

Abstract
Recent large-scale sequencing studies have revealed that cancer genomes contain variable numbers of somatic point mutations distributed across many genes. These somatic mutations most likely include passenger mutations that are not cancer causing and pathogenic driver mutations in cancer genes. Establishing a significant presence of driver mutations in such data sets is of biological interest. Whereas current techniques from phylogeny are applicable to large data sets composed of singly mutated samples, recently exemplified with a p53 mutation database, methods for smaller data sets containing individual samples with multiple mutations need to be developed. By constructing distinct models of both the mutation process and selection pressure upon the cancer samples, exact statistical tests to examine this problem are devised. Tests to examine the significance of selection toward missense, nonsense, and splice site mutations are derived, along with tests assessing variation in selection between functional domains. Maximum-likelihood methods facilitate parameter estimation, including levels of selection pressure and minimum numbers of pathogenic mutations. These methods are illustrated with 25 breast cancers screened across the coding sequences of 518 kinase genes, revealing 90 base substitutions in 71 genes. Significant selection pressure upon truncating mutations was established. Furthermore, an estimated minimum of 29.8 mutations were pathogenic.
RECENTLY, a number of large-scale screens for somatic mutations in human cancers have been started. The primary aim of these screens is to identify the driver mutations in cancer genes that are causally implicated in cancer development (Futreal et al. 2004). Identification of these cancer genes provides major insights into the biology of neoplastic change. Moreover, the proteins encoded by some of these mutated cancer genes have recently proven to be tractable targets for new anticancer drug development. However, analysis of the results of genomic screens for somatic mutations can be complicated by a background noise of mutations that confer no clonal growth advantage (passenger mutations). For the identification of some driver mutations and cancer genes, the problems caused by background passenger mutations are minor. In a cancer gene that is frequently involved in cancer development, the somatic mutation frequency per nucleotide of coding sequence in a set of cancer samples is clearly higher than in other genes and is often characterized by distinctive patterns of mutation type and/or position. For example, mutations observed in BRAF mostly cluster in exons 11 and 15, with a large subset of these consisting of the single mutation V600E (Davies et al. 2002). However, such features will not easily be detected in cancer genes that are infrequently involved in cancer causation, unless very large numbers of cancer samples are analyzed. For example, in a recent screen of 518 kinase genes of 25 breast cancers (Stephens et al. 2005), 90 base substitutions were discovered in 71 genes. The number of mutations per gene was highly correlated with the coding sequence length, and no gene with a clear elevation in its mutation rate per coding nucleotide presented itself as a candidate cancer gene. Since 14 of these mutations were silent and hence likely to be passenger mutations (see Table 1) this opens the question of determining whether any of the remaining 76 mutations are pathogenic, which would provide evidence that some of the 71 mutated genes are involved in the formation of cancer.
TABLE 1.
The distribution of screened base pairs and mutations observed in Stephens et al. (2005)
| Mutation category
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Silent base pairs
|
Missense base pairs
|
Nonsense base pairs
|
Splice site base pairs
|
|||||||||
| Mutation type | Prekinase | Kinase | Postkinase | Prekinase | Kinase | Postkinase | Prekinase | Kinase | Postkinase | Prekinase | Kinase | Postkinase |
| C:G > G:C | 30,449 | 26,511 | 23,184 | 148,021 | 125,625 | 109,293 | 2,321 | 2,870 | 1,350 | 6,623 | 7,774 | 5,159 |
| C:G > G:C, TpC | 9,068 | 8,902 | 5,955 | 53,235 | 47,318 | 38,499 | 2,184 | 1,221 | 2,095 | 1,311 | 1,120 | 867 |
| C:G > A:T | 32,695 | 28,741 | 25,073 | 140,852 | 119,407 | 104,734 | 7,244 | 6,858 | 4,020 | 6,623 | 7,774 | 5,159 |
| C:G > A:T, TpC | 12,856 | 13,424 | 8,150 | 36,972 | 32,380 | 26,697 | 14,659 | 11,637 | 11,702 | 1,311 | 1,120 | 867 |
| C:G > T:A | 40,269 | 38,284 | 30,681 | 57,809 | 45,968 | 40,714 | 6,658 | 5,572 | 4,677 | 5,644 | 6,692 | 4,166 |
| C:G > T:A, TpC | 20,385 | 20,841 | 14,001 | 35,985 | 28,911 | 26,561 | 2,159 | 1,733 | 1,587 | 1,276 | 1,108 | 859 |
| C:G > T:A, CpG | 26,434 | 23,181 | 20,352 | 46,351 | 39,335 | 34,573 | 3,270 | 2,666 | 2,830 | 979 | 1,082 | 993 |
| C:G > T:A, TpC, CpG | 1,508 | 1,754 | 993 | 4,178 | 3,915 | 3,209 | 272 | 287 | 198 | 35 | 12 | 8 |
| A:T > T:A | 40,331 | 30,816 | 27,818 | 180,932 | 163,668 | 121,926 | 19,294 | 18,125 | 13,538 | 5,965 | 6,704 | 4,583 |
| A:T > C:G | 41,272 | 30,865 | 29,146 | 195,741 | 178,076 | 131,060 | 3,544 | 3,668 | 3,076 | 5,965 | 6,704 | 4,583 |
| A:T > G:C | 76,405 | 61,277 | 51,944 | 164,150 | 151,325 | 110,480 | 2 | 7 | 858 | 5,965 | 6,704 | 4,583 |
| Mutation category
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Silent mutations
|
Missense mutations
|
Nonsense mutations
|
Splice site mutations
|
|||||||||
| Mutation type | Prekinase | Kinase | Postkinase | Prekinase | Kinase | Postkinase | Prekinase | Kinase | Postkinase | Prekinase | Kinase | Postkinase |
| C:G > G:C | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C:G > G:C, TpC | 1 | 1 | 0 | 9 | 10 | 5 | 0 | 1 | 2 | 1 | 1 | 2 |
| C:G > A:T | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| C:G > A:T, TpC | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 4 | 1 | 0 | 0 |
| C:G > T:A | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| C:G > T:A, TpC | 2 | 1 | 3 | 3 | 4 | 2 | 2 | 0 | 2 | 0 | 0 | 0 |
| C:G > T:A, CpG | 2 | 0 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| C:G > T:A, TpC, CpG | 0 | 0 | 0 | 2 | 3 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| A:T > T:A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| A:T > C:G | 0 | 0 | 1 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| A:T > G:C | 0 | 1 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
Counts are separated by mutation type, the category of mutation, and the domain.
A similar problem was considered by Yang et al. (2003). Phylogenetic techniques from evolution analyses were adapted to analyze a large database containing p53 mutations, and missense and nonsense mutations were successfully shown to have different effects across distinct functional domains of biological interest. However, this data set is marked by characteristics that distinguish it from the protein kinase data set provided in Stephens et al. (2005), implying that alternative analyses may be more appropriate for the latter. Samples in the p53 data set typically contain a single mutation and were modeled as such in Yang et al. (2003). This is not the case for the protein kinase data set, where some cancers with mutator phenotypes contain several mutations, which need to be modeled accordingly. The protein kinase data set is of moderate size, meaning exact tests are more desirable than the asymptotic likelihood-ratio tests applied to the large p53 data set.
Current methods (Yang et al. 2003) can establish the existence of pathogenic mutations, without indicating the proportion of nonsynonymous mutations that are pathogenic rather than passenger in nature. This parameter is a desirable quantity as it is indicative of the proportion of mutated genes that are implicated in the development of oncogenesis. The methods described below incorporate these differences into the modeling techniques.
