

Abstract
Sibships are commonly used in genetic dissection of complex diseases, particularly for late-onset diseases. Haplotype-based association studies have been advocated as powerful tools for fine mapping and positional cloning of complex disease genes. Existing methods for haplotype inference using data from relatives were originally developed for pedigree data. In this study, we proposed a new statistical method for haplotype inference for multiple tightly linked single-nucleotide polymorphisms (SNPs), which is tailored for extensively accumulated sibship data. This new method was implemented via an expectation-maximization (EM) algorithm without the usual assumption of linkage equilibrium among markers. Our EM algorithm does not incur extra computational burden for haplotype inference using sibship data when compared with using unrelated parental data. Furthermore, its computational efficiency is not affected by increasing sibship size. We examined the robustness and statistical performance of our new method in simulated data created from an empirical haplotype data set of human growth hormone gene 1. The utility of our method was illustrated with an application to the analyses of haplotypes of three candidate genes for osteoporosis.
THE human genome has been portrayed as a series of high linkage disequilibrium (LD) regions with limited haplotype diversity (Patil et al. 2001; Gabriel et al. 2002). Several common haplotypes that can be captured by a few tagging single-nucleotide polymorphisms (SNPs) usually account for a majority of genetic variation in genomic regions or candidate genes (Johnson et al. 2001; Patil et al. 2001; Gabriel et al. 2002). Such haplotype patterns observed in empirical studies have triggered the development of the International HapMap Project that aims to determine the common patterns of DNA sequence variation in the human genome (Gibbs et al. 2003). Focusing on these common haplotypes greatly facilitates LD-based mapping analyses, such as those for fine mapping and positional cloning of complex disease genes (Johnson et al. 2001). However, linkage phase information in diploids, such as humans, is usually unknown from genotype data. The determination of haplotypes by experimental methods in large samples is currently time consuming and expensive. Therefore, computational algorithms and statistical methods have been used for large-scale haplotype determination.
Sibships are commonly used with increasing emphasis on genetic studies of complex diseases, particularly those late-onset ones such as Alzheimer's and Parkinson's diseases, for which genotype data of parents of affected individuals are usually not available (Freimer and Sabatti 2004). Extensively accumulated sibship data call for algorithmic and methodological advances in haplotype inference for subsequent fine mapping and positional cloning studies. Currently available computational algorithms and statistical methods of haplotype inference from relatives were originally developed for pedigree data (Lander and Green 1987; Sobel and Lange 1996; O'Connell 2000; Qian and Beckmann 2002). It is unclear how these methods perform in haplotype inference without founder information in sibship data.
Existing methods for pedigree haplotype inference can be broadly classified into two categories: rule-based and likelihood-based haplotyping methods. The rule-based approaches (Wijsman 1987; O'Connell 2000; Tapadar et al. 2000; Qian and Beckmann 2002; Li and Jiang 2003; Gao et al. 2004) are deterministic and fast and thus can handle large pedigrees with dense markers. However, they do not normally provide numerical assessments of the reliability of their results. On the other hand, likelihood-based methods (Lander and Green 1987; Sobel and Lange 1996; Lin and Speed 1997; Abecasis et al. 2002) are typically stochastic and flexible in tackling complex pedigrees, but they are time consuming and thus may not be suitable for large data sets. In particular, most likelihood-based methods for pedigree haplotype inference implicitly assume linkage equilibrium among markers. However, this assumption is contradicted by haplotype block structures in human genomes, as observed ubiquitously in recent empirical data (Patil et al. 2001; Gabriel et al. 2002). The potential problem of using linkage-based software that assumes linkage equilibrium among markers in haplotype inference has been nicely addressed in the analysis of haplotypes within the HPC1 gene (Schaid et al. 2002). Two recent methods, FAMHAP and FBAT, are exceptions in that they can be used to infer haplotype from nuclear families without the assumption of linkage equilibrium among markers (Becker and Knapp 2004; Horvath et al. 2004).
In this study, we proposed a maximum-likelihood method for haplotype inference for multiple tightly linked SNPs, tailored for sibship data. We implemented our method via a well-known expectation-maximization (EM) algorithm without the assumption of linkage equilibrium among markers. The utility of the new method was validated by using a wide variety of simulated and real data sets. We compared our method with commonly used software for haplotype inference, Genehunter (Kruglyak et al. 1996) and FAMHAP (Becker and Knapp 2004).
