blocked by zinc, although the action of zinc is confusing, as it can also complex with Aβ, prevent aggregation and so prevent insertion into the membrane or prevent pore formation rather than blocking the formed channel (see Arispe et al. 1996; Abramov et al. 2003; Bush 2003). In our hands also, zinc could block the calcium response in astrocytes if applied before Aβ, but could not stop calcium signals once established. The channels are formed by Aβ 25–35 or 1–40, but not by the reverse peptide, 35–25, confirming that this is specific to the toxic peptides and is not a non-specific general disruption of membrane structure by addition of peptides.
The calcium signals in astrocytes were blocked by the drug clioquinol, a heavy metal chelator which has been used as an antimalarial agent, and that acts to prevent Aβ aggregation and so prevents its insertion into membranes (see Abramov et al. 2003). Encouragingly, perhaps, clioquinol has been shown to improve defects in transgenic models of AD and is being used currently in clinical trials in patients (Cherny et al. 2001; Helmuth 2002; Melov 2002), possibly putting our observations into a clinical context.
A very surprising and rather puzzling aspect to this model remains the selectivity of the effect of Aβ for astrocytes while having negligible effects on [Ca2+]c signals in adjacent neurons. If Aβ acts by inserting into lipid bilayers and forming a channel, why should there be a difference between different cell types? The answer may lie in the lipid composition of the cell membranes, as the pore forming activity of the peptide in lipid bilayers appears to be critically dependent on the composition of the lipid membrane, particularly in terms of the cholesterol content. The pore forming activity of Aβ in bilayers appears to be inversely related to the cholesterol content of the lipid mixture, and depletion of cholesterol using agents such as cyclodextrin or inhibition of cholesterol synthesis in PC12 cells increases Aβ toxicity (Arispe & Doh 2002). Similarly, Kawahara & Kuroda (2001) showed that the [Ca2+]c increase in cells was attenuated by pre-treament of the cells to increase membrane cholesterol content. We have no data at present about the differences in cholesterol content of different cell types in the CNS, but this seems a simple mechanism that might account for the differences in the responses of different cell types that we have described.
Surprisingly, we found that after 18–24 h, Aβ caused significant neuronal death but only modest death among astrocytes (Abramov et al. 2003). Neuronal death was calcium dependent and was blocked by clioquinol, providing a direct link with the astrocytic calcium signals. We have then tried to address the issue of how calcium signals in astrocytes might lead to neuronal death. One possibility is that astrocytes might release glutamate and so cause glutamate mediated excitotoxicity. This seems unlikely as we did not see neuronal calcium signals following the astrocytic signals as would be expected for such a mechanism. Furthermore, neurons were not protected by glutamate antagonists (see Abramov et al. 2004b), strongly suggesting that this is not a major mechanism in this model.
3. Effects of Aβ on mitochondrial function in astrocytes
As cells were dying, we also chose to monitor mitochondrial function in the cultures, using the lipophilic cationic dye rhodamine123, which acts as a reliable indicator of changing mitochondrial potential (Δψm). Essentially, Rh123 partitions selectively into mitochondria in response to their large membrane potentials. A loss of potential causes the redistribution of the dye into the cytosol. If the dye is loaded above a critical ‘quench’ threshold, the fluorescence signal from the dye concentrated within polarized mitochondria is less than expected for the dye concentration, because of a phenomenon known as autoquenching. Redistribution of the dye into the cytosol in this instance causes an increase in fluorescence signal which, therefore, reflects a loss of mitochondrial potential (figure 2).
Figure 2.

The changes in mitochondrial potential caused by Aβ were attributable to the activation of an NADPH oxidase. (a) The NADPH oxidase inhibitor apocynin (1 mM) prevented any significant change in mitochondrial potential in response to Aβ but had no effect at all on the Aβ induced calcium signals (n=84 cells) in mouse cortical astrocytes. Similar responses were seen to the other NADPH oxidase inhibitors AEBSF (20 μM, n=44 cells) and DPI (0.5 μM; n=111 cells). (b) Astrocytes cultured from the cortex of transgenic mice with knockout of gp91phox expression also showed no mitochondrial response to Aβ while the calcium response was unaffected (n=97 cells).