Finally we note that current methods (Yang et al. 2003) incorporate selection toward certain mutation types as multiplicative weighting factors in codon substitution models. We present a method whereby selection is explicitly described as a process separate from mutation, from which the full model of observables can be constructed accordingly. This allows any model of selection in cancer to be developed and explored.
In this article we use a similar approach to phylogenetic methods to evaluate the evidence that the observed data set contains pathogenic mutations. The basic principle behind this approach is that silent (synonymous) somatic mutations are passenger mutations. Although a minority of apparently synonymous mutations may encode exonic splice enhancers or other cryptic elements that affect the translated product of a DNA sequence, in general this assumption is likely to be correct. The set of silent mutations can therefore be used as a control group to estimate the number of nonsilent mutations that would be expected to occur by chance, under the null hypothesis of no association between mutations and cancer development. Tests of significance can then be derived by comparing the observed number of nonsilent (nonsynonymous) mutations to the expected number. It is also possible to derive estimates of the minimum proportion of mutations likely to be pathogenic. We also show that there is a strong analogy with standard analyses of epidemiological case–control or cohort studies to evaluate risk factors.
To develop the approach, we consider an experiment in which a number of tumors are screened through a particular coding sequence. Suppose that l silent mutations and n nonsilent mutations are observed, with 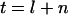 mutations in total. We further assume that there are T possible mutations across the sequence (T is three times the length of the sequence), of which L are silent and N are nonsilent. Thus a randomly positioned mutation will be silent with probability
mutations in total. We further assume that there are T possible mutations across the sequence (T is three times the length of the sequence), of which L are silent and N are nonsilent. Thus a randomly positioned mutation will be silent with probability 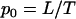 . If tumor samples exhibit no preference toward either silent or nonsilent mutations, then
. If tumor samples exhibit no preference toward either silent or nonsilent mutations, then 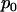 also represents the probability that a randomly chosen mutation from a tumor sample will be silent. The odds ratio
also represents the probability that a randomly chosen mutation from a tumor sample will be silent. The odds ratio 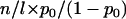 is a measure of the strength of the association or selection toward nonsilent mutations. Values greater than unity would indicate positive selection (that is, nonsilent mutations occurring more often than would be expected by chance and, therefore, to some degree related to cancer development), while values less than unity would indicate negative selection (nonsilent mutations occurring less often than expected, perhaps because they result in cell death). However, any deviation from unity may be due to the random nature of mutation rather than to any underlying positive or negative selection by cancer. Alternatively, if any of the N nonsilent mutations either promote or inhibit the development of cancer, the probability p that a randomly chosen mutation observed in cancer is silent may differ from
is a measure of the strength of the association or selection toward nonsilent mutations. Values greater than unity would indicate positive selection (that is, nonsilent mutations occurring more often than would be expected by chance and, therefore, to some degree related to cancer development), while values less than unity would indicate negative selection (nonsilent mutations occurring less often than expected, perhaps because they result in cell death). However, any deviation from unity may be due to the random nature of mutation rather than to any underlying positive or negative selection by cancer. Alternatively, if any of the N nonsilent mutations either promote or inhibit the development of cancer, the probability p that a randomly chosen mutation observed in cancer is silent may differ from 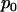 . These two scenarios can be summarized by null and alternative hypotheses
. These two scenarios can be summarized by null and alternative hypotheses 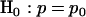 and
and 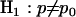 . Note that p is unobservable, so inference statistics are required to compare the hypotheses. The significance level of a suitable statistic, such as the odds ratio, would enable such a comparison. To obtain this, the expected distribution of the odds ratio under the null hypothesis is required. If we condition upon the total number of observed mutations t we note that under the null hypothesis
. Note that p is unobservable, so inference statistics are required to compare the hypotheses. The significance level of a suitable statistic, such as the odds ratio, would enable such a comparison. To obtain this, the expected distribution of the odds ratio under the null hypothesis is required. If we condition upon the total number of observed mutations t we note that under the null hypothesis 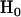 the number of silent mutations l would be drawn from a Binomial (t, p0) distribution. The expected distribution can then be estimated by simulating l from this distribution and calculating numerous odds ratios (p0 is a function of DNA sequence and so fixed throughout). Comparing the observed odds ratio to this distribution will then provide a significance level. This will help determine whether any of the observed nonsilent mutations are likely to have contributed to oncogenesis in the sampled tumors.
the number of silent mutations l would be drawn from a Binomial (t, p0) distribution. The expected distribution can then be estimated by simulating l from this distribution and calculating numerous odds ratios (p0 is a function of DNA sequence and so fixed throughout). Comparing the observed odds ratio to this distribution will then provide a significance level. This will help determine whether any of the observed nonsilent mutations are likely to have contributed to oncogenesis in the sampled tumors.
In practice, however, this approach is too simplistic. There are six different categories of base substitution, namely C:G > G:C, C:G > A:T, C:G > T:A, A:T > T:A, A:T > C:G, and A:T > G:C, where, for example, C:G > A:T implies that a cytosine nucleotide is replaced by an adenine on one DNA strand and a guanine is replaced by a thymine on the complementary strand. Note that although there are, theoretically, 12 possible single-base changes, these reduce to the 6 types listed above, as each mutation cannot be distinguished from the corresponding mutation on the complementary strand. The consequence of a specific mutation at a specific position will depend on the genetic code, and so the probability that a random point mutation is silent will therefore be a function of precise DNA sequence being examined. It will also vary depending upon the mutation type. For example, across the coding sequences of 518 kinase genes studied in Stephens et al. (2005), the proportion of possible C:G > A:T substitutions that lead to a silent mutation is 0.18, whereas the corresponding probability for a C:G > T:A mutation is 0.36. In addition, different mutation types will occur at different rates, depending on the cell type and its environment. In statistical terms, therefore, mutation type is a confounding factor that must be adjusted for. This is accomplished most simply by considering each mutation type as a separate stratum.
The mutation rate may depend not only on the mutated base but also on the neighboring sequence. For example, C:G > T:A mutations occur at an increased rate due to deamination of cytosine at CpG dinucleotides. In principle, such neighborhood effects can be handled by introducing larger numbers of strata, although the required number of strata may be large (for example, if all combinations of bases on both sides of the mutated base are considered, there are 192 possible mutation types). Such finer stratification will reduce the risk of bias but will also reduce power. For example, a C:G > G:C mutation at the central nucleotide of an ApGpG trinucleotide cannot be silent. Any selection pressure on such mutations will elevate the mutation rate of this stratum. However, this cannot be distinguished formally from an inherently different mutation rate at these sequences, so such mutations are uninformative. In practice, a compromise between representative stratification and statistical power is required. Deamination at CpG dinucleotides is well established, and a strong C:G > {T:A, A:T, or G:C} mutation rate at TpC dinucleotides was observed in Stephens et al. (2005). For our main analyses, we have used the 11 strata given in Table 1.
The degree of selection by cancer upon specific mutations may depend on the type of amino acid change adopted by the protein. In particular, nonsense and splice site mutations can lead to a truncated or reduced protein, respectively, or indeed to total absence of protein through nonsense-mediated decay, which may remove domains of functional importance. If any such mutations occur in the presence of loss of heterozygosity (LOH) on the homologous chromosome, function will be lost. This is the mechanism by which many tumor suppressor genes are involved in carcinogenesis. For example, the mutation data set of the RB1 tumor suppressor gene examined in Valverde et al. (2005) contains more nonsense and splice mutations than are typical for the pattern of mutations observed in hereditary diseases. Conversely, a dominant change in function is more likely to be achieved through missense mutations. Furthermore, proteins containing multiple domains of functional necessity are less likely to tolerate deletions induced by nonsense and splice variants, as exemplified by the p53 gene, where most of the recorded variants are missense, as can be found in the database described by Béroud and Soussi (2003). These potential differences in selection by cancer can be incorporated by separating nonsilent mutations into missense, nonsense, and splice site categories.
