

Abstract
The aim of this article is to develop an integrated-likelihood (IL) approach to estimate the genetic differentiation between populations. The conventional maximum-likelihood (ML) and pseudolikelihood (PL) methods that use sample counts of alleles may cause severe underestimations of FST, which means overestimations of θ = 4Nm, when the number of sampling localities is small. To reduce such bias in the estimation of genetic differentiation, we propose an IL method in which the mean allele frequencies over populations are regarded as nuisance parameters and are eliminated by integration. To maximize the IL function, we have developed two algorithms, a Monte Carlo EM algorithm and a Laplace approximation. Our simulation studies show that the method proposed here outperforms the conventional ML and PL methods in terms of unbiasedness and precision. The IL method was applied to real data for Pacific herring and African elephants.
THE study of population structures arises in many contexts in population genetics, and a wide variety of patterns of population structures have been modeled, including the stepping-stone, island, and mixed population models (e.g., Kimura and Weiss 1964; Wright 1969; Millar 1987; Rannala and Hartigan 1995). Statistical methods have also advanced, reflecting the discovery of fine-scale markers and the development of modeling for population structures (e.g., Weir 1996; Balding et al. 2003).
To study the genetic structures of natural populations, samples are usually taken from several localities. However, in many situations, there are no specific boundaries or obvious regional units, and therefore subpopulations cannot be well defined beforehand. In the case of mixing populations, it may be possible to apply individual assignment methods based on multilocus genotypes (Pritchard et al. 2000; Falush et al. 2003; Manel et al. 2005) to identify a finite number of subpopulations. However, if the population has a continuous structure and consists of a large number of subpopulations, assuming a metapopulation or an infinite-island model is a natural way to estimate the genetic differentiation between subpopulations (Pannell and Charlesworth 2000; Rousset 2003; Hanski and Gaggiotti 2004).
The metapopulation model considers a universe of demes (subpopulations) that have peculiar genetic structures and introduces a distribution of allele frequencies among these demes. When the allele frequencies are distributed as a Dirichlet distribution, the probability distribution for the sampled counts of alleles at each locus can be explicitly expressed as a Dirichlet-multinomial distribution (Rannala and Hartigan 1996; Weir 1996; Holsinger 1999; Kitada et al. 2000; Corander et al. 2003; Rousset 2003). In this model, the variance of the allele frequencies among sampling localities is closely related to FST or θ = 4Nm (Wright 1969; Rannala and Hartigan 1996; Kitada et al. 2000; Balding 2003; Kitada and Kishino 2004). The parameter FST or θ is explicitly included in the statistical model and can therefore be estimated using likelihood methods. The maximum-likelihood (ML) estimation has been discussed by Lange (1995), Kitada et al. (2000), and Balding (2003). Furthermore, a pseudolikelihood (PL) estimation was proposed by Rannala and Hartigan (1996) as a variant of the ML method.
Because the parameters that express population differentiation, FST and θ, are related to the variance, as discussed above, the difference in the sampled counts of alleles among sampling localities contains information about these parameters. A larger number of sampling localities improve the precision of the estimates with these parameters (Kitada and Kishino 2004). However, sampling from many localities is often difficult in genetic studies of wildlife populations. In such cases, the ML and PL methods cause difficulties, as in the estimation of the variance in a normal distribution. The ML estimator of σ2 based on a sample of size n, 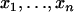 , from a normal distribution N(μ, σ2) is given by
, from a normal distribution N(μ, σ2) is given by 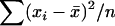 , whereas an unbiased estimator is given by
, whereas an unbiased estimator is given by 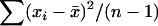 . The only difference is in the denominator. However, if the sample size n is small, the amount of bias in the ML estimator cannot be ignored. This might be the case for the estimation of FST or θ in the metapopulation model.
. The only difference is in the denominator. However, if the sample size n is small, the amount of bias in the ML estimator cannot be ignored. This might be the case for the estimation of FST or θ in the metapopulation model.
An unbiased estimation of σ2 described above can be achieved using several different likelihood adjustments, known as conditioning, marginalization, and integration (Lindsey 1996; Berger et al. 1999). Unfortunately, the conditional- and marginal-likelihood methods cannot be applied to a Dirichlet-multinomial distribution because no appropriate statistics that separate the likelihood can be obtained. On the other hand, the integrated-likelihood (IL) method does not require such separation. Instead, integration with respect to nuisance parameters must be undertaken. Typically, the IL function does not have a closed form. However, the recent development of computational methods allows us to deal with it: for example, the EM algorithm (McLachlan and Krishnan 1997), the Markov chain Monte Carlo (MCMC) method (Robert and Casella 2004), and the Laplace approximation (Pawitan 2001).
In this article, we use an IL approach to the metapopulation model and develop a new method to precisely estimate important parameters of interest, FST and θ. Two algorithms, a Monte Carlo EM algorithm with an MCMC and the Laplace approximation, are applied to maximize the IL function. The performance of the IL method is evaluated with numerical simulations and compared with the ML and PL estimation methods. Microsatellite allele frequencies in Pacific herring and mtDNA restriction fragment length polymorphism (RFLP) haplotypes of African elephants are analyzed. The impact of bias on the inference of genetic differentiation is also discussed.