STATISTICAL METHODS
Suppose that there is a sample of n sibships, and each sibship contains ni individuals. Let 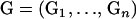 denote the observed genotypes for the n sibships, where
denote the observed genotypes for the n sibships, where 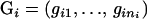 and
and 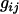 is the genotype of individual j in the sibship i. Let
is the genotype of individual j in the sibship i. Let 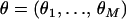 denote population haplotype frequencies, where M is the number of all possible haplotypes in the sample. Let
denote population haplotype frequencies, where M is the number of all possible haplotypes in the sample. Let 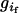 and
and 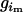 denote the unobserved father and mother genotypes in the sibship i, respectively. The likelihood function of the data can be expressed as
denote the unobserved father and mother genotypes in the sibship i, respectively. The likelihood function of the data can be expressed as
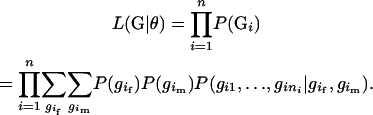 |
(1) |
Suppose that the marker loci are tightly linked so that the recombination in the parental generation can be safely ignored and Hardy–Weinberg equilibrium (HWE) holds true. Then,
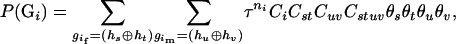 |
(2) |
where the notion 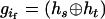 denotes a parental zygote configuration in which the two haplotypes,
denotes a parental zygote configuration in which the two haplotypes, 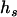 and
and 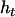 , are compatible with the genotype
, are compatible with the genotype 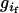 ;
; 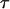 is the probability of offspring genotypes given different mating design and equals 1 when both parents are homozygotes,
is the probability of offspring genotypes given different mating design and equals 1 when both parents are homozygotes, 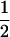 when only one of the parents is a homozygote,
when only one of the parents is a homozygote, 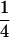 when both parents are two different heterozygotes, and
when both parents are two different heterozygotes, and 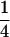 and
and 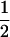 for homozygous and heterozygous offspring, respectively, when both parents are identical heterozygotes;
for homozygous and heterozygous offspring, respectively, when both parents are identical heterozygotes; 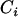 has values of 1 or 0 according to whether parental genotypes
has values of 1 or 0 according to whether parental genotypes 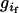 and
and 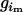 are compatible with their offspring genotypes (
are compatible with their offspring genotypes (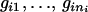 ) or not;
) or not; 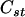 is the combination coefficient of two haplotypes and has values of 1 or 2 according to whether
is the combination coefficient of two haplotypes and has values of 1 or 2 according to whether 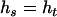 or not;
or not; 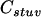 is the combination coefficient of two parental genotypes and has values of 1 or 2 according to whether
is the combination coefficient of two parental genotypes and has values of 1 or 2 according to whether 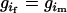 or not; and
or not; and 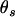 ,
, 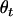 ,
, 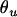 , and
, and 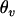 are frequencies of haplotypes
are frequencies of haplotypes 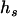 ,
, 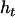 ,
, 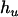 , and
, and 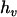 , respectively.
, respectively.
The maximum-likelihood estimate (MLE) of 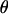 can be found analytically by solving a system of regular equations with a Lagrange multiplicator
can be found analytically by solving a system of regular equations with a Lagrange multiplicator 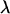 ,
,
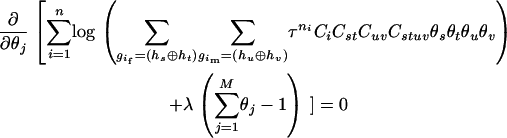 |
(3) |
and
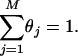 |
(4) |
However, solving a set of M equations is tedious when M is large, and the number M is often unknown a priori. Alternatively, a wide variety of iterative algorithms for solving MLE equations can be used. Among them, the EM method is very general for missing data problems, with fairly simple forms and well-established statistical properties (Dempster et al. 1977). In our method, in the expectation step, the probability of each of the parental zygote configurations that are compatible with their offspring genotypes is calculated by using haplotype frequencies obtained in the previous iteration step. In the maximization step, haplotype frequencies are estimated by counting copies of a specific haplotype in parental zygote configurations, which maximizes the likelihood of Equation 1. Here we combined the expectation and maximization steps and obtained the following expectation-maximization recursion,
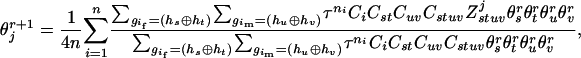 |
(5) |
where 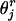 and
and 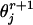 are the estimated frequencies at steps r and r+1, respectively, and
are the estimated frequencies at steps r and r+1, respectively, and 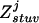 counts how often (zero, one, two, three, or four times) haplotype j occurs in the two haplotype pairs (
counts how often (zero, one, two, three, or four times) haplotype j occurs in the two haplotype pairs (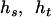 ) and (
) and (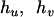 ). Within the EM framework, the approximate variances of the estimates of haplotype frequencies can be obtained by inverting the estimated information matrix (appendix a) (Louis 1982).