The nucleotides most likely to induce splice variants under mutation are 1, 2, or 5 bp 3′ to an exon or 1 or 2 bp 5′ to an exon. Although other nucleotides may be sources of splice variants under mutation, they have been ignored in this analysis.
This idea may be extended to consider separately different types of amino acid substitutions. For example, conservative and nonconservative changes could be differentiated. Alternatively, the heterogeneity of variants could be distinguished to reflect the idea that pathogenic homozygous variants are likely to occur in recessive cancer genes, whereas pathogenic heterozygous variants will occur in dominant cancer genes.
The functional domains of the genes under investigation may also be subject to different selection pressures. For example, tumor cells may not tolerate mutations within some highly conserved regions, possibly inducing apoptosis, which will be selected against by cancer. Alternatively, mutations in functional domains key to mitotic pathways may enhance the clonal growth rate, such as within the kinase domains of certain protein kinases. These mutations will be under positive selection pressure by cancer. Establishing differences in selection across distinct functional domains within the screened genes thus becomes a question of biological interest.
This article is organized as follows. The next section introduces the Poisson processes used to model the random nature of mutations under no selection pressure. To describe the pattern of mutations observed in tumor samples, the subsequent section models the selection pressure of cancer upon mutations, from which methods to estimate the numbers of pathogenic mutations are introduced. Likelihood-ratio and score statistics are then developed to assess whether selection pressure upon the screened genome exists and hence whether a subset of the observed mutations is implicated in the development of cancer. Next, these methods are adapted to test for variation in selection across different functional domains. Finally, these techniques are illustrated and discussed with the breast cancer data set of Stephens et al. (2005).
MODELING MUTATIONS
Various models of the mutation process have been defined and explored, frequently to model the evolutionary development of species (see Goldman and Yang 1994 for an example) but also to model cancer (Yang et al. 2003). These models are known as codon substitution models and account for potential factors in mutation processes. These include differences in mutation rates between transversions and transitions, as well as between silent and nonsilent mutations. Selection pressures are generally incorporated as multiplicative factors. These models are based on continuous-time Markov (Poisson) processes, reflecting the hypothesis that mutations are random events that occur independently of one another. Although there is some evidence that mutations are not entirely random in nature (Hall 1990), Poisson processes provide a natural basis from which to derive an analytic approach.
Suppose we have J tumor samples to analyze. Suppose furthermore that tumor sample j has undergone mj mitoses and that, at the rth mitosis, the number of mutations in the screened coding sequence (CDS) of type k is a Possion process with rate 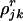 , where the mutation type k represents 1 of 11 strata indicated in Table 1. The number of mitoses and mutation rates may vary among samples, and the mutation rates may vary between mitoses. It is assumed throughout that these intensities are small and that all processes are independent. Note that these mutation intensities are unobservable. Furthermore, although these are of biological interest, the main motivation of the present analysis is to evaluate the evidence for pathogenicity. In the current context, the mutation intensities are nuisance parameters to be eliminated by estimation or conditioning.
, where the mutation type k represents 1 of 11 strata indicated in Table 1. The number of mitoses and mutation rates may vary among samples, and the mutation rates may vary between mitoses. It is assumed throughout that these intensities are small and that all processes are independent. Note that these mutation intensities are unobservable. Furthermore, although these are of biological interest, the main motivation of the present analysis is to evaluate the evidence for pathogenicity. In the current context, the mutation intensities are nuisance parameters to be eliminated by estimation or conditioning.
For each mutation type 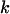 we calculate the number of base pairs in the CDS that can give rise to silent, missense, nonsense, or splice mutations. These counts are denoted
we calculate the number of base pairs in the CDS that can give rise to silent, missense, nonsense, or splice mutations. These counts are denoted 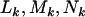 , and
, and 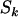 , respectively, with totals
, respectively, with totals 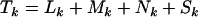 . These observables can be calculated precisely from the sequence of DNA in the region screened, available from any database containing the human genome sequence, and the genetic code. The values for the kinase data set of Stephens et al. (2005) are provided in the top half of Table 1.
. These observables can be calculated precisely from the sequence of DNA in the region screened, available from any database containing the human genome sequence, and the genetic code. The values for the kinase data set of Stephens et al. (2005) are provided in the top half of Table 1.
Some genes may have multiple transcripts, possibly out of frame, making such counts ambiguous. In such cases average counts weighted by protein frequency would be appropriate, if possible. However, for application to the protein kinase genes in Stephens et al. (2005), multiple transcripts varied little and no frameshifts were observed, so only the longest transcript was used in application of these methods.
Single-nucleotide polymorphisms (SNPs) in the samples will result in some differences between the sample CDSs and a database reference CDS. These are normally detected when wild-type samples are screened against the cancers to distinguish SNPs from somatic mutations. In principle, values of Lk, Mk, Nk, and Sk could be adjusted to take account of these SNPs. However, since they typically occur at an average frequency of about one every kilobase, errors involved in using the reference sequence were assumed negligible.
The aim of this article is to distinguish pathogenic mutations from passenger ones. It is thus natural to divide each of missense, nonsense, and splice mutations into two groups, those associated with cancer (driver or pathogenic) and those not associated with the growth rate of cells (passenger or neutral). As such, we partition the counts 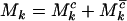 ,
, 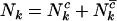 , and
, and 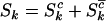 , where superscript c indicates a count across bases pathogenic under mutation, and
, where superscript c indicates a count across bases pathogenic under mutation, and 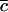 indicates counts across bases neutral to cancer. Although only the totals
indicates counts across bases neutral to cancer. Although only the totals 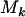 ,
, 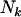 , and
, and 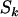 can be observed, the division of counts into pathogenic and neutral counts serves the problem twofold. First, selection pressure induced by cancer will apply only to the pathogenically mutable bases. Second, this will allow us to estimate the number of pathogenic mutations.
can be observed, the division of counts into pathogenic and neutral counts serves the problem twofold. First, selection pressure induced by cancer will apply only to the pathogenically mutable bases. Second, this will allow us to estimate the number of pathogenic mutations.
Suppose that 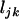 ,
, 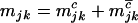 ,
, 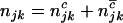 , and
, and 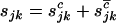 are the numbers of silent, missense, nonsense, and splice site mutations actually observed in sample j. Again, the missense, nonsense, and splice site counts are partitioned into pathogenic or passenger mutations. The total counts are denoted
are the numbers of silent, missense, nonsense, and splice site mutations actually observed in sample j. Again, the missense, nonsense, and splice site counts are partitioned into pathogenic or passenger mutations. The total counts are denoted 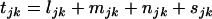 . Then assuming that mutations are independent random events, the counts
. Then assuming that mutations are independent random events, the counts 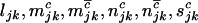 , and
, and 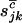 will be drawn from independent Poisson distributions with expectations
will be drawn from independent Poisson distributions with expectations 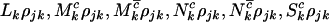 , and
, and 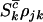 , respectively, where
, respectively, where 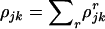 represents the overall intensity across all mitoses of sample j for mutation type k. Although each mutation may change
represents the overall intensity across all mitoses of sample j for mutation type k. Although each mutation may change 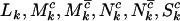 , or
, or 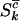 slightly, the total number of mutations is typically small enough that these sizes can be regarded as fixed. For example, the breast kinase screen of Stephens et al. (2005) detected only 90 base substitutions out of 3.2 × 107 bp screened.
slightly, the total number of mutations is typically small enough that these sizes can be regarded as fixed. For example, the breast kinase screen of Stephens et al. (2005) detected only 90 base substitutions out of 3.2 × 107 bp screened.