MODELS AND METHODS
Statistical modeling:
Consider a sample taken from multiple localities in a metapopulation having an infinite-island model. Suppose that K demes or subpopulations are drawn from the metapopulation and 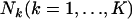 alleles of individuals are counted for L loci. Let
alleles of individuals are counted for L loci. Let 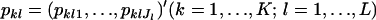 be a vector of the true allele frequencies at the lth locus in the kth subpopulation, where
be a vector of the true allele frequencies at the lth locus in the kth subpopulation, where 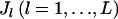 is the number of different alleles at the lth locus, and
is the number of different alleles at the lth locus, and 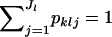 . Let
. Let 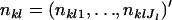 denote a vector of observed allele frequencies at the lth locus at the kth subpopulation. Under the assumption of random sampling at each locality, the distribution of nkl, given pkl, can be assumed to be multinomial with the probability function
denote a vector of observed allele frequencies at the lth locus at the kth subpopulation. Under the assumption of random sampling at each locality, the distribution of nkl, given pkl, can be assumed to be multinomial with the probability function
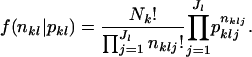 |
Here, 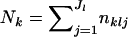 . Note that the allele counts at each locus, given the true allele frequencies, are independent among subpopulations. We also assume linkage equilibrium. Therefore, the allele counts are also independent over all loci within each subpopulation. We assume that the distribution of pkl at the lth locus in the kth subpopulation follows a Dirichlet distribution, with the probability density function
. Note that the allele counts at each locus, given the true allele frequencies, are independent among subpopulations. We also assume linkage equilibrium. Therefore, the allele counts are also independent over all loci within each subpopulation. We assume that the distribution of pkl at the lth locus in the kth subpopulation follows a Dirichlet distribution, with the probability density function
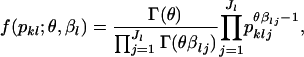 |
where θ is a scale parameter that is a vector common to all loci, and 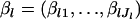 are the mean allele frequencies at the lth locus satisfying
are the mean allele frequencies at the lth locus satisfying 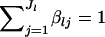 . This distribution was originally derived by Wright (1945, 1951) as a diffusion approximation to the discrete generation Wright's model. A justification for the continuous generation model has been given by Rannala and Hartigan (1996).
. This distribution was originally derived by Wright (1945, 1951) as a diffusion approximation to the discrete generation Wright's model. A justification for the continuous generation model has been given by Rannala and Hartigan (1996).
In this model, the variance of the jth allele frequency for the lth locus, pklj, is given by
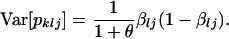 |
(1) |
The larger the value of θ, the smaller the genetic differentiation among subpopulations, and vice versa. In fact, genetic differentiation is expressed as
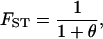 |
(2) |
which was given by Wright (1969), Rannala and Hartigan (1996), Balding (2003), and Kitada and Kishino (2004). Hence, θ is a parameter that controls the degree of genetic differentiation among subpopulations.
Estimation by the ML method:
The marginal distribution of the observed random vector nkl is given by a Dirichlet-multinomial distribution as follows:
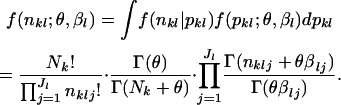 |
(3) |
The parameters to be estimated are θ and 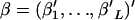 . Because the mutual independence of data is assumed, the overall likelihood for these parameters based on Equation 3 is defined as
. Because the mutual independence of data is assumed, the overall likelihood for these parameters based on Equation 3 is defined as
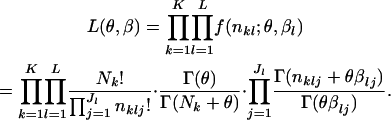 |
The ML estimator of θ is given by maximizing the likelihood function L(θ, β) (Lange 1995; Weir 1996; Kitada et al. 2000). The parameter FST can be estimated with Equation 2.
Estimation by the PL method:
Rannala and Hartigan (1996) used a PL approach to reduce computational time in optimizing the original likelihood L(θ, β). In their method, the vector of the nuisance parameter 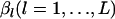 is replaced with the average values for observed allele frequencies throughout the population,
is replaced with the average values for observed allele frequencies throughout the population,
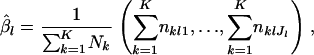 |
and 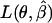 is maximized as if
is maximized as if 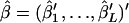 were the true parameters.
were the true parameters.
Estimation by the IL method:
As an alternative to the two likelihood estimations described above, we propose an IL approach to estimate θ. This means that the free parameters in 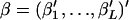 are regarded as nuisance parameters and are eliminated from L(θ, β) by integration. We use an integrated-likelihood function for θ, defined as
are regarded as nuisance parameters and are eliminated from L(θ, β) by integration. We use an integrated-likelihood function for θ, defined as
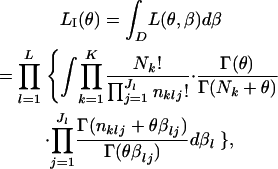 |
(4) |
where D is the parameter space of β. This treatment can be regarded as a kind of Bayesian estimation using a noninformative prior for β. In fact, when the prior 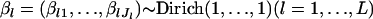 is assumed, the density function is
is assumed, the density function is
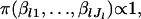 |
and therefore 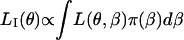 holds. Unfortunately, an explicit form for LI(θ) cannot be obtained in the metapopulation model.
holds. Unfortunately, an explicit form for LI(θ) cannot be obtained in the metapopulation model.
We consider two alternative algorithms for the maximization of LI(θ). One is the Monte Carlo EM (MCEM) algorithm (Wei and Tanner 1990). The EM algorithm converges on the maximum value of LI(θ). However, as is well known, the convergence of the EM algorithm is slow. For this reason, we use mainly the Laplace approximation (Pawitan 2001). The program for the latter method is available from the authors upon request. Detailed descriptions of both of the methods are given in appendixes a and b.
SIMULATION STUDIES
Simulation scenarios:
We evaluated the estimation performance of the ML, PL, and IL methods with numerical simulations. For simplicity, we considered only cases in which the same number of individuals are collected from each of the sampled subpopulations and the different loci share the same mean allele frequencies.