In response to Aβ, we saw complex changes in Δψm in astrocytes and not in neurons. These changes consisted of apparently two separate components—a slow progressive loss of potential on which were superimposed large transient depolarizations the like of which we have never seen before. These could be very large abrupt depolarizing transients that recovered over the space of a few minutes (figure 1). That the recovery of signal was a genuine mitochondrial repolarization and not simply the loss of dye from the cell was demonstrated by application of the uncoupler FCCP at the end of the experiment to dissipate Δψm and so release any dye retained within the mitochondria—clearly a response will only be seen if mitochondria repolarized and reaccumulated dye (see figure 1b). Large transient depolarizations were seen in a large proportion but not all cells, generally correlated with the calcium transients, and were abolished in the absence of external calcium, leaving a slow progressive loss of potential—developing over tens of minutes or hours—largely unchanged. In some cells, only the slow depolarization was seen. The large transient responses appear to reflect transient openings of the mitochondrial permeability transition pore, and were blocked by ciclosporin A, especially when used in combination with antioxidants (see below and Abramov et al. 2003).
We have found previously that 24 h exposure of neuronal cultures causes a loss of mitochondrial respiratory chain activity (Casley et al. 2002a). We thought, therefore, that the slow depolarization might reflect a failure of mitochondrial respiration. However, we found that giving the cells additional substrates that enter at any point along the respiratory chain could reverse or prevent the loss of mitochondrial potential (see figure 3). These included methyl-succinate, a membrane permeant form of succinate which enters at complex II, and glutamate or pyruvate, which supply electrons to complex I. Therefore, the defect caused by Aβ in astrocytes in the short term must be upstream from complex I and may reflect changes in glycolysis or even in glucose uptake.
Figure 3.
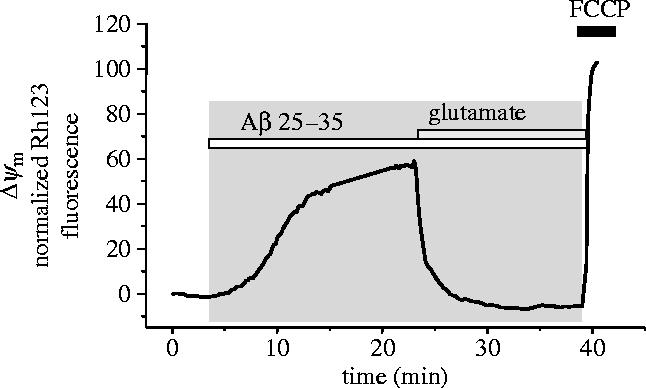
The slow loss of mitochondrial potential was reversed by mitochondrial substrate. In some cells, the slow progressive collapse of mitochondrial potential was the dominant response to Aβ. This loss of potential was reversed by provision of mitochondrial substrate, in the case illustrated, by glutamate (1 mM).
4. Aβ and oxidative stress
There is an extensive literature suggesting that Aβ causes oxidative stress which we have reviewed recently (see Canevari et al. 2004). Thus, many of the downstream changes in cellular metabolism that follow Aβ exposure and also post-mortem changes in enzyme activities seen in AD patients are also generated by exposure of tissues to oxygen free radical donors. In particular, changes have been described in the activities of aconitase and α-ketoglutarate dehydrogenase, enzymes with iron–sulphur centres (Blass & Gibson 1991; Longo et al. 2000; Casley et al. 2002b), in combination with widespread evidence of lipid peroxidation and glutathione depletion. Sources of oxidative stress that have been suggested include mitochondria and activation of microglia with concomitant activation of the microglial NADPH oxidase. It has even been suggested that Aβ alone can generate oxygen radical species in solution possibly through a reaction of Aβ with trace levels of Cu2+.