In the absence of any selection pressure, the probability of observing a given set of mutations in sample j then takes the form of the following product of Poisson distributions:
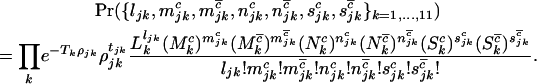 |
(1) |
Note that the ratios 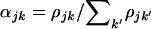 represent the probability that a random mutation is of type
represent the probability that a random mutation is of type  . These values are commonly referred to as the mutation spectra.
. These values are commonly referred to as the mutation spectra.
MODELING SELECTION
To model the effects of selection, Equation 1 needs to be modified to incorporate the effect that a specific distribution of mutations will have upon the development of cancer. This effect is modeled as a probability as there are multiple sources of uncertainty in the development of cancer for a specific distribution of mutations. First, the positioning of the pathogenic mutations within the region of screened genome will vary between samples. As some positions of pathogenic mutation are likely to confer more clonal growth advantage than others, any values describing selection pressures should be viewed as an average across the screened genome. Also, unless the entire coding genome is analyzed, samples will possibly contain undetected pathogenic mutations within unscreened regions that confer varying degrees of growth advantage to the cells. Finally, we note that there will be natural variation in the effects of mutations from person to person; gene expression levels may vary between samples, for example. For these reasons, it is natural to model the development of cancer for a given mutation set stochastically rather than deterministically.
If  denotes the event that a given cell lineage j develops cancer, the distribution of observed mutations can be resolved through Bayes' theorem into the following product,
denotes the event that a given cell lineage j develops cancer, the distribution of observed mutations can be resolved through Bayes' theorem into the following product,
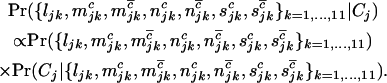 |
The final term 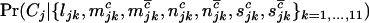 models the probability that a DNA sample with the given set of mutations across the region screened will be cancerous.
models the probability that a DNA sample with the given set of mutations across the region screened will be cancerous.
The form of these latter terms will depend on the assumed model of cancer. Some models, for example, assume that a fixed number of mutations are observed. This is a general fixed parameter in Little and Wright (2003). The work of Yang et al. (2003) fit numerous models to data arising from experiments where one or a few genes are screened across numerous samples, exemplified by their analysis upon a p53 mutation data set. However, the typical data set considered here contains the results of a screen across several genes in relatively few tumor samples, such as that described in Stephens et al. (2005). As such, we assume that for all samples, each additional pathogenic mutation of a given category confers the same relative increase in the probability of developing cancer. Although this may not be strictly true, it provides an intuitive model for developing tests and estimates. The final term will thus be of the form,
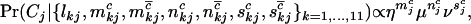 |
(2) |
where 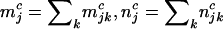 , and
, and 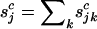 denote the total numbers of pathogenic missense, nonsense, and splice mutations in sample j, respectively.
denote the total numbers of pathogenic missense, nonsense, and splice mutations in sample j, respectively.
We note that this probability is independent of mutation counts within each mutation type, k. This may be expected, as the likelihood of developing cancer is likely to depend upon the category of amino acid change (i.e., missense, nonsense, or splice) rather than the type of source mutation, k.
The terms η,μ, and ν represent the relative change in probability of developing a tumor conferred by a pathogenic missense, nonsense, or splice mutation, respectively. Values greater than unity indicate an increase in this likelihood, whereas values less than unity represent a decrease. These terms are analogous to rate ratios or relative risks in epidemiology, where the mutations are analogous to exposures. The resulting likelihood is then of the form
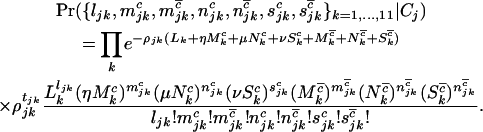 |
(3) |
This is equivalent to a product of independent Poisson distributions where 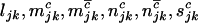 , and
, and 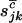 have intensities given by
have intensities given by 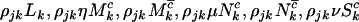 , and
, and 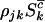 , respectively. If
, respectively. If 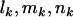 , and
, and 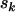 denote the total number of silent, missense, nonsense, and splice variants across all samples, we have
denote the total number of silent, missense, nonsense, and splice variants across all samples, we have 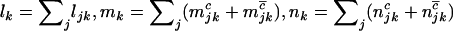 , and
, and 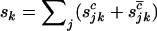 . As mutations are independent events, the counts
. As mutations are independent events, the counts 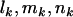 , and
, and 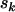 will also follow independent Poisson distributions, with intensities given by
will also follow independent Poisson distributions, with intensities given by 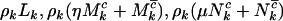 , and
, and 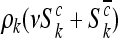 , respectively, where
, respectively, where 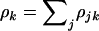 . This can be summarized by
. This can be summarized by
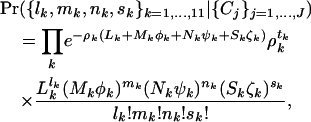 |
(4) |
where 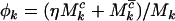 ,
, 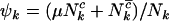 , and
, and 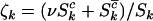 are the rate ratios and represent selection pressures. Values
are the rate ratios and represent selection pressures. Values 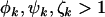 increase the probability of observing pathogenic mutations and thus represent positive selection pressure, whereas
increase the probability of observing pathogenic mutations and thus represent positive selection pressure, whereas 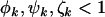 indicate negative selection pressure.
indicate negative selection pressure.
It is worth observing that although the parameters η, μ, and ν, used to define the probability that a cell lineage adopts cancer (Equation 2), are independent of mutation type k, differences in the proportions of potentially pathogenic mutations (i.e., 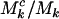 ,
, 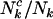 , or
, or 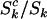 ), could still lead to selection pressures
), could still lead to selection pressures 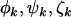 that depend upon k. We also note that, in the present context, these parameters are assumed to be the same for all mutations of each type. In practice this may not be true, in which case they can be regarded as a representative “average” effect. Later, we consider approaches to evaluating variation in these parameters, for example, by domain.
that depend upon k. We also note that, in the present context, these parameters are assumed to be the same for all mutations of each type. In practice this may not be true, in which case they can be regarded as a representative “average” effect. Later, we consider approaches to evaluating variation in these parameters, for example, by domain.
ESTIMATING THE NUMBER OF PATHOGENIC MUTATIONS
One question that naturally arises is how to estimate the proportions of observed mutations that are actually pathogenic. These are denoted 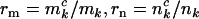 , and
, and 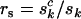 for missense, nonsense, and splice variants, respectively, which can be estimated by taking the ratio of the relevant rates from Equation 3. This gives
for missense, nonsense, and splice variants, respectively, which can be estimated by taking the ratio of the relevant rates from Equation 3. This gives 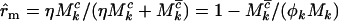 for missense mutations, with similar estimates
for missense mutations, with similar estimates 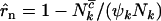 and
and 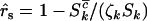 for nonsense and splice site mutations. In practice, of course these cannot be determined since the counts
for nonsense and splice site mutations. In practice, of course these cannot be determined since the counts 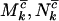 , and
, and 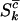 are unobservable. However, in the limit where
are unobservable. However, in the limit where 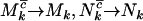 , and
, and 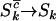 (i.e., most base pairs are neutral under mutation) and assuming positive selection, these probabilities converge to
(i.e., most base pairs are neutral under mutation) and assuming positive selection, these probabilities converge to 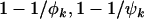 , and
, and 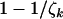 , respectively. Since these expressions are also the fractions of mutations that occur in excess of expectation under no selection, this is perhaps the natural answer to the question as to what fraction of mutations can be attributed to the cancer. However, because
, respectively. Since these expressions are also the fractions of mutations that occur in excess of expectation under no selection, this is perhaps the natural answer to the question as to what fraction of mutations can be attributed to the cancer. However, because 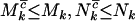 , and
, and 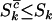 , these expressions are lower bounds on the proportion of mutations actually involved in the disease process. At the opposite extreme one might have
, these expressions are lower bounds on the proportion of mutations actually involved in the disease process. At the opposite extreme one might have 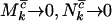 , and
, and 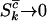 , implying that all nonsilent mutations are (more weakly) pathogenic. More strictly, therefore, provided selection pressure is positive, the estimated proportions of missense, nonsense, and splice site mutations that are pathogenic are described by the inequalities
, implying that all nonsilent mutations are (more weakly) pathogenic. More strictly, therefore, provided selection pressure is positive, the estimated proportions of missense, nonsense, and splice site mutations that are pathogenic are described by the inequalities
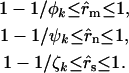 |
These ranges reflect the idea that one cannot distinguish between a small number of mutations conferring strong selection and a larger number of mutations conferring weak selection.