Simulation data were generated as follows. A simulation scenario was specified by the number of subpopulations sampled (K), sample size (Nk), the number of loci (L), the number of alleles (Jl), mean allele frequencies (βl), and the degree of genetic differentiation (FST). The parameters used in this study are as follows:
Number of subpopulations sampled: K = 2, 3, 4, 5, 7, 10, 15
Sample size: Nk ≡ N = 50, 100, 200
Number of loci: L = 1, 3, 5, 10, 20
Number of alleles: Jl ≡ J = 2, 5, 10
Mean allele frequencies:
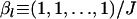 and
and 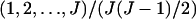
Genetic differentiation: FST = 0.01, 0.05, 0.1, 0.4.
Once specifying a simulation scenario, the observed allele frequencies, 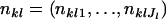 , were generated from a Dirichlet-multinomial distribution in Equation 3 for each locus. After the data were generated, we estimated the parameter θ using the ML, PL, and IL methods, and then we estimated FST with the relationship FST = 1/(1 + θ). The number of simulation replicas was fixed at 1000 throughout each simulation scenario. Then, the mean and standard deviation of 1000 estimates of FST were assessed for each method. In the IL method, we used the Laplace approximation because its computation cost is lower than that of the MCEM algorithm. However, before undertaking more comprehensive simulations, we checked the consistency of the two algorithms by using some of the simulation data sets.
, were generated from a Dirichlet-multinomial distribution in Equation 3 for each locus. After the data were generated, we estimated the parameter θ using the ML, PL, and IL methods, and then we estimated FST with the relationship FST = 1/(1 + θ). The number of simulation replicas was fixed at 1000 throughout each simulation scenario. Then, the mean and standard deviation of 1000 estimates of FST were assessed for each method. In the IL method, we used the Laplace approximation because its computation cost is lower than that of the MCEM algorithm. However, before undertaking more comprehensive simulations, we checked the consistency of the two algorithms by using some of the simulation data sets.
Simulation results:
We first examined the efficacy of the Laplace approximation for our IL approach. Figure 1 compares the estimates based on the two different algorithms used to find the maximum value of the IL function. Simulation data were generated for the case where K = 5, N = 50, L = 10, J = 5, and FST = 0.1. As shown in Figure 1, estimates made with the Laplace approximation tended to be close to those made with the MCEM sequence. The difference of computation times between the two algorithms was noteworthy. For the situation above, the estimation with the Laplace approximation took ∼6 sec, which was comparable to those with the ML and PL methods (∼3 and 1 sec, respectively), while MCEM spent ∼40 min up to the 40th step on a Pentium-IV 3.2-GHz PC. Note that the computing time varied little among data sets utilized within a same scenario, but depended much on the number of subpopulations sampled, loci, and alleles.
Figure 1.—

Comparison of estimates for FST by the IL function, based on the two different algorithms, the MCEM and Laplace approximation. Simulation data were generated for K = 5, N = 50, L = 10, J = 5, and FST = 0.1.
Figure 2 shows the estimation performance of the ML, PL, and IL methods for FST = 0.05 for various values of K, the number of loci L, the number of alleles J, and the sample size N. We used the set of parameters K = 5, N = 50, L = 10, and J = 5 as baseline values and changed each of them. Only the results for the case of 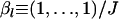 are shown here, because differences in mean allele frequencies between scenarios had little impact on estimation performance. As expected, severe underestimation of FST was observed with both the ML and PL methods, especially for small values of K. In contrast, less bias was observed with the IL method. These results demonstrate that the IL method outperforms the ML and PL procedures in terms of minimizing bias in all cases.
are shown here, because differences in mean allele frequencies between scenarios had little impact on estimation performance. As expected, severe underestimation of FST was observed with both the ML and PL methods, especially for small values of K. In contrast, less bias was observed with the IL method. These results demonstrate that the IL method outperforms the ML and PL procedures in terms of minimizing bias in all cases.
Figure 2.—




Means of ML, PL, and IL estimates with the 5th and 95th percentiles (vertical lines). The true FST was fixed at 0.05. The baseline scenario was K = 5, L = 10, J = 5, and N = 50 with 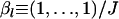 . x-Axes are different for each part: the number of sampling localities (K), the number of loci (L), the number of alleles (J), and the sample size common to sampling localities (N).
. x-Axes are different for each part: the number of sampling localities (K), the number of loci (L), the number of alleles (J), and the sample size common to sampling localities (N).
With all three methods, increasing the number of subpopulations sampled, K, reduced both bias and variance. On the other hand, increasing the number of loci, the number of alleles per locus, and the sample size had little effect on bias, but led to a reduction in variance. The latter phenomenon has been reported by Balding (2003) for the ML method. These results demonstrate that the number K is very important for the precise estimation of FST.
More detailed results for N = 50, L = 10, and J = 5 are shown in Table 1. All the estimates were negatively biased. The ML and PL estimates were very similar in all cases and had large negative biases, especially when FST was <0.1 and the number of sampling localities was less than five. On the other hand, the PL estimates had a much smaller bias under these conditions. When FST was as large as 0.4, the bias of the ML and PL estimates was moderate and similar to that of the IL estimate.
TABLE 1.