). Within the EM framework, the approximate variances of the estimates of haplotype frequencies can be obtained by inverting the estimated information matrix (appendix a) (Louis 1982).
We adopted the following two strategies to improve the computation speed of our method. First, we used an efficient approach of starting iteration suggested by Rohde and Fuerst (2001). That is, at the first iteration step, we took into account only the most likely haplotype pairs in the sample and then estimated haplotype frequencies by counting these most likely haplotype pairs. This approach usually generated the best haplotype estimates while obviating multiple starting iterations. Second, the possible unknown parental genotypes have three to the power of the number of loci in haplotype inference. These possible parental genotypes in Equation 5 were analytically collapsed and reduced given different mating designs. As an example, this collapse approach for sibship data with two siblings is detailed in appendix b. It can be seen from appendix b that haplotype inference using sibship data does not increase the computational burden of our EM algorithm compared with that using unrelated parental data. Also, the increasing sibship size does not result in a larger computational burden since the number of compatible parental genotypes is reduced and more easily resolved given the larger sibship size.
SIMULATIONS
Simulation data were sampled from empirical haplotype data of human growth hormone gene 1 (GH1) (Horan et al. 2003), where GH1 underwent a combination of events including mutation, recombination, and gene conversion. These complexities inherent in the GH1 gene are common in real data applications. Haplotypes of the GH1 gene were experimentally determined in a sample of 154 male British Caucasians. The 15 SNP sites spanned 535 nucleotides in the promoter of the GH1, with minor allele frequencies ranging from 0.3 to 41.2%. Six of these SNPs can be considered as rare variants with minor allele frequencies <5% (0.3–3.6%). Standardized linkage disequilibrium measured by |D′| (Lewontin 1964) among the remaining 9 common SNPs ranged from complete LD (i.e., sites −301 and −308) to effective linkage equilibrium (i.e., sites −1 and +59).
To simplify simulation and comparison of the simulation results, we here considered each sibship with two siblings. In the simulations, in each nuclear family, both parents' haplotypes were randomly selected from the haplotype distribution of the GH1 gene as shown in Table 1 of Horan et al. (2003). Two offspring (i.e., a sib pair) were then randomly generated from their parents. The following six cases were studied:
Our newly proposed EM algorithm using sib-pair data (EM-Sib): We treated parents' genotypes as missing data and used only the sib-pair data for haplotype inference.
The conventional EM algorithm using singleton data (EM-Single): To avoid related subjects, we randomly selected one individual from each sib pair and used this singleton data for haplotype inference by applying the conventional EM algorithm that was designed for random population samples (Excoffier and Slatkin 1995).
Genehunter using sib-pair data (GH-Sib): Genehunter Version 2.1 (http://www.fhcrc.org/labs/kruglyak/Downloads/index.html) was used for haplotype inference for the sib-pair data without using parent information.
Genehunter using family data (GH-Family): Genehunter was used for haplotype inference for the whole-family data in which each nuclear family had both parents and two offspring. To keep the genotyping cost the same for each method, the number of nuclear families used was halved in this case and thus the total number of subjects used for haplotype inference was the same as in the cases using the sib-pair data.
FAMHAP using sibship data (FAMHAP-Sib) without reordering SNP data: The FAMHAP software was obtained from Tim Becker's website (http://www.uni-bonn.de/%7Eumt70e/becker.html).
FAMHAP using sibship data with reordered SNP data (FAMHAP-Sib-R): To obtain the best estimates, we tried several orders of genotype data and took estimates with the largest likelihood.
TABLE 1.