In the case of negative selection, no information upon 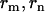 , or
, or 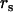 can be provided. However, since η,μ,ν>0, lower bounds for any negative selection pressures
can be provided. However, since η,μ,ν>0, lower bounds for any negative selection pressures 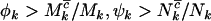 , and
, and 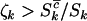 result. Therefore, under negative selection pressure,
result. Therefore, under negative selection pressure, 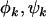 , and
, and 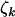 provide upper bounds on the proportion of base pairs that are selected against by cancer.
provide upper bounds on the proportion of base pairs that are selected against by cancer.
TESTS OF SELECTION
So far we have developed a model (summarized by Equation 4) for mutations across a section of screened genome, which incorporates selection pressures. The observable parameters include the silent, missense, nonsense, and splice site mutation counts 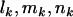 , and
, and 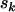 , along with base pair counts
, along with base pair counts 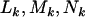 , and
, and 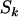 , respectively. The unobservable parameters include the selection pressures, represented in the model by the terms φk, ψk, and
, respectively. The unobservable parameters include the selection pressures, represented in the model by the terms φk, ψk, and 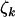 , and the parameters
, and the parameters 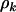 , which represent a mutation rate for each type k.
, which represent a mutation rate for each type k.
The principal aim of the analysis is to examine evidence for selection. That is, determine if any of φk, ψk, and 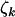 are distinct from unity, indicating that a subset of the point mutations detected across the screened genome are related to the genesis of cancer in the sampled tumors. Unfortunately the selection pressures cannot be directly measured, and we have to rely on the observables to derive estimates, denoted
are distinct from unity, indicating that a subset of the point mutations detected across the screened genome are related to the genesis of cancer in the sampled tumors. Unfortunately the selection pressures cannot be directly measured, and we have to rely on the observables to derive estimates, denoted 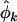 ,
, 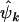 , and
, and 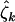 . Although these estimates may differ from unity, the population values φk, ψk, and
. Although these estimates may differ from unity, the population values φk, ψk, and 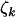 could still be unity, differences arising due to natural random fluctuation in the observed mutation counts, rather than a genuine underlying effect. This is an inference problem, typically examined by considering two alternatives, the null hypothesis (neutral selection) and an alternative hypothesis (selection by cancer), where a significance value provides a measure of the strength of evidence supporting the alternative hypothesis.
could still be unity, differences arising due to natural random fluctuation in the observed mutation counts, rather than a genuine underlying effect. This is an inference problem, typically examined by considering two alternatives, the null hypothesis (neutral selection) and an alternative hypothesis (selection by cancer), where a significance value provides a measure of the strength of evidence supporting the alternative hypothesis.
For the problem in hand, there are many possible alternative hypotheses, and it is desirable to know which one is most likely. We can express the possible scenarios in terms of the four hypotheses described in Table 2. 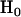 is the global null hypothesis of no selection pressure.
is the global null hypothesis of no selection pressure. 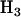 is the most general alternative, allowing for positive or negative selection pressures, heterogeneous across mutation types. The alternatives
is the most general alternative, allowing for positive or negative selection pressures, heterogeneous across mutation types. The alternatives 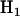 and
and 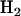 assume that the selection pressures, as measured by φ, ψ, and ζ, are common across mutation types. These reflect the notion that different mutation types are equally likely to be associated with disease. H1 differs from H2 in further assuming that the selection pressures for missense, nonsense, and splice site mutations are equal.
assume that the selection pressures, as measured by φ, ψ, and ζ, are common across mutation types. These reflect the notion that different mutation types are equally likely to be associated with disease. H1 differs from H2 in further assuming that the selection pressures for missense, nonsense, and splice site mutations are equal.
TABLE 2.
Properties of the four hypotheses H0, H1, H2, and H3
| Model | Nesting structure | Selection pressures across strata | Selection pressures across mutation category | No. selection pressures | Description |
|---|---|---|---|---|---|
| H0 | H0 | Unity | Unity | 0 | 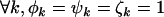 |
| H1 | H1 H0 H0
|
Equal | Equal | 1 | 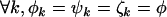 |
| H2 | H2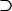 H1 H1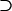 H0 H0
|
Equal | Distinct | 3 | 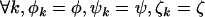 |
| H3 | H3 H2 H2 H1 H1 H0 H0
|
Distinct | Distinct | 33 | 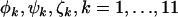 |
Nesting structures of the four models, variation in selection pressure between strata (mutation type), and variation between mutation categories (missense, nonsense, or splice) are indicated. The number of resulting selection pressures follows. Parameterizations are indicated in the final column.
To develop estimates of the selection pressures and statistical tests to evaluate the above hypotheses, we must first eliminate the unknown nuisance parameters 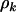 . A natural approach is to argue conditionally on the total number of mutations tk of each type. These are the minimal sufficient statistics for
. A natural approach is to argue conditionally on the total number of mutations tk of each type. These are the minimal sufficient statistics for 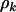 , which are consequently eliminated. This leads (from Equation 4) to a product of multinomial distributions:
, which are consequently eliminated. This leads (from Equation 4) to a product of multinomial distributions:
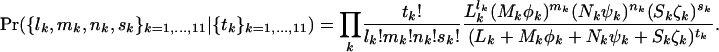 |
(5) |
Again, it is interesting to note that there is a strong analogy with the analysis of cohort studies in epidemiology. The mutation types are equivalent to different strata in a cohort analysis, while the terms Lk, Mk, Nk, and Sk are equivalent to the “person years” at risk.
Several different tests for selection can now be developed, on the basis of the possible alternative hypotheses. Likelihood methods provide a natural framework for hypothesis tests, and maximum-likelihood estimation can also be used to estimate the various parameters. One standard approach to hypothesis testing is to use a likelihood-ratio test (LRT), on the basis of the ratio of the maximum likelihoods under the two hypotheses. In large samples LRT statistics are distributed, asymptotically, as chi-square distributions, provided that the hypotheses are nested. However, the likelihood ratio may not be analytically tractable, in which case numerical methods may need to be applied. An alternative class of test statistics is score tests, based on the first derivative U of the log-likelihood at the null. The score statistic is of the form 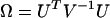 , where V is the covariance of U, and this also has an asymptotic chi-square distribution, provided that the null hypothesis is nested in the alternative hypothesis. The LRT and score test also have similar efficiency (Cox and Hinkley 1974). If the data set is small (e.g., Stephens et al. 2005), or hypotheses are not nested, the chi-square approximations may not be reliable. In these cases, the significance levels may be estimated by simulation, using permutation arguments in which mutations are randomly permuted among tumors. Permutation arguments could be applied to LRTs but are more easily applied to score tests since they can often be computed without iteration. Further details of these methods can be found in Sorensen and Gianola (2002).