Mean and standard deviation (SD) of FST from 1000 simulations at various levels of FST for different numbers of sampling localities
| ML
|
PL
|
IL
|
|||||
|---|---|---|---|---|---|---|---|
| FST | K | Mean | SD | Mean | SD | Mean | SD |
| 0.01 | 2 | 0.00014 (−98.6) | 0.0007 | 0.00008 (−99.2) | 0.0006 | 0.00691 (−30.9) | 0.0051 |
| 5 | 0.00421 (−57.9) | 0.0026 | 0.00415 (−58.5) | 0.0026 | 0.00952 (−4.8) | 0.0031 | |
| 10 | 0.00690 (−31.0) | 0.0021 | 0.00686 (−31.4) | 0.0021 | 0.00962 (−3.8) | 0.0022 | |
| 15 | 0.00802 (−19.8) | 0.0017 | 0.00799 (−20.1) | 0.0017 | 0.00984 (−1.6) | 0.0018 | |
| 0.05 | 2 | 0.0154 (−69.2) | 0.0082 | 0.0134 (−73.2) | 0.0093 | 0.0356 (−28.7) | 0.0105 |
| 5 | 0.0368 (−26.5) | 0.0061 | 0.0365 (−27.0) | 0.0061 | 0.0461 (−7.9) | 0.0066 | |
| 10 | 0.0434 (−13.3) | 0.0045 | 0.0432 (−13.5) | 0.0045 | 0.0481 (−3.7) | 0.0046 | |
| 15 | 0.0457 (−8.5) | 0.0037 | 0.0457 (−8.7) | 0.0037 | 0.0489 (−2.1) | 0.0038 | |
| 0.1 | 2 | 0.0431 (−56.9) | 0.0140 | 0.0408 (−59.2) | 0.0135 | 0.0704 (−29.6) | 0.0162 |
| 5 | 0.0784 (−21.6) | 0.0101 | 0.0779 (−22.1) | 0.0101 | 0.0902 (−9.8) | 0.0104 | |
| 10 | 0.0887 (−11.3) | 0.0072 | 0.0885 (−11.5) | 0.0072 | 0.0946 (−5.4) | 0.0073 | |
| 15 | 0.0929 (−7.1) | 0.0060 | 0.0927 (−7.3) | 0.0060 | 0.0968 (−3.2) | 0.0061 | |
| 0.4 | 2 | 0.260 (−35.1) | 0.515 | 0.231 (−43.2) | 0.475 | 0.313 (−21.6) | 0.467 |
| 5 | 0.352 (−12.1) | 0.272 | 0.341 (−14.7) | 0.271 | 0.367 (−8.3) | 0.264 | |
| 10 | 0.378 (−5.5) | 0.188 | 0.373 (−6.8) | 0.189 | 0.383 (−4.3) | 0.185 | |
| 15 | 0.385 (−3.8) | 0.150 | 0.382 (−4.6) | 0.151 | 0.388 (−3.1) | 0.148 | |
The bias (percentage) is shown in parentheses. The sample size is N = 50, and the number of loci and alleles are L = 10 and J = 5, respectively. The mean allele frequencies are fixed at 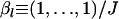 .
.
APPLICATIONS TO REAL DATA
Pacific herring:
The first example, a marine fish species, shows a case with a low FST-value. A total of 1263 fish were taken from three localities, Akkeshi, Yudounuma Lake, and Funkawan Bay, which are located on the east coast of Hokkaido in Japan. Table 2 shows the observed allele frequencies at five microsatellite loci, which were in Hardy–Weinberg equilibrium in each sample (Sato 2004). A large value for the gene flow rate, which means a small value for FST, was observed (Table 3). In this case, the estimates made with the ML and PL methods were much smaller than that made with the IL method. This result is consistent with a phenomenon observed in the simulation studies; that is, there is a clear difference between the IL and ML or PL estimates when the FST-value is small.
TABLE 2.
Allele frequencies at five loci in Pacific herring
| Locus 1
|
Locus 2
|
Locus 3
|
Locus 4
|
Locus 5
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alleles | AK | YD | FK | AK | YD | FK | AK | YD | FK | AK | YD | FK | AK | YD | FK |
| 1 | 0 | 10 | 1 | 14 | 26 | 5 | 0 | 3 | 1 | 3 | 0 | 4 | 0 | 0 | 8 |
| 2 | 0 | 1 | 1 | 5 | 4 | 5 | 3 | 15 | 6 | 11 | 1 | 1 | 0 | 0 | 12 |
| 3 | 0 | 16 | 0 | 2 | 0 | 1 | 0 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | 7 |
| 4 | 8 | 9 | 2 | 38 | 27 | 34 | 34 | 40 | 19 | 37 | 4 | 31 | 0 | 0 | 8 |
| 5 | 3 | 7 | 8 | 43 | 29 | 63 | 0 | 9 | 5 | 179 | 148 | 169 | 0 | 0 | 43 |
| 6 | 5 | 20 | 14 | 155 | 103 | 134 | 0 | 0 | 66 | 11 | 11 | 55 | 0 | 0 | 9 |
| 7 | 1 | 16 | 12 | 133 | 165 | 128 | 21 | 9 | 18 | 3 | 4 | 7 | 3 | 0 | 44 |
| 8 | 0 | 0 | 3 | 177 | 197 | 149 | 71 | 66 | 71 | 241 | 300 | 257 | 0 | 0 | 14 |
| 9 | 25 | 28 | 22 | 68 | 73 | 128 | 11 | 4 | 20 | 56 | 76 | 46 | 0 | 3 | 37 |
| 10 | 63 | 42 | 59 | 33 | 27 | 45 | 8 | 4 | 4 | 16 | 5 | 72 | 1 | 0 | 8 |
| 11 | 64 | 57 | 59 | 41 | 28 | 75 | 45 | 19 | 49 | 3 | 8 | 6 | 16 | 6 | 9 |
| 12 | 157 | 121 | 99 | 57 | 44 | 47 | 27 | 61 | 48 | 4 | 6 | 11 | 0 | 12 | 11 |
| 13 | 9 | 32 | 35 | 4 | 0 | 54 | 1 | 1 | 8 | 7 | 41 | 35 | 25 | 51 | 43 |
| 14 | 23 | 10 | 16 | 0 | 3 | 10 | 202 | 128 | 135 | 0 | 5 | 33 | 36 | 31 | 39 |
| 15 | 15 | 11 | 25 | 11 | 50 | 13 | 62 | 68 | 58 | 42 | 34 | 36 | 9 | 28 | 67 |
| 16 | 6 | 89 | 37 | 10 | 8 | 8 | 162 | 160 | 124 | 2 | 0 | 10 | 23 | 21 | 43 |
| 17 | 131 | 57 | 48 | 7 | 6 | 8 | 34 | 32 | 66 | 9 | 12 | 17 | 64 | 29 | 35 |
| 18 | 12 | 16 | 49 | 1 | 1 | 0 | 49 | 122 | 77 | 0 | 0 | 7 | 25 | 22 | 23 |
| 19 | 127 | 55 | 157 | 7 | 2 | 8 | 28 | 20 | 63 | 20 | 24 | 54 | 21 | 26 | 15 |