We, therefore, examined glutathione status in our culture model following exposure to Aβ. We used monochlorobimane as a semi-quantitative measure of GSH status in the mixed cultures. This is a non-fluorescent compound which is conjugated to GSH through a reaction catalysed by glutathione-S-transferase and so gives a ‘snapshot’ of GSH content (Keelan et al. 2001). In mixed cultures, confocal imaging with a UV laser allows measurements of the GSH levels in both neurons and astrocytes side by side in the same culture, a differentiation aided by the tendency of neurons to grow in a focal plane above that of the astrocyte layer. This kind of analysis would be quite impossible using other biochemical approaches and permits direct measurements in each cell type in the mixed culture (see also Canevari et al. 2004). Both Aβ 1–42 and 25–35 caused significant depletion of GSH in both astrocytes and neurons—the first sign of any pathological change that we could detect in neurons—at 24 h after exposure to Aβ. The GSH depletion was calcium dependent in both neurons and astrocytes, even though no changes in calcium were detectable in neurons, suggesting a direct link with the calcium signals that we had described in the astrocytes. Furthermore, neuronal cell death could be reduced significantly by supplementing the antioxidant defences of the cells by giving GSH precursor γ-glutamyl cysteine (see Abramov et al. 2004a), which increases cellular GSH levels.
Together these data strongly suggest that the astrocyte calcium signals initiate an oxidative stress that is somehow transmitted to the neurons. The astrocyte mitochondria and astrocytic intermediary metabolism is also a target of the oxidative stress, but the neurons seem more vulnerable and die. These considerations led us to measure free radical generation from the astrocytes directly and to seek the source of the oxygen radical generation. Direct measurements of reactive oxygen species (ROS) generation from the cultures using a variety of different probes, including hydroethidine or difluoro-dicarboxyfluorescein, both non-fluorescent derivatives of fluorescent compounds that are oxidized to fluorescent molecules by oxygen free radical species, showed that Aβ increases the rate of ROS generation in astrocytes (figure 4a; see Abramov et al. 2004a). This was consistent in pure astrocyte cultures and was clearly not a reflection of the production of ROS from occasional microglial contaminants, as it was seen in fields of cells in which there were no microglia. The increased rate of ROS generation was calcium dependent, again linking this response to the primary observation of calcium signals in astrocytes.
Figure 4.
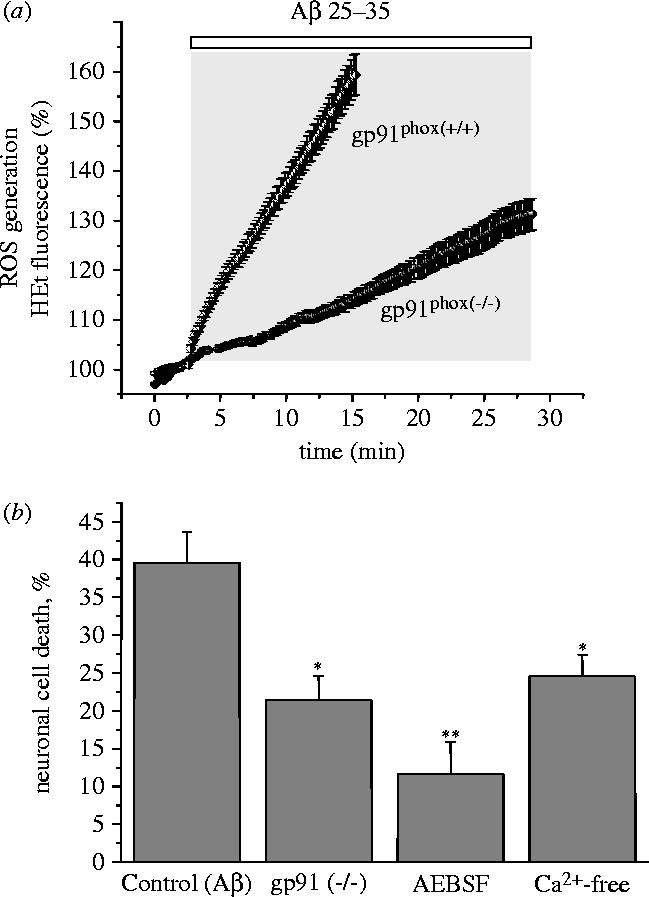
Aβ induced generation of oxygen free radicals and cell death are largely attributable to NADPH oxidase. (a) The rate of ROS generation was measured using hydroethidine (HEt), which is oxidized to a fluorescent product by ROS. The rate of appearance of the fluorescent product was clearly increased in mouse astrocytes exposed to Aβ 1–42 (2.5 μM; n=59) which caused a 3.6 fold increase in the rate of rise of the HEt signal (from 1.1±0.12 to 3.98±0.21 arb. fluorescence U min−1; n=179 cells), in cortical astrocytes of both rats and mice. In astrocytes cultured from gp91phox knockout transgenic mice, no significant increase in ROS generation was detectable (n=116). (b) Neuronal death of cells from gp91phox knockout mice following 24 h exposure to Aβ was significantly reduced from control (39.6±4.1%) to 21.4±3.2% (*p<0.05). The inhibitor of NADPH oxidase AEBSF and removal of external Ca2+ both also reduced neuronal cell death in mouse cultures, (cell death was reduced from 39.6±4.1% in control to 11.6±4.3% (AEBSF, **p<0.001) and to 24.6±2.8% (Ca2+-free, *p<0.05).