Comparison of VDR haplotype frequencies estimated from different methods
| Haplotypes | EM-Parent | EM-Sib | EM-Single | GH-Sib | GH-Family | FAMHAP-Sib |
|---|---|---|---|---|---|---|
| GTAT | 0.0024 | 0.0010 | — | 0.0738 | 0.0046 | — |
| GTAC | 0.1257 | 0.1208 | 0.1337 | 0.0705 | 0.1147 | 0.1197 |
| GTGT | 0.1453 | 0.1421 | 0.1143 | 0.1066 | 0.1479 | 0.1498 |
| GTGC | — | — | 0.0028 | 0.0246 | 0.0149 | — |
| GCAT | 0.0104 | 0.0068 | 0.0073 | 0.0721 | 0.0321 | 0.0062 |
| GCAC | 0.2180 | 0.2089 | 0.1839 | 0.1197 | 0.1686 | 0.2135 |
| GCGT | 0.2298 | 0.2355 | 0.2865 | 0.1754 | 0.2167 | 0.2436 |
| GCGC | 0.0025 | — | — | 0.0754 | 0.0149 | — |
| ATAT | 0.0021 | 0.002 | 0.0023 | 0.0066 | 0.0046 | — |
| ATAC | 0.0173 | 0.0278 | 0.0001 | 0.0131 | 0.0103 | 0.0204 |
| ATGT | 0.0885 | 0.0826 | 0.0799 | 0.0393 | 0.0768 | 0.0830 |
| ATGC | 0.0021 | 0.0027 | 0.0041 | 0.0213 | 0.0057 | — |
| ACAT | 0.0010 | 0.006 | 0.0018 | 0.0033 | 0.0092 | — |
| ACAC | 0.0404 | 0.0472 | 0.0759 | 0.0443 | 0.0585 | 0.0480 |
| ACGT | 0.1144 | 0.1053 | 0.1075 | 0.0885 | 0.1147 | 0.1061 |
| ACGC | — | — | — | 0.0656 | 0.0057 | — |
| Discrepancy | — | 0.0342 | 0.1059 | 0.3260 | 0.0922 | 0.0338 |
In Tables 1–3, haplotype frequencies that are estimated from parental samples using the conventional EM algorithm (i.e., EM-Parent) are treated as “true” haplotype frequencies in the population. The discrepancy of haplotype frequencies was calculated using Equation 7. Values in italics are those haplotypes whose frequencies deviated >5% from those estimated from the EM-Parent. FAMHAP-Sib and FAMHAP-Sib-R yielded identical results for haplotypes with small numbers of loci.
To compare the performance of each statistical method, we evaluated the following commonly used measurements:
- The discrepancy between the estimated and true sample haplotype frequencies,
where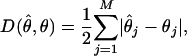
(6) 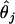 and
and 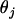 denote, respectively, the estimated and true frequencies of the jth haplotype in the sample.
denote, respectively, the estimated and true frequencies of the jth haplotype in the sample. Error rate—namely, the proportion of individuals with ambiguous phase whose haplotypes are not correctly inferred (Niu et al. 2002).
- Identification rate: A good statistical method for haplotype inference should meet two criteria: (i) high proportion of true haplotypes identified to total true haplotypes in the sample and (ii) low proportion of false (nonexistent) haplotypes to total estimated haplotypes in the sample. The haplotype identification rate is an integrative index measuring these two criteria (Excoffier and Slatkin 1995). We consider that a haplotype is identified as being present in the true sample if its frequency is estimated to be above the threshold value of 1/(2n) (n is sample size). The identification rate can be defined as
where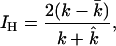
(7) 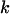 is the number of haplotypes in the true sample,
is the number of haplotypes in the true sample, 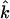 is the number of estimated haplotypes with frequencies above the threshold, and
is the number of estimated haplotypes with frequencies above the threshold, and 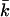 is the number of true haplotypes not identified in the sample.
is the number of true haplotypes not identified in the sample.
SIMULATION RESULTS
We simulated six sample sizes: n = 50, 100, 150, 200, 250, and 300. Simulations were replicated 100 times for each case and the mean results of the 100 simulations were summarized. Comparisons of discrepancies, error rates, and identification rates among different methods are shown in Figures 1 and 2. Our new method for haplotype inference using sibship data (i.e., EM-Sib) has the smallest discrepancy and error rate and the highest identification rate. The FAMHAP does not work well for inferring haplotype with a large number of loci without reordering SNP genotype data. However, the performance of FAMHAP is improved when running several orders of SNP data. The Genehunter method for haplotype inference using sibship data (i.e., GH-Sib) produces the largest discrepancy (>20%) and error rate (>30%) and the lowest identification rate (<70%) for haplotype analyses. However, when using the whole family data Genehunter performs dramatically better than that using sibship data, but is still not as good compared to the EM methods. The EM-Single method that only uses singleton data is less accurate for haplotype inference compared with our EM-Sib method. This is because the EM-Single method does not completely explore sibship information for haplotype inference.