| 20 | 2 | 14 | 98 | 0 | 0 | 2 | 33 | 19 | 21 | 38 | 12 | 7 | 35 | 35 | 35 |
| 21 | 25 | 50 | 43 | 0 | 0 | 1 | 0 | 1 | 31 | 119 | 60 | 35 | 54 | 43 | 10 |
| 22 | 29 | 21 | 32 | 0 | 1 | 1 | 10 | 9 | 18 | 5 | 29 | 14 | 21 | 20 | 18 |
| 23 | 2 | 6 | 28 | 0 | 0 | 2 | 3 | 2 | 7 | 0 | 8 | 17 | 35 | 82 | 33 |
| 24 | 11 | 8 | 15 | 0 | 0 | 4 | 1 | 0 | 3 | 0 | 5 | 0 | 31 | 26 | 21 |
| 25 | 2 | 9 | 4 | 0 | 0 | 1 | 0 | 1 | 2 | 0 | 0 | 1 | 45 | 39 | 86 |
| 26 | 1 | 1 | 0 | 1 | 0 | 0 | 46 | 68 | 23 | ||||||
| 27 | 6 | 12 | 6 | 0 | 0 | 1 | 56 | 34 | 28 | ||||||
| 28 | 5 | 1 | 4 | 0 | 0 | 2 | 52 | 18 | 12 | ||||||
| 29 | 3 | 3 | 7 | 0 | 0 | 1 | 25 | 46 | 41 | ||||||
| 30 | 21 | 10 | 5 | 84 | 33 | 40 | |||||||||
| 31 | 4 | 8 | 3 | 21 | 39 | 22 | |||||||||
| 32 | 4 | 5 | 3 | 25 | 12 | 21 | |||||||||
| 33 | 2 | 5 | 10 | 29 | 22 | 15 | |||||||||
| 34 | 1 | 4 | 2 | 3 | 4 | 7 | |||||||||
| 35 | 5 | 3 | 4 | 4 | 2 | 1 | |||||||||
| 36 | 7 | 11 | 4 | 6 | 10 | 9 | |||||||||
| 37 | 9 | 7 | 7 | 7 | 8 | 4 | |||||||||
| 38 | 2 | 2 | 1 | 1 | 8 | 12 | |||||||||
| 39 | 7 | 1 | 0 | 0 | 6 | 5 | |||||||||
| 40 | 3 | 0 | 1 | 0 | 5 | 6 | |||||||||
| 41 | 4 | 0 | 1 | 1 | 4 | 0 | |||||||||
| 42 | 1 | 1 | 0 | 1 | 0 | 1 | |||||||||
| 43 | 0 | 8 | 0 | 1 | 1 | 0 | |||||||||
| 44 | 0 | 3 | 0 | 0 | 0 | 1 | |||||||||
| 45 | 1 | 4 | 1 | ||||||||||||
AK, YD, and FK refer to Akkeshi, Yudounuma Lake, and Funkawan Bay, respectively.
TABLE 3.
Estimation results for case studies
| Case | Method | θ | FST |
|---|---|---|---|
| Pacific herring | ML | 156.6 (13.4) | 0.0063 (0.00054) |
| PL | 167.9 (13.8) | 0.0059 (0.00049) | |
| IL | 91.7 (9.2) | 0.0108 (0.00109) | |
| African elephants | ML | 1.86 (0.486) | 0.350 (0.109) |
| PL | 1.97 (0.503) | 0.337 (0.103) | |
| IL | 1.67 (0.433) | 0.374 (0.114) |
The values in parentheses are the standard errors.
African elephants:
Next, we used the mtDNA RFLP haplotype data given in Table 3 of Rannala and Hartigan (1996), which are a modification of the original data of Georgiadis et al. (1994). In this study, 10 haplotypes were evaluated in 270 elephants from 10 populations in Kenya, Zimbabwe, Botswana, and South Africa. As shown in Table 3, the relative differences in the estimates of θ and FST were smaller than those observed in the Pacific herring. This is due to the greater number of sampling localities and the higher value of FST compared with those of the Pacific herring population.
DISCUSSION
In this article, we have proposed a new method for the estimation of genetic differentiation between populations, based on the IL function, to reduce the bias observed in the ML and PL methods, which have been used previously in this field. Our simulation results demonstrate that the estimation performance of the IL method is much better than that of the conventional ML and PL methods in terms of unbiasedness and precision. Differences in estimation performance were observed, especially when the number of subpopulations sampled, K, was small or the level of genetic differentiation, FST, was low. In fact, the number of subpopulations sampled is often restricted in studies of the population genetics of wildlife. Furthermore, FST could be small for species that have no specific boundaries, such as birds and fishes, as shown in the data analysis for Pacific herring. In this sense, the IL method is strongly recommended for the estimation of FST.
We focused on the estimation of FST and θ for the metapopulation models. Thus, we regarded the mean allele frequencies as nuisance parameters. The elimination of nuisance parameters has been one of the central problems in statistics. As shown in our simulation studies, the large degree of bias in the ML and PL estimates of FST did not decrease even when the number of loci was increased. These phenomena are typical cases of so-called Neyman–Scott problems (Neyman and Scott 1948), in which the dimension of the nuisance parameter increases with the number of observations. There are several possible ways to eliminate nuisance parameters: the conditional, marginal, and IL methods. Of these, we used the IL method because no suitable statistics for conditional- or marginal-likelihood methods were available in our model. Although a small amount of bias was still observed in the IL estimates, the improvement in estimation performance using the IL method is considerable.