5. Aβ and NADPH oxidase
Aβ activates an NADPH oxidase in microglia. A number of studies have suggested more generally that Aβ can increase the rate of ROS generation in immuno-competent cells, although the models vary considerably. Aβ activates neutrophils directly, and enhances neutrophil activation by other stimuli such as phorbol myristate acetate (PMA), which activates the enzyme through activation of protein kinase C (PKC) (Wautier et al. 2001) and to enhance ROS generation in response to activation by P2x receptors (which raise [Ca2+]c) of neutrophils from patients with familial AD (Yan et al. 1996). It remains unclear to what extent these effects relate to neurodegeneration in AD patients. Neuritic plaques are enriched in microglia and microglial activation seems a more likely contributor to neurodegeneration than the activation of peripheral neutrophils, although of course alterations in the periphery may reflect interesting changes in cell biology in these patients.
Neuritic plaques are also enriched in reactive astrocytes, and as we saw ROS generation in pure astrocyte cultures, we wondered whether there was any equivalent response in astrocytes, which were not known to express an NADPH oxidase. In fact, we found that the Aβ induced increase in ROS generation and the downstream effect of a loss of mitochondrial potential were inhibited by three different inhibitors of the phagocytic NADPH oxidase—apocynin, diphenylene iodonium (DPI) and 4-(2-aminoethyl)-benzenesulphonylfluoride (AEBSF) (figure 3a). These drugs also prevented the Aβ induced GSH depletion and protected cells from Aβ induced cell death, placing the NADPH oxidase firmly at the heart of Aβ induced neuronal death. Consistent with this observation, we have since found evidence for the expression of the phagocyte enzyme subunits gp91phox, p67phox, p22, p40 and p47 in pure cultures of astrocytes both by Western blots and by immunofluorescence (Abramov et al. 2005). The importance of the enzyme as a determinant of the response to Aβ is emphasized by the almost complete absence of any response to Aβ in cortical astrocytes cultured from gp91phox knockout transgenic mice both in terms of changing mitochondrial potential (figure 2b) and in terms of an increase in ROS generation (figure 4a). Furthermore, in the transgenic mouse cultures, Aβ induced neuronal cell death was also dramatically reduced (figure 4b).
One question that remains is the role of the oxidase in dictating the changes in mitochondrial function in astrocytes that we described above. The enzyme is activated by the phorbol ester, PMA, the classical activator of the neutrophil enzyme, which causes an increase in the rate of ROS generation in astrocytes that is prevented by inhibitors of the NADPH oxidase. We found that PMA also caused a slow loss of mitochondrial potential which was inhibited by DPI and apocynin, which was reversed by application of substrate such as glutamate or methyl succinate (figure 5).
Figure 5.