Figure 1.—

Comparisons of discrepancies, error rates, and identification rates among different methods using 15-locus haplotype simulated data.
Figure 2.—

Comparisons of discrepancies, error rates, and identification rates among different methods using nine-locus haplotype simulated data.
Generally, the discrepancy and the error rate for the EM methods accumulate with increased number of loci for haplotype inference. However, the performance of Genehunter depends largely on the LD among markers and is not affected by the number of loci for haplotype inference. For instance, in our simulations, the error rate of the GH-Sib is 33 and 44% for inferring 15- and 9-locus haplotypes, respectively. Another remarkable feature is that increasing sample sizes does not reduce the error rate or raise the identification rate when using the GH-Sib and GH-Family methods (Figures 1 and 2). The EM algorithms including the EM-Sib, EM-Single, FAMHAP-Sib, and FAMHAP-Sib-R are favored by increasing sample sizes. Larger sample sizes result in more accurate haplotype estimates and smaller standard errors in these EM methods.
APPLICATION EXAMPLES
To demonstrate the utility of our new method, we selected ∼200 Caucasian nuclear families (each with both parents and two offspring) from our studies that aim to search genes underlying the risk to osteoporosis in the Creighton University Osteoporosis Research Center. Three important candidate genes for osteoporosis were chosen for estimation of haplotype frequencies. These were the vitamin D receptor (VDR), apolipoprotein E (APOE), and parathyroid hormone (PTH)/PTH-related peptide receptor type 1 (PTHR1). Four SNPs were genotyped for each of these three genes in these subjects (Long et al. 2004). For the VDR gene, the SNPs spanned 63.4 kb and pairwise LD (|D′|) (Lewontin 1964) ranged from 0.049 to 0.980 with an average of 0.345. For the APOE gene, the SNPs spanned 3.6 kb and pairwise LD ranged from 0.745 to 0.999 with an average of 0.898. For the PTHR1 gene, the SNPs spanned 20.3 kb and pairwise LD ranged from 0.947 to 0.985 with an average of 0.970. The nuclear families were divided into two subsamples (with the same number of subjects): unrelated parental sample and sibship sample. We estimated haplotype frequencies at the VDR (n = 221), APOE (n = 197), and PTHR1 (n = 218) genes using the above six strategies: EM-Sib, EM-Single, GH-Sib, GH-Family, FAMHAP-Sib, and FAMHAP-Sib-R. To evaluate the performance of the above strategies in the real data application, we applied the conventional EM algorithm for estimation of haplotype frequencies to the parental samples (denoted as EM-Parent). The conventional EM algorithm is effective for unrelated parental samples and should yield a high accuracy of haplotype frequency estimates in a sample of ∼400 unrelated subjects (Fallin and Schork 2000). Therefore, haplotype frequencies estimated from the EM-Parent are treated as references of “true” haplotype frequencies in the population. We then compared haplotype frequencies from the EM-Parent with those from the other six strategies (Tables 1–3).
TABLE 3.
Comparison of PTHR1 haplotype frequencies estimated from different methods
| Haplotypes | EM-Parent | EM-Sib | EM-Single | GH-Sib | GH-Family | FAMHAP-Sib |
|---|---|---|---|---|---|---|
| ACAAa | — | — | — | 0.1839 | 0.0422 | — |
| ACAG | 0.0012 | 0.0016 | 0.0023 | 0.0088 | 0.0060 | — |
| ACGAa | — | — | — | 0.0035 | 0.0030 | — |
| ACGG | 0.5767 | 0.5834 | 0.5641 | 0.3993 | 0.5241 | 0.5915 |
| ATAA | 0.0023 | 0.0032 | 0.0023 | 0.0123 | 0.0075 | — |
| ATAG | — | — | — | — | — | — |
| ATGA | 0.0174 | 0.021 | 0.0253 | 0.0105 | 0.0286 | 0.0195 |
| ATGG | 0.0010 | 0.0016 | 0.0023 | 0.0070 | 0.0015 | — |
| GCAA | 0.0057 | 0.0064 | 0.0069 | 0.0070 | 0.0030 | 0.0061 |
| GCAG | — | 0.0016 | 0.0024 | 0.0018 | — | — |
| GCGA | 0.0011 | 0.0016 | — | — | 0.0030 | — |
| GCGG | 0.0058 | 0.0064 | 0.0023 | 0.0140 | 0.0045 | — |
| GTAA | 0.3806 | 0.3751 | 0.3829 | 0.1506 | 0.3102 | 0.3829 |
| GTAG | 0.0024 | 0.0016 | 0.0023 | — | 0.0090 | — |
| GTGA | 0.0034 | 0.0049 | 0.0046 | 0.0158 | 0.0136 | — |
| GTGG | 0.0023 | 0.0016 | 0.0023 | 0.1856 | 0.0437 | — |
| Discrepancy | — | 0.0121 | 0.0174 | 0.4178 | 0.1270 | 0.0196 |
See Table 1 legend for details.