The resources for sampling surveys in the field are always limited. Given the total sample size, it is recommended that the number of sampling localities be as large as possible by sacrificing the number of individuals sampled at each locality (Figure 2). Increased sample size at each locality yielded little reduction in bias in the estimation of FST, although it could decrease the estimation variance. To better understand the role played by the sampling scheme of this study, we conducted further simulation studies, in which the total sample size over all sampling localities was fixed. The simulation scenario was set at FST = 0.05, L = 10, and J = 5, with 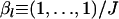 . The total sample size, K · N, was fixed at 600. As shown in Figure 3, an increase in K improved estimation performance. Furthermore, for small K, the variance was small because of the large sample size. However, the bias was still large when the values of K were small, although the sample size was considerably larger than that in the case of a large K. In the field, it is often much more difficult to visit many sites than to study many individuals at each site. Therefore, our IL method may be especially valuable in such circumstances.
. The total sample size, K · N, was fixed at 600. As shown in Figure 3, an increase in K improved estimation performance. Furthermore, for small K, the variance was small because of the large sample size. However, the bias was still large when the values of K were small, although the sample size was considerably larger than that in the case of a large K. In the field, it is often much more difficult to visit many sites than to study many individuals at each site. Therefore, our IL method may be especially valuable in such circumstances.
Figure 3.—

Estimation performance for a fixed total sample size of K · N = 600. The number of loci and alleles are L = 10 and J = 5, respectively.
Our statistical estimation assumes the Dirichlet distribution for allele frequencies. This assumption is well suited to the metapopulation with an island structure. On the other hand, examination of robustness of the IL estimation based on such an assumption is of interest. For this purpose, we conducted a small simulation study assuming a simple scenario for non-Dirichlet with biallelic loci. Now consider sampling from a metapopulation that consists of a large number of subpopulations, and suppose that allele frequencies at a locus in a subpopulation are either (0.6, 0.4) or (0.4, 0.6). The proportion of those two kinds of subpopulations in the metapopulation is assumed to be 1:1, which means that allele frequencies at a locus of a sampled subpopulation are (0.6, 0.4) with the probability 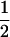 . In this case, the mean allele frequencies over the metapopulations are (0.5, 0.5), and the variance is calculated as 0.5(0.4 − 0.5)2 + 0.5(0.6 − 0.5)2 = 0.01. Hence, FST is 0.04 (= 0.01/0.25). Under this scenario with K = 5, N = 50, and L = 10, we estimated FST using the IL method. The results are summarized in Table 4. Although the estimation performances are slightly different between the cases of Dirichlet and non-Dirichlet, the IL method based on the Dirichlet assumption still performed well in both cases. Note that the ML and PL methods were still heavily biased in this case (not shown here). This simulation is not comprehensive, but it suggests that our method may be useful for other genetic models.
. In this case, the mean allele frequencies over the metapopulations are (0.5, 0.5), and the variance is calculated as 0.5(0.4 − 0.5)2 + 0.5(0.6 − 0.5)2 = 0.01. Hence, FST is 0.04 (= 0.01/0.25). Under this scenario with K = 5, N = 50, and L = 10, we estimated FST using the IL method. The results are summarized in Table 4. Although the estimation performances are slightly different between the cases of Dirichlet and non-Dirichlet, the IL method based on the Dirichlet assumption still performed well in both cases. Note that the ML and PL methods were still heavily biased in this case (not shown here). This simulation is not comprehensive, but it suggests that our method may be useful for other genetic models.
TABLE 4.
The IL estimation of FST in cases of Dirichlet and non-Dirichlet allele frequencies
| Dirichlet
|
Non-Dirichlet
|
|||
|---|---|---|---|---|
| K | Mean | SD | Mean | SD |
| 5 | 0.0388 (−2.88) | 0.0123 | 0.0379 (−5.37) | 0.0094 |
| 10 | 0.0396 (−0.94) | 0.0082 | 0.0383 (−4.24) | 0.0065 |
The bias (percentage) is shown in parentheses. The true value of FST is 0.04. The sample size is N = 50, and the number of loci and alleles are L = 10 and J = 5, respectively.
In this article, we assumed that the parameter θ is common throughout all loci. However, as discussed by Balding (2003), θ can vary across loci because of different mutation rates and/or selection effects. Even if the mutation rates are incorporated into the model and assumed to be different among loci, the distributional form of the Dirichlet holds through θl = 4Nm + 4Nμl or FST,l = 1/(1 + 4Nm + 4Nμl), where m is a migration rate and μl is a mutation rate at the lth locus (Wright 1969). In this case, it is possible to estimate the value of θ locus by locus. However, assuming a random effect model for θ over all loci is also a promising alternative method. This assumption can make the estimation not only easier but also more efficient. The first advantage is the reduction in the dimension of the parameter. The second advantage arises from the concept of “borrowing strength” across loci in the context of an empirical Bayes procedure (Beaumont and Rannala 2004). Incorporating such random effects leads to further hierarchical modeling. Our IL method, however, can be extended to such modeling. Furthermore, Bayesian computational methods and algorithms are also possible candidate methods (Beaumont and Rannala 2004). Meanwhile, the presence of the selection effects unfortunately breaks down the Dirichlet assumption for the allele frequencies, and even the variance of allele frequency does not have a simple form as in Equation 1 (Wright 1969). Hence, the extension of our method may not be straightforward. These topics are problems to be investigated in the future.