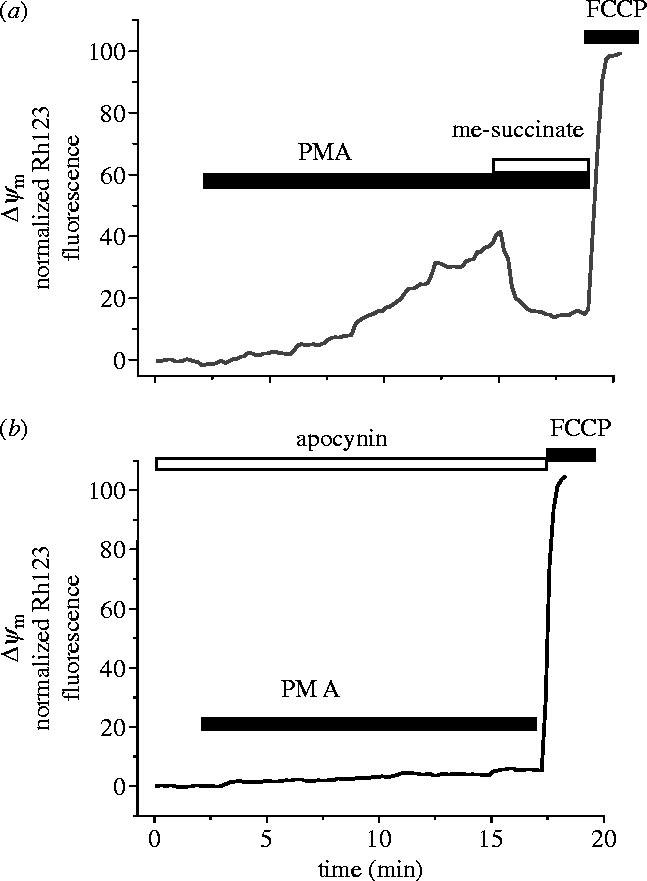
Activation of the NADPH oxidase by PMA causes a slow loss of mitochondrial potential that is reversed by mitochondrial substrate. The ‘classical’ activator of the NADPH oxidase in neutrophils, PMA, increases ROS generation in astrocytes (not shown) and produced a slow but sustained mitochondrial depolarization in the majority of cortical astrocytes tested (n=91 of 98 cells). B. This effect was inhibited by apocynin (n=65) or DPI (0.5 μM; n=48, not shown). Further similarity between the Aβ- and PMA-induced changes in Δψm can be seen by the reversal of the PMA induced response by delivery of mitochondrial substrate—illustrated here for methyl-succinate. Similar data were obtained for glutamate (1 mM; n=62 for PMA and n=218 for Aβ) and methyl-succinate (n=66 for PMA and n=145 for Aβ).
6. Conclusion
Our observations lead to the novel and surprising conclusion that the neurotoxicity of Aβ is mediated primarily by the calcium dependent activation of an NADPH oxidase in astrocytes. This generates an oxidative stress of which the changes in astrocyte mitochondrial function are a consequence. We do not yet fully understand how that oxidative stress is communicated to the neurons, which die. A simple possibility is that neurons require a supply of glutathione precursors which are provided by the extracellular cleavage of glutathione released from astrocytes (e.g. see Keelan et al. 2001; Dringen & Hirrlinger 2003). It will, therefore, follow inevitably that glutathione depletion in astrocytes will lead to glutathione depletion in neurons, which seem far more vulnerable to oxidative injury than astrocytes. It remains perfectly possible that there are other mediators that communicate between astrocytes and neurons and promote neuronal damage; however, the NADPH oxidase does appear to play a central role in Aβ neurotoxicity, raising exciting possibilities for future therapeutic strategies.
Acknowledgments
We thank Drs Laura Canevari and Frans Wientjes for their contributions to the background work that is described and Prof. Ajay Shah and Dr Alison Cave for help in obtaining gp91phox knockout transgenic mice. The work was funded by the Wellcome Trust.
Footnotes
One contribution of 18 to a Theme Issue ‘Reactive oxygen species in health and disease’.