, where V is the covariance of U, and this also has an asymptotic chi-square distribution, provided that the null hypothesis is nested in the alternative hypothesis. The LRT and score test also have similar efficiency (Cox and Hinkley 1974). If the data set is small (e.g., Stephens et al. 2005), or hypotheses are not nested, the chi-square approximations may not be reliable. In these cases, the significance levels may be estimated by simulation, using permutation arguments in which mutations are randomly permuted among tumors. Permutation arguments could be applied to LRTs but are more easily applied to score tests since they can often be computed without iteration. Further details of these methods can be found in Sorensen and Gianola (2002).
These statistics can be used to test the null hypothesis H0 against alternatives H1, H2, or H3. Which test is to be preferred depends on the likely alternative hypothesis and comes down to a trade-off between generality of the alternative and number of unknown parameters in the test (respectively 1, 3, and 33). In general, our preference is for a primary test of H0 vs. H2 rather than H0 vs. H1, since this reflects the fact that missense, nonsense, and splice mutations are likely to behave differently. This is reinforced by results of Yang et al. (2003), where differences between missense and nonsense selection pressures were observed. A test to compare H1 vs. H2 would also help resolve this issue. The more general test of H0 vs. H3 would be expected to lack power given the large number of parameters, unless there is strong reason to suspect a marked tendency for mutations of a particular type to be pathogenic. One would then want to conduct a separate test of H2 vs. H3 to evaluate whether there is evidence of heterogeneity in selection pressure across mutation types.
We note that the hypotheses H0, H1, H2, and H3 are nested, as indicated in Table 2. The LRT comparing any pair of hypotheses then has a standard chi-square distribution, provided data sets are of sufficient size. The likelihoods used in this ratio are derived from the conditional likelihood in Equation 5, maximized with respect to all free parameters (selection pressures) within each hypothesis. The number of degrees of freedom required to implement the LRT is simply the difference between the numbers of free parameters for each hypothesis, as indicated in Table 2. However, for data sets of moderate size, the resulting significance levels may not be accurate, so exact score tests can be derived to provide more reliable information.
The score test for comparison of H0 vs. H2 is based on the first derivatives U of the log-likelihood with respect to the selection pressures φ, ψ, and ζ, evaluated at the null hypothesis 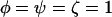 . Then using Equation 5, this leads to a test statistic of the form
. Then using Equation 5, this leads to a test statistic of the form 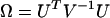 , where
, where
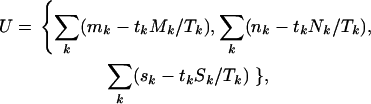 |
and covariance
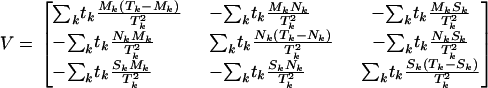 |
In the epidemiological literature this is just the Mantel–Haenszel test for cohort studies. The terms 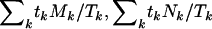 , and
, and 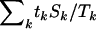 can be thought of as the expected numbers of missense, nonsense, and splice site mutations under the null hypothesis of no selection, given the total number of mutations observed. Exact significance levels can be computed by simulating the null distribution of the test statistic, randomly reallocating the tk mutations of type k to the four categories in the ratios Lk:Mk:Nk:Sk.
can be thought of as the expected numbers of missense, nonsense, and splice site mutations under the null hypothesis of no selection, given the total number of mutations observed. Exact significance levels can be computed by simulating the null distribution of the test statistic, randomly reallocating the tk mutations of type k to the four categories in the ratios Lk:Mk:Nk:Sk.
A similar test for H0 vs. H1 can be constructed by combining missense, nonsense, and splice site mutations into a single category for each mutation type.
For the more general test of H0 vs. H3,
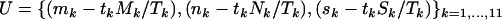 |
and
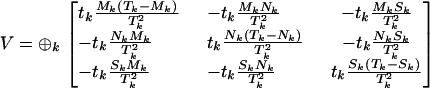 |
For this comparison the relevant likelihoods can be maximized without iteration, so that exact tests based on a LRT are also straightforward. The likelihood-ratio statistic in this case can be obtained by maximizing the conditional likelihood in Equation 5 with respect to all the selection pressures:
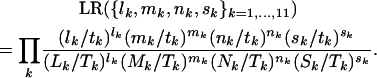 |
It is also desirable to derive comparisons between the different alternatives. The most straightforward test for H1 vs. H3 or H2 vs. H3 is a likelihood-ratio test. In this case there is no straightforward exact test and numerical iterative methods are required (only H2 vs. H3 was implemented in application). However, an exact test for H1 vs. H2 can be achieved with the methods described in functional domains below. Viabilities of likelihood-ratio and score tests for comparisons between the different hypotheses are summarized in Table 3.
TABLE 3.
Preferential comparisons between hypotheses H0, H1, H2, and H3
| H0 | H1 | H2 | H3 | |
|---|---|---|---|---|
| H0 | — | Score | Score | Score, LRT |
| H1 | Score | — | Scorea | LRTb |
| H2 | Score | Scorea | — | LRTb |
| H3 | Score, LRT | LRTb | LRTb | — |
All comparisons can be implemented as exact tests.
The more general score statistic described in the domains section is required.
The exact test requires an optimization routine to maximize the likelihood, as the likelihood ratio does not take a closed form.
We note that the tests described above can be applied individually to missense, nonsense, or splice site mutations. For example, one may want to examine the significance of the selection pressure upon nonsense mutations 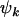 , irrespective of the missense and splice site selection pressures
, irrespective of the missense and splice site selection pressures 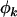 and
and 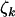 . That is, test null hypothesis
. That is, test null hypothesis 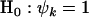 against
against 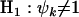 . This can be achieved by simply removing all terms involving
. This can be achieved by simply removing all terms involving 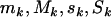 from the expressions above, redefining the totals
from the expressions above, redefining the totals 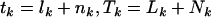 in terms of silent and nonsense counts only, and proceeding as before. Tests specific to missense or splice variants are achieved similarly.
in terms of silent and nonsense counts only, and proceeding as before. Tests specific to missense or splice variants are achieved similarly.
Parameter estimation under the various alternative hypotheses can be found by implementing maximum-likelihood methods. Under the most general model H3, these are given (from Equation 5) by the usual odds ratios:
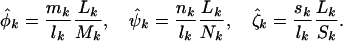 |
Under the more restrictive models H1 and H2, maximum-likelihood estimates can be obtained only iteratively. However, from Equation 4, counts 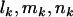 , and
, and 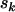 are Poisson with means ρkLk, ρkMkφk, ρkNkψk, and ρkSkζk, respectively. Thus estimation for the various unknown parameters, including the nuisance parameters, can be obtained by implementing Poisson regression with a log link function in one of the standard packages capable of fitting generalized linear models (for example, Matlab, Stata, or S plus). The terms log(Lk), log(Mk), log(Nk), and log(Sk) are handled as offsets in the analysis. Confidence intervals for the parameters, based on standard asymptotic arguments, are also produced by these routines.
are Poisson with means ρkLk, ρkMkφk, ρkNkψk, and ρkSkζk, respectively. Thus estimation for the various unknown parameters, including the nuisance parameters, can be obtained by implementing Poisson regression with a log link function in one of the standard packages capable of fitting generalized linear models (for example, Matlab, Stata, or S plus). The terms log(Lk), log(Mk), log(Nk), and log(Sk) are handled as offsets in the analysis. Confidence intervals for the parameters, based on standard asymptotic arguments, are also produced by these routines.