The underlined haplotypes likely do not exist in the samples.
TABLE 2.
Comparison of APOE haplotype frequencies estimated from different methods
| Haplotypes | EM-Parent | EM-Sib | EM-Single | GH-Sib | GH-Family | FAMHAP-Sib |
|---|---|---|---|---|---|---|
| CGTC | 0.3721 | 0.3789 | 0.3855 | 0.3599 | 0.3939 | 0.3763 |
| CGTG | — | 0.0015 | — | — | 0.0058 | — |
| CGCC | 0.0094 | 0.0075 | 0.0010 | 0.0130 | 0.0044 | — |
| CGCG | — | — | — | — | — | — |
| CATC | 0.0018 | 0.0046 | 0.0069 | 0.0093 | 0.0087 | 0.0096 |
| CATG | 0.0013 | — | — | — | — | — |
| CACC | — | — | — | — | — | — |
| CACG | — | — | — | — | — | — |
| GGTC | 0.0272 | 0.0275 | 0.0409 | 0.1262 | 0.0392 | 0.0352 |
| GGTG | 0.0615 | 0.063 | 0.0508 | 0.0353 | 0.0538 | 0.0512 |
| GGCC | 0.1215 | 0.108 | 0.0979 | 0.0557 | 0.1279 | 0.1330 |
| GGCG | 0.0010 | — | — | 0.0130 | — | — |
| GATC | 0.3982 | 0.3953 | 0.4170 | 0.3210 | 0.3576 | 0.3914 |
| GATG | 0.0060 | 0.0055 | — | 0.0204 | 0.0044 | 0.0055 |
| GACC | 0.0001 | 0.0006 | — | 0.0464 | 0.0044 | — |
| GACG | — | — | — | — | — | — |
| Discrepancy | — | 0.0173 | 0.0511 | 0.1826 | 0.0573 | 0.0304 |
See Table 1 legend for details.
Haplotype frequencies estimated from the EM-Sib and FAMHAP methods are most similar to those from the EM-Parent. The total discrepancy for these methods is ∼2.5%. The performance of the EM-Single ranks fourth among the six strategies with an average discrepancy of 5.8%. The total discrepancies for the GH-Sib method are 32.6, 41.8, and 18.3% for the VDR, PTHR1, and APOE genes, respectively. The GH-Family performs reasonably better than the GH-Sib with an average discrepancy of 9.2%. There are three to six haplotypes whose frequency differences between the EM-Parent and the GH-Sib are >5%. Such a large estimated frequency difference is also observed between the EM-Parent and GH-Family methods. Furthermore, the GH methods falsely declared several pseudohaplotypes. In the PTHR1 gene, two haplotypes (ACAA and ACGA) whose frequencies were estimated to be zero in all of the EM methods were detected by both the GH-Sib and GH-Family methods. These two haplotypes likely do not exist in the sample since the EM methods are very robust in random samples (Fallin and Schork 2000). It is also worth noting that the FAMHAP methods tend to miss rare haplotypes. For example, four VDR haplotypes with frequencies of ∼2% (>1/4n) that were detected by the EM-Parent method were not found in the FAMHAP methods.