References
- Abramov A.Y, Canevari L, Duchen M.R. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 2003;23:5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov A.Y, Canevari L, Duchen M.R. Amyloid β peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004a;23:5088–5095. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov A.Y, Canevari L, Duchen M.R. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta. 2004b;6:81–87. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Abramov A.Y, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen M.R. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J. Neurosci. 2005;25:9176–9184. doi: 10.1523/JNEUROSCI.1632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, Doh M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer's disease AβP (1–40) and (1–42) peptides. FASEB J. 2002;16:1526–1536. doi: 10.1096/fj.02-0829com. 10.1096/fj.02-0829com [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard H.B, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1–40)] in bilayer membranes. Proc. Natl Acad. Sci. USA. 1993;90:10 573–10 577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, Pollard H.B, Rojas E. Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc. Natl Acad. Sci. USA. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. 10.1073/pnas.93.4.1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia R, Lin H, Lal R. Fresh and globular amyloid β-protein (1–42) induces rapid cellular degeneration: evidence for AβP channel-mediated cellular toxicity. FASEB J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- Blass J.P, Gibson G.E. The role of oxidative abnormalities in the pathophysiology of Alzheimer's disease. Rev. Neurol. (Paris) 1991;147:513–525. [PubMed] [Google Scholar]
- Bush A.I. The metallobiology of Alzheimer's disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. 10.1016/S0166-2236(03)00067-5 [DOI] [PubMed] [Google Scholar]
- Canevari L, Abramov A.Y, Duchen M.R. Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 2004;29:637–650. doi: 10.1023/b:nere.0000014834.06405.af. 10.1023/B:NERE.0000014834.06405.af [DOI] [PubMed] [Google Scholar]
- Casley C.S, Land J.M, Sharpe M.A, Clark J.B, Duchen M.R, Canevari L. Beta-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 2002a;10:258–267. doi: 10.1006/nbdi.2002.0516. 10.1006/nbdi.2002.0516 [DOI] [PubMed] [Google Scholar]
- Casley C.S, Canevari L, Land J.M, Clark J.B, Sharpe M.A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002b;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. 10.1046/j.0022-3042.2001.00681.x [DOI] [PubMed] [Google Scholar]
- Cherny R.A, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. 10.1016/S0896-6273(01)00317-8 [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol. Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. 10.1515/BC.2003.059 [DOI] [PubMed] [Google Scholar]
- Helmuth L. New therapies. New Alzheimer's treatments that may ease the mind. Science. 2002;23:1260–1262. doi: 10.1126/science.297.5585.1260. 10.1126/science.297.5585.1260 [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y. Intracellular calcium changes in neuronal cells induced by Alzheimer's β-amyloid protein are blocked by estradiol and cholesterol. Cell. Mol. Neurobiol. 2001;21:1–13. doi: 10.1023/A:1007168910582. 10.1023/A:1007168910582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelan J, Allen N.J, Antcliffe D, Pal S, Duchen M.R. Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J. Neurosci. Res. 2001;66:873–884. doi: 10.1002/jnr.10085. 10.1002/jnr.10085 [DOI] [PubMed] [Google Scholar]
- Lin H, Bhatia R, Lal R. Amyloid β-protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. 10.1096/fj.01-0377com [DOI] [PubMed] [Google Scholar]
- Longo V.D, Viola K.L, Klein W.L, Finch C.E. Reversible inactivation of superoxide-sensitive aconitase in Abeta1-42-treated neuronal cell lines. J. Neurochem. 2000;75:1977–1985. doi: 10.1046/j.1471-4159.2000.0751977.x. 10.1046/j.1471-4159.2000.0751977.x [DOI] [PubMed] [Google Scholar]
- Melov S. ‘…and C is for Clioquinol’—the AbetaCs of Alzheimer's disease. Trends Neurosci. 2002;25:121–123. doi: 10.1016/s0166-2236(00)02086-5. 10.1016/S0166-2236(00)02086-5 [DOI] [PubMed] [Google Scholar]
- Näslund J, Haroutunian V, Mohs R, Davis K.-L, Davies P, Greengard P, Buxbaum J.-D. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. J. Am. Med. Assoc. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. 10.1001/jama.283.12.1571 [DOI] [PubMed] [Google Scholar]
- Rhee S.K, Quist A.P, Lal R. Amyloid β-protein (1–42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem. 1998;273:13 379–13 382. doi: 10.1074/jbc.273.22.13379. 10.1074/jbc.273.22.13379 [DOI] [PubMed] [Google Scholar]
- Tanzi R.E, Kovacs D.M, Kim T.W, Moir R.D, Guenette S.Y, Wasco W. The gene defects responsible for familial Alzheimer's disease. Neurobiol. Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. 10.1006/nbdi.1996.0016 [DOI] [PubMed] [Google Scholar]
- Wautier M.P, Chappey O, Corda S, Stern D.M, Schmidt A.M, Wautier J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- Yan S.D, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. 10.1038/382685a0 [DOI] [PubMed] [Google Scholar]