The terms 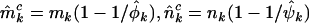 , and
, and 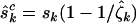 estimate the minimum number of pathogenic missense, nonsense, and splice site mutations, respectively (assuming positive selection pressure). That is, they estimate the minimum number of mutations attributable to the disease process. It may be preferable to replace
estimate the minimum number of pathogenic missense, nonsense, and splice site mutations, respectively (assuming positive selection pressure). That is, they estimate the minimum number of mutations attributable to the disease process. It may be preferable to replace 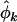 ,
, 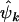 , and
, and 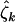 by common estimates across mutation types (i.e., assume hypothesis H2) if there is no evidence of heterogeneity.
by common estimates across mutation types (i.e., assume hypothesis H2) if there is no evidence of heterogeneity.
Although the main interest is in the selection parameters associating mutation with disease, it is also possible to make simultaneous inferences about the mutation spectra 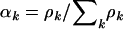 . The maximum-likelihood estimates for
. The maximum-likelihood estimates for 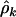 from Equation 4 are given by
from Equation 4 are given by 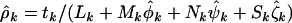 . These mutation rates are generated naturally in the Poisson regression analyses. The estimates
. These mutation rates are generated naturally in the Poisson regression analyses. The estimates 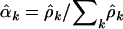 can then be calculated for any of the alternative hypotheses.
can then be calculated for any of the alternative hypotheses.
Finally, we observe that a parameter of common biological interest is the probability that a mutation is silent. By writing 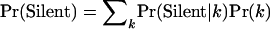 , this can readily be estimated by
, this can readily be estimated by
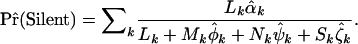 |
(6) |
Note that this estimated probability is a function of DNA sequence, selection pressure, and mutation spectra, under the alternative hypotheses. This will correct for any biases arising from the heuristic ratio 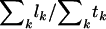 .
.
FUNCTIONAL DOMAINS
The above models can be extended to evaluate the possibility of differential selection according to additional covariates. This might include, specifically, functional domains or subsets of genes. Suppose that the DNA sequence screened is divided into H domains of interest. Suppose furthermore that we are interested in detecting differential selection toward missense mutations between these domains. Nonsense and splice site variants are (for the moment) ignored. The rate of silent mutations per nucleotide will be constant across these domains by the hypothesis of neutral selection. We are therefore interested only in missense mutations throughout these domains. By defining domain-specific selection pressures, mutation counts, and base pair counts, the density  can be expressed as the following product of Poisson distributions:
can be expressed as the following product of Poisson distributions:
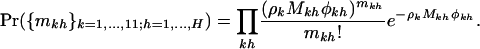 |
This term is essentially Equation 4 restricted to just missense mutations, where all terms except 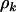 have an additional subscript h referring to the domain. The mutation rates
have an additional subscript h referring to the domain. The mutation rates 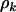 are assumed constant across domains, implying that significant differences in mutation counts between domains are due to variation in selection pressure alone.
are assumed constant across domains, implying that significant differences in mutation counts between domains are due to variation in selection pressure alone.
Assuming independence of mutations between domains, the total number of mutations across all domains for each mutation type k will also have a Poisson distribution, with the rate equal to the sum of individual rates across domains, so that
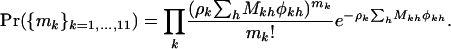 |
As we wish to compare domain-specific selection pressures irrespective of the overall selection pressure, it is natural to consider the distribution of the domain- and type-specific mutation counts, conditional on the total type-specific counts:
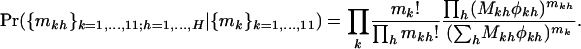 |
(7) |
We now assume that either model H1 or H2 applies. That is, we assume in what follows that selection pressures are unrelated to mutation type k. Thus we can write 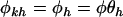 . We impose the additional constraint
. We impose the additional constraint 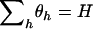 to ensure that the overall selection pressure
to ensure that the overall selection pressure 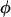 is uniquely specified. By summing across domains we note that
is uniquely specified. By summing across domains we note that 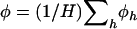 represents the mean selection pressure across domains. Note furthermore that the null hypothesis of constant selection across domains is represented by
represents the mean selection pressure across domains. Note furthermore that the null hypothesis of constant selection across domains is represented by 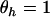 . The conditional likelihood in Equation 7 then reduces to
. The conditional likelihood in Equation 7 then reduces to
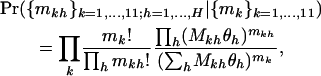 |
which is dependent only on the interaction parameters 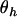 and not on the nuisance parameter
and not on the nuisance parameter 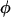 . Although this expression contains H unknown parameters, the constraint
. Although this expression contains H unknown parameters, the constraint 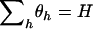 means that there are only
means that there are only 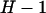 d.f. We thus define the following
d.f. We thus define the following 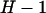 parameters,
parameters,
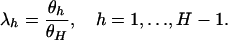 |
These substitute into the conditional likelihood through the inverse transformation,
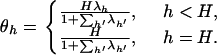 |
A likelihood-ratio test may be used to the test for differences by domain, but it will require iteration and a score test again provides a simpler alternative. The test statistic is of the form Ω = UTV−1U, where 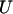 denotes the partial differentials of the log-likelihood
denotes the partial differentials of the log-likelihood 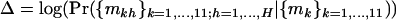 evaluated at the null hypothesis,
evaluated at the null hypothesis,
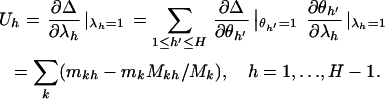 |
This is a natural statistic, summing the difference between observed and expected counts across the mutation types k. The term 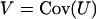 is then a matrix of multinomial covariances summed across the mutation types; that is,
is then a matrix of multinomial covariances summed across the mutation types; that is,
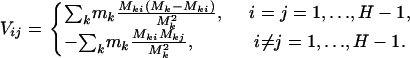 |
To apply the test, the null distribution of the statistic Ω is generated by simulating the counts mkh across the domains in the ratios Mk1:Mk2: … :MkH.
Score statistics for domain effects upon nonsense or splice site variants can be constructed analogously.
We note that this approach can be used to derive an analogous test of H1 vs. H2, by regarding missense, nonsense, and splice site mutations as arising from three distinct domains. That is 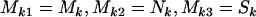 .
.
If 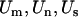 and
and 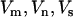 represent the statistics for missense, nonsense, and splice terms, a test
represent the statistics for missense, nonsense, and splice terms, a test 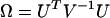 for domain effects for all mutations can be constructed, where
for domain effects for all mutations can be constructed, where 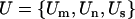 and
and 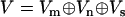 .
.
A test for domain effects under hypothesis H1 can be constructing by combining the missense, nonsense, and splice site information into single counts for each mutation and domain type, kh.
Under alternative hypothesis H3 the selection pressures 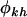 can be redefined in terms of the domain-averaged pressure and interaction terms. That is,
can be redefined in terms of the domain-averaged pressure and interaction terms. That is, 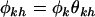 , where
, where 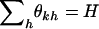 . The likelihood in Equation 7 again has a redundancy of parameters. Defining the transformation
. The likelihood in Equation 7 again has a redundancy of parameters. Defining the transformation 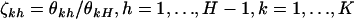 gives
gives 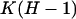 parameters. The likelihood-ratio statistic in this case can be calculated directly as
parameters. The likelihood-ratio statistic in this case can be calculated directly as
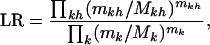 |
from which a LRT can be applied to test for missense mutation domain effects. Nonsense and splice site variants are tested similarly. A combined single test for all missense, nonsense, and splice site domain effects follows by multiplying their respective likelihood ratios together into one statistic.