DISCUSSION
There is an increasing consensus among human geneticists to use sib-pair linkage and association approaches to map disease-susceptibility genes (Freimer and Sabatti 2004). Many large sibship data sets have already been accumulated, calling for due applications. For late-onset diseases, sibship data without parents are common. One of the applications for these sibship data is haplotype analysis, which has shown some distinct advantages over single-marker analysis in genetic studies of common diseases. In the context of association studies, when the disease association of a specific allele is dependent on cis-acting interactions with other loci, the disease association may not be detected by testing a single allele unless the whole functional haplotype itself is analyzed. This has been demonstrated through both empirical studies (Drysdale et al. 2000; Martin et al. 2000) and simulation studies (Zhang et al. 2002, 2003). Computational algorithms and statistical methods are currently the preferred means, particularly for large-scale haplotype determination. In this study, we developed a new statistical method for haplotype inference for multiple tightly linked SNPs, which is specially designed for sibship data. We examined the robustness and statistical performance of the new method in our simulated data that were created from empirical haplotype data of the GH1 gene (Horan et al. 2003). The utility of our method was demonstrated with an application to the inferences of haplotypes at several candidate genes underlying osteoporosis.
The accuracy of the proposed method was improved over that of existing methods, as demonstrated in both simulated and real data. Our EM algorithm did not increase the computational burden for haplotype inference using sibship data, compared with that using unrelated parental data. Its computational efficiency was not affected by increasing sibship size. FAMHAP did not perform well for inferring haplotype with a large number of loci in single SNP genotype data and its performance was dramatically improved when running several orders of SNP data. In addition, FAMHAP tended to miss rare haplotypes in the sample. However, Genehunter estimated haplotype frequencies with large biases and falsely declared nonexistent haplotypes in the sample. This is because Genehunter assumes linkage equilibrium among markers on the basis of the Lander–Green algorithm (Lander and Green 1987). This assumption was apparently violated for dense SNP data that were used in our simulations and real data analyses. Similar results were also observed in the haplotype analysis of the HPC1 gene in a recent study (Schaid et al. 2002).
Haplotype inference can be viewed as a missing data problem. In our method, specifically developed for sibship data without parents, marker data from all of the sibships are observed data, whereas parental zygote configurations [i.e., 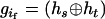 ] are unobservable or missing data. The principle of our method is to resolve sibship genotype data into their parental zygote configurations, from which haplotype inference can then be conducted. This circumvents the problem of related subjects that arises in the direct application of the conventional EM algorithm to sibship data. In our method, parental genotype data, if available, can reduce the number of potential zygote configurations and thus increase the accuracy of haplotype inference and accelerate the computation. Incorporating parental data into our method is straightforward and simply discards those zygote configurations that are not compatible with their observed parental genotypes. This is somewhat similar to a recent study of haplotype inference using nuclear family information (Rohde and Fuerst 2001). We therefore encourage researchers to collect available parent data if possible in addition to sibship data in their respective studies.
] are unobservable or missing data. The principle of our method is to resolve sibship genotype data into their parental zygote configurations, from which haplotype inference can then be conducted. This circumvents the problem of related subjects that arises in the direct application of the conventional EM algorithm to sibship data. In our method, parental genotype data, if available, can reduce the number of potential zygote configurations and thus increase the accuracy of haplotype inference and accelerate the computation. Incorporating parental data into our method is straightforward and simply discards those zygote configurations that are not compatible with their observed parental genotypes. This is somewhat similar to a recent study of haplotype inference using nuclear family information (Rohde and Fuerst 2001). We therefore encourage researchers to collect available parent data if possible in addition to sibship data in their respective studies.
Finally, two assumptions underlying our method should be acknowledged. One is that no recombination occurs among dense SNP markers in the parental generation. This simplifies the variable 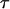 in Equation 2 as a constant for different mating types. If
in Equation 2 as a constant for different mating types. If 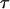 takes into account marker interval distance as a function of recombination, it will lead to complexity of the likelihood of the data and require further investigation. However, this assumption should hold in real applications, especially for inferring haplotypes in haplotype blocks where recombination is highly restricted in human genomes. There is little evidence for historical recombination in past generations in haplotype blocks (Patil et al. 2001; Gabriel et al. 2002), let alone recombination occurring in a given single generation (e.g., the parental generation). The other is that HWE holds at haplotypes. Fallin and Schork (2000) have recently reported a simulation study to assess the effects of departure from HWE on estimation of haplotype frequency by the EM algorithm in random population samples. They demonstrated that the EM algorithm is reasonably robust to departure from HWE and there is no increase in error with extreme departure from HWE toward excess homozygosity.
takes into account marker interval distance as a function of recombination, it will lead to complexity of the likelihood of the data and require further investigation. However, this assumption should hold in real applications, especially for inferring haplotypes in haplotype blocks where recombination is highly restricted in human genomes. There is little evidence for historical recombination in past generations in haplotype blocks (Patil et al. 2001; Gabriel et al. 2002), let alone recombination occurring in a given single generation (e.g., the parental generation). The other is that HWE holds at haplotypes. Fallin and Schork (2000) have recently reported a simulation study to assess the effects of departure from HWE on estimation of haplotype frequency by the EM algorithm in random population samples. They demonstrated that the EM algorithm is reasonably robust to departure from HWE and there is no increase in error with extreme departure from HWE toward excess homozygosity.
Acknowledgments
We thank two anonymous reviewers for their helpful criticisms and comments. Investigators of this work were partially supported by grants from the National Institutes of Health and the state of Nebraska. This study also benefited by grants from the National Natural Science Foundation of China, Huo Ying Dong Education Foundation, the Ministry of Education of China, Xi'an Jiaotong University, and Hunan Normal University.
APPENDIX A
The approximate variances of the estimates of haplotype frequencies can be obtained by inverting the estimated information matrix 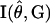 ,
,
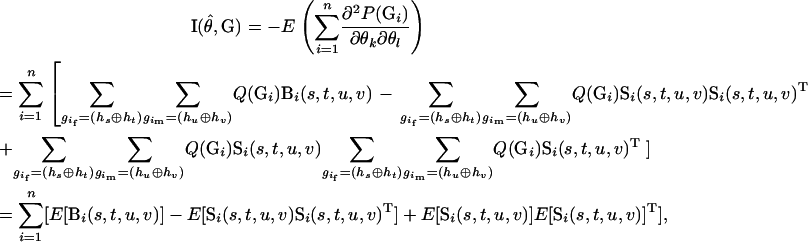 |
where
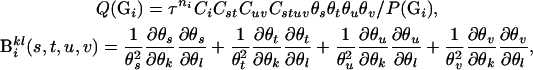 |
and
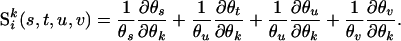 |
The last term of the above equation equals zero when evaluated at 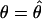 . The above equation can also be regarded as the observed information for the EM algorithm (Louis 1982).
. The above equation can also be regarded as the observed information for the EM algorithm (Louis 1982).
APPENDIX B
The possible unknown parental genotypes in Equation 5 were analytically collapsed given different mating designs, which could greatly reduce the EM computation. In our method, we first found the possible parental zygote configurations that are compatible with the sibling's genotypes. We then derived the explicit form of Equation 5 in each configuration. Below, we illustrate this collapse technique for sibship data with two siblings. The two siblings could take four, three, two, or one possible unobserved haplotypes.
- Suppose that the two siblings take four haplotypes and their haplotype configuration is (
 ,
,  ). Accordingly, their compatible parental genotypes should be (
). Accordingly, their compatible parental genotypes should be (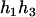 ,
, 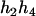 ) and (
) and (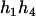 ,
, 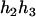 ). For the parental genotype (
). For the parental genotype (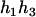 ,
, 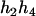 ), the denominator of Equation 5 can be expressed as
), the denominator of Equation 5 can be expressed as
Then we counted the number of haplotype j that occurred in the two haplotype pairs (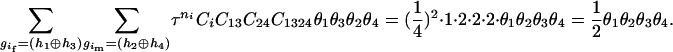
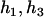 ) and (
) and (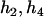 ). The numerator of Equation 5 can be expressed as
). The numerator of Equation 5 can be expressed as
The same results can be obtained from the parental genotype (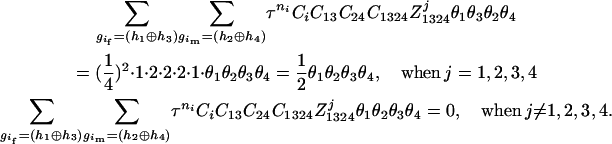
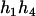 ,
, 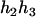 ).
). - Suppose that the two siblings take three haplotypes and their possible haplotype configurations are (
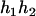 ,
, 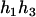 ) and (
) and (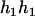 ,
, 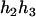 ). Accordingly, their compatible parental genotypes should be (
). Accordingly, their compatible parental genotypes should be (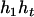 ,
, 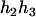 ) and (
) and (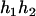 ,
, 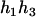 ), respectively, where
), respectively, where 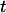 can be any haplotype