Linkage disequilibrium is an important factor in practice. Under linkage disequilibrium, a probability distribution of composite genotypes for biallelic cases is given in Kitada and Kishino (2004). In this article, a simulation study was conducted for assessing the estimation performance of FST under various levels of linkage disequilibrium. The results (shown in Table 3 in Kitada and Kishino 2004) suggested that the maximum-likelihood estimation based on smaller numbers of sampling localities tended to cause underestimation of FST, which is a similar phenomenon observed in our article under linkage equilibrium. Meanwhile, they showed that the level of linkage disequilibrium had little impact on the amount of estimation bias although it slightly affected the estimation variance for FST. Therefore, we expect that our integrated-likelihood approach is also effective to improve the estimation performance in the linkage disequilibrium model, and the amount of reduction of bias is not related to the level of linkage disequilibrium. Although the likelihood function is complicated in the model, this topic also warrants further investigation.
Acknowledgments
The authors are grateful to two anonymous reviewers for their constructive comments on the original version of this manuscript.
APPENDIX A: ESTIMATION WITH THE MCEM ALGORITHM
At first, we briefly illustrate a typical case of the EM algorithm (McLachlan and Krishnan 1997). Let Y and Z be the observed and latent/missing random vectors, respectively. Let f(y, z; ω) denote the joint probability density function, where ω is a vector of parameters. The log-likelihood function for ω, based on the complete data (Y, Z), is given by lc(ω) = log f(y, z; ω). To find the maximum-likelihood estimator, the likelihood based on the observed data y, 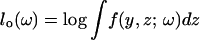 must be maximized. However, some models do not have closed forms for lo(ω). Instead of directly maximizing lo(ω), the EM algorithm alternates between the following two steps:
must be maximized. However, some models do not have closed forms for lo(ω). Instead of directly maximizing lo(ω), the EM algorithm alternates between the following two steps:
E step: compute Q(ω|ω[t]) = E[lc(ω)|Y, ω[t]].
M step: define ω[t+1] as a value of ω that maximizes Q(ω|ω[t]).
In our metapopulation model, the observed data are the set 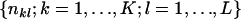 , and the latent variables are
, and the latent variables are 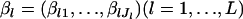 . Although βl is originally a parameter vector, it can be regarded as a random vector with a flat prior in the context of Bayesian estimation. The complete log-likelihood function is given by
. Although βl is originally a parameter vector, it can be regarded as a random vector with a flat prior in the context of Bayesian estimation. The complete log-likelihood function is given by
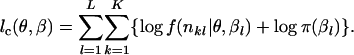 |
Note that the second term is ignorable because π(βl) ∝ 1. The original EM algorithm expects an explicit formula for the conditional expectation Q in the E-step. However, Q in our model does not have a closed form because the log-likelihood is complicated by the Dirichlet-multinomial distribution. To evaluate the function Q, we use a Monte Carlo integration via the MCMC method.
We calculate 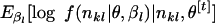 . To generate the random variables
. To generate the random variables 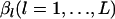 from the conditional distribution of βl, given
from the conditional distribution of βl, given 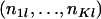 and θ[t],
and θ[t],
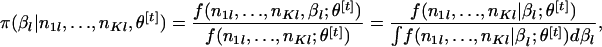 |
the following Metropolis–Hasting sampling method (e.g., Robert and Casella 2004) is used.
Sample a candidate vector βl* from a proposal distribution
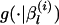 .
.- Compute an acceptance ratio defined by
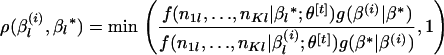
- Set
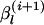 as
as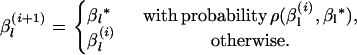
Let mt be the sample size of the Monte Carlo integration at the tth step in the EM algorithm. Precision of the Monte Carlo integration depends on the sample size. To obtain better precisions at later steps in the E-step, which affect convergence of θ[t], we increase mt by t as
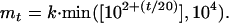 |
The first mt/5 samples are discarded as constituting the burn-in period to make the MCMC sequences independent of initial values of β. The value k is the length of thinning; that is, every kth simulation draw is kept as an output of the Monte Carlo data. This is to reduce the autocorrelation in each sequence. We set k = 10. The function Q(θ|θ[t]) is approximately assessed as
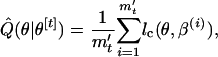 |
where m′t is the length of the effective MCMC sequence. If the standard error of the parameter estimate is required, it can be evaluated using the observed information as follows:
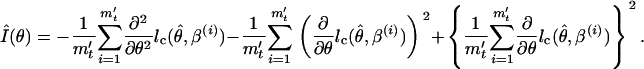 |
APPENDIX B: ESTIMATION WITH THE LAPLACE APPROXIMATION
Our integrated likelihood can be expressed as
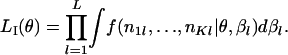 |
(B1) |
We illustrate the approximation by considering the contribution made by the lth locus [say 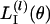 ] to LI(θ). The Laplace approximation is often used in statistics. The approximation depends on a second-order Taylor expansion of
] to LI(θ). The Laplace approximation is often used in statistics. The approximation depends on a second-order Taylor expansion of 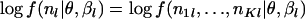 . Let
. Let 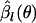 be the value of βl that maximizes log f(nl | θ, βl) for fixed θ. Then,
be the value of βl that maximizes log f(nl | θ, βl) for fixed θ. Then, 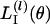 can be approximated (ignoring factors not depending on θ) as
can be approximated (ignoring factors not depending on θ) as
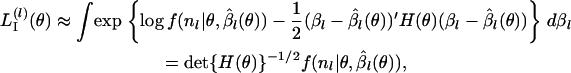 |
where
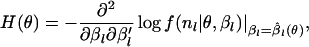 |
and det{H(θ)} denotes the determinant of H(θ). Taking the logarithm of 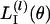 , we get
, we get
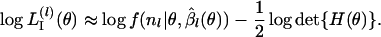 |
(B2) |
The first term on the right-hand side of Equation B2 is the profile log-likelihood of θ. In fact, Equation B2 can be regarded as an approximate modified profile log-likelihood of θ to reduce the estimation bias (Lindsey 1996; Pawitan 2001). The estimates 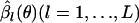 have of course uncertainty due to their own nature. However, if only the first term of the right-hand side of Equation B2 is present,
have of course uncertainty due to their own nature. However, if only the first term of the right-hand side of Equation B2 is present, 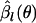 acts as if the true value, and therefore the uncertainty in
acts as if the true value, and therefore the uncertainty in 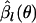 is not taken into account. This is a reason why the IL method outperforms the conventional ML and PL methods.
is not taken into account. This is a reason why the IL method outperforms the conventional ML and PL methods.
In the metapopulation model, it is not possible to obtain an exact formula for H(θ) or 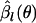 . To maximize
. To maximize 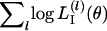 , we use an iterative method based on automatic differentiation (Skaug and Fournier 2006), which is implemented with the statistical software ADMB-RE (http://otter-rsch.com/admbre/admbre.html). To improve the accuracy of the Laplace approximation, we changed the variables in the integral of Equation B1 using the logistic transformation
, we use an iterative method based on automatic differentiation (Skaug and Fournier 2006), which is implemented with the statistical software ADMB-RE (http://otter-rsch.com/admbre/admbre.html). To improve the accuracy of the Laplace approximation, we changed the variables in the integral of Equation B1 using the logistic transformation 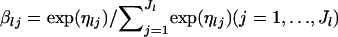 with the constraint
with the constraint 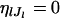 . The computation time is much less than that required for the MCEM algorithm.
. The computation time is much less than that required for the MCEM algorithm.
References
- Balding, D. J., 2003. Likelihood-based inference for genetic correlation coefficients. Theor. Popul. Biol. 63: 221–230. [DOI] [PubMed] [Google Scholar]
- Balding, D. J., M. Bishop and C. Cannings, 2003. Handbook of Statistical Genetics, Ed. 2. John Wiley & Sons, Chichester, UK.
- Beaumont, M. A., and B. Rannala, 2004. The Bayesian revolution in genetics. Nat. Rev. Genet. 5: 251–261. [DOI] [PubMed] [Google Scholar]
- Berger, J. O., L. Brunero and R. L. Wolpert, 1999. Integrated likelihood methods for eliminating nuisance parameters. Stat. Sci. 14: 1–22. [Google Scholar]
- Corander, J., P. Waldmann and M. J. Sillanpää, 2003. Bayesian analysis of genetic differentiation between populations. Genetics 163: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadis, N., L. Bischof, A. Templeton, J. Patton, W. Karesh et al., 1994. Structure and history of African elephant populations. I. Eastern and southern Africa. J. Hered. 85: 100–104. [DOI] [PubMed] [Google Scholar]
- Hanski, I., and O. E. Gaggiotti, 2004. Ecology, Genetics, and Evolution of Metapopulations. Academic Press, San Diego.
- Holsinger, K. E., 1999. Analysis of genetic diversity in geographically structured populations: a Bayesian perspective. Hereditas 130: 245–255. [Google Scholar]
- Kimura, T., and G. H. Weiss, 1964. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, S., and H. Kishino, 2004. Simultaneous detection of linkage disequilibrium and genetic differentiation of subdivided populations. Genetics 167: 2003–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, S., T. Hayashi and H. Kishino, 2000. Empirical Bayes procedure for estimating genetic distance between populations and effective population size. Genetics 156: 2063–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, K., 1995. Application of the Dirichlet distribution to forensic match probabilities. Genetica 96: 107–117. [DOI] [PubMed] [Google Scholar]
- Lindsey, J. K., 1996. Parametric Statistical Inference. Oxford University Press, New York.
- Manel, S., O. E. Gaggiotti and R. S. Waples, 2005. Assignment methods: matching biological questions with appropriate techniques. Trends Ecol. Evol. 20: 136–142. [DOI] [PubMed] [Google Scholar]
- McLachlan, G. J., and T. Krishnan, 1997. The EM Algorithm and Extensions. John Wiley & Sons, New York.
- Millar, R. B., 1987. Maximum likelihood estimation of mixed stock fishery composition. Can. J. Fish. Aquat. Sci. 44: 583–590. [Google Scholar]
- Neyman, J., and E. L. Scott, 1948. Consistent estimates based on partially consistent observations. Econometrica 16: 1–32. [Google Scholar]
- Pannell, J. R., and B. Charlesworth, 2000. Effects of metapopulation processes on measures of genetic diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355: 1851–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawitan, Y., 2001. In All Likelihood: Statistical Modelling and Inference Using Likelihood. Oxford University Press, New York.
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala, B., and J. A. Hartigan, 1995. Identity by descent in island–mainland populations. Genetics 139: 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala, B., and J. A. Hartigan, 1996. Estimating gene flow in island populations. Genet. Res. 67: 147–158. [DOI] [PubMed] [Google Scholar]
- Robert, C. P., and G. Casella, 2004. Monte Carlo Statistical Methods, Ed. 2. Springer-Verlag, New York.
- Rousset, F., 2003. Inferences from spatial population genetics, pp. 681–712 in Handbook of Statistical Genetics, Ed. 2, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Sato, M., 2004. A study of genetic population structure and impacts of hatchery release of Pacific herrings. M.S. Thesis, Tokyo University of Marine Science and Technology, Tokyo (in Japanese).
- Skaug, H. J., and D. A. Fournier, 2006. Automatic approximation of the marginal likelihood in non-Gaussian hierarchical models. Comp. Stat. Data Anal. (in press).
- Wei, G. C. G., and M. A. Tanner, 1990. A Monte Carlo implementation of the EM algorithm and the poor man's data augmentation algorithm. J. Am. Stat. Assoc. 85: 699–704. [Google Scholar]
- Weir, B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
- Wright, S., 1945. The differential equation of the distribution of gene frequencies. Proc. Natl. Acad. Sci. USA 31: 383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1951. The general structure of populations. Ann. Eugen. 15: 323–354. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1969. Evolution and the Genetics of Populations: The Theory of Gene Frequencies, Vol. 2. University of Chicago Press, Chicago.