Finally, we note that these tests will lack power if the number of domains is large, unless domains can be grouped in a biologically meaningful manner.
APPLICATION TO PROTEIN KINASE GENE MUTATIONS IN BREAST CANCER
These methods were applied to the screen of 25 breast tumors through ∼32 Mb of DNA from 518 protein kinase genes (Stephens et al. 2005). The results of this experiment are summarized in Table 1, and the results of various tests are given in Table 4. Significance levels for score tests were based on 100,000 Monte Carlo simulations, except for the H2 vs. H3 comparisons, which were based on either asymptotic assumptions or exact LRTs using 10,000 simulations, due to the time constraints of iterative methods. Parameter estimates are given in Table 5.
TABLE 4.
Significance levels comparing hypotheses H0, H1, H2, and H3
| Mutation types | H0vs. H1 | H0vs. H2 | H0vs. H3 | H1vs. H2 | H2vs. H3 |
|---|---|---|---|---|---|
| All nonsynonymous point mutations | 0.0943 | 0.00029 | 0.0046 | 0.00096 | 0.6368a |
| Missense mutations | NA | 0.2684 | 0.3905 | NA | 0.0829b |
| Nonsense mutations | NA | 0.0013 | 0.0105 | NA | 0.4164b |
| Splice site mutations | NA | 0.0068 | 0.0067 | NA | 0.0824b |
Significances are split into missense, nonsense, splice, and combined effects. Exact tests were based upon 100,000 Monte Carlo simulations, unless otherwise indicated.
An asymptotic likelihood-ratio test.
Based upon 10,000 simulations.
TABLE 5.
Parameter estimates
| Parameter | Missense | Nonsense | Splice |
|---|---|---|---|
Selection pressure estimates 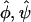 , and , and 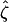
|
1.37 | 4.48 | 4.59 |
| Selection pressure confidence intervals (95%) | 0.76–2.49 | 2.00–9.99 | 1.75–12.02 |
No. of mutations 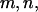 and and 
|
58 | 12 | 6 |
Estimated minimum no. of pathogenic mutations 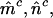 and and 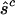
|
15.8 | 9.3 | 4.7 |
Selection pressure estimates under hypothesis H2 with 95% confidence intervals. The minimum numbers of pathogenic mutations are provided for missense, nonsense, and splice site, variants. Mutation counts are summed across mutation types k.
There was no significant evidence of heterogeneity in selection pressure by mutation type, as determined by the test of H2 vs. H3 (P = 0.64). H2 provided a superior fit than H1 (P = 0.00096), indicating variation in selection pressure between missense, nonsense, and splice site effects. The proposed test of H0 vs. H2 provided strong evidence of selection (P = 0.00029). In fact, the selection pressure estimates were similar for nonsense and splice mutations (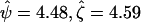 ) but substantially lower for missense mutations (
) but substantially lower for missense mutations (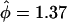 ), although the value for missense mutations is still greater than unity. The nonsense selection pressure was significantly different from unity (P = 0.0013). Similarly, the splice site selection pressure was significant (P = 0.0068), but the missense selection pressure was not (P = 0.27). These observations suggest that the breast cancers exhibit stronger selection toward the protein-truncating mutations.
), although the value for missense mutations is still greater than unity. The nonsense selection pressure was significantly different from unity (P = 0.0013). Similarly, the splice site selection pressure was significant (P = 0.0068), but the missense selection pressure was not (P = 0.27). These observations suggest that the breast cancers exhibit stronger selection toward the protein-truncating mutations.
From Table 5, an estimated minimum of 29.8 of the 76 nonsynonymous mutations are pathogenic (39%), including 9.3 of the 12 nonsense mutations. In fact, 10 of the nonsense mutations arose from one sample, PD0119 (see Stephens et al. 2005 for details), suggesting that, at least for this tumor, cells accumulate growth advantage from multiple mutations, similar to the model of colorectal cancer evolution given by Little and Wright (2003).
The silent rate under the null hypothesis was estimated using Equation 6 to be 0.2460, suggesting that the silent:nonsilent ratio in the absence of selection will typically be ∼1:3. Different mutation spectra or genome composition could substantially alter this, however.
Protein kinase domains are involved in the phosphorylation of proteins in signaling pathway cascades. These are highly conserved, implying that mutations within these regions are likely to affect protein function. Such mutations may enhance cell division and offer good candidate oncogenic variants. Conversely, the cell may not tolerate mutations in such important regions, possibly inducing apoptosis, in which case protein-changing mutations will be avoided. Either scenario is indicative of selection pressures that vary according to the relative position of mutations with respect to the kinase domains. As such, all genes were split into three regions: prekinase, kinase, and postkinase, with each region having a separate selection parameter. Further details of the set of kinase genes can by found in the supplementary information in Stephens et al. (2005). The exact positioning of these domains within the coding sequence can be found from a variety of database sources such as Ensembl (see Hubbard et al. 2005). Significant deviation of these parameters between the domains was then examined. The selection pressure for nonsense mutations varied significantly by position (P = 0.0053), with 9/12 mutations lying 3′ to the kinase domain. This variation might suggest truncation of a regulatory domain or domains, while leaving the kinase domain intact, free to drive the cancer.
In summary, these methods provide a straightforward and robust statistical approach to evaluating the impact of mutations identified in genomic screens on the development of cancer. Practical application to the kinase data has shown that the methods were able to demonstrate significant selection that varied among missense, nonsense, and splice variants. The use of such methods will be increasingly important in large-scale screens for somatic mutations in cancer.
Acknowledgments
We thank the Wellcome Trust and the Institute of Cancer Research for their support. D.F.E. is a Principal Research Fellow of Cancer Research United Kingdom.
References
- Béroud, C., and T. Soussi, 2003. The UMD-p53 database: new mutations and analysis tools. Hum. Mutat. 21: 176–181. [DOI] [PubMed] [Google Scholar]
- Cox, D. R., and D. V. Hinkley, 1974. Theoretical Statistics. Chapman & Hall, London.
- Davies, H., G. R. Bignell, C. Cox, P. Stephens, S. Edkins et al., 2002. Mutations of the BRAF gene in human cancer. Nature 417: 949–954. [DOI] [PubMed] [Google Scholar]
- Futreal, P. A., L. Coin, M. Marshall, T. Down, T. Hubbard et al., 2004. A census of human cancer genes. Nat. Rev. Cancer 4: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, N., and Z. Yang, 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11(5): 725–736. [DOI] [PubMed] [Google Scholar]
- Hall, B. G., 1990. Spontaneous point mutations that occur more often when advantageous than when neutral. Genetics 126: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, T., D. Andrews, M. Caccamo, G. Cameron, Y. Chen et al., 2005. Ensembl 2005. Nucleic Acids Res. 33: D447–D453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, M. P., and E. G. Wright, 2003. A stochastic carcinogenesis model incorporating genomic instability fitted to colon cancer data. Math. Biosci. 183: 111–134. [DOI] [PubMed] [Google Scholar]
- Sorensen, D., and D. Gianola, 2002. Likelihood, Bayesian and MCMC Methods in Quantitative Genetics. Springer, Berlin/Heidelberg, Germany/New York.
- Stephens, P., S. Edkins, H. Davies, C. Greenman, C. Cox et al., 2005. A screen of the complete protein kinase gene family reveals diverse patterns of somatic mutations in human breast cancer. Nat. Genet. 37: 590–592. [DOI] [PubMed] [Google Scholar]
- Valverde, J. R., J. Alonso, I. Palacious and A. Pestana, 2005. RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet. 6: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., S. Ro and B. Rannala, 2003. Likelihood models of somatic mutation and codon substitution in cancer genes. Genetics 165: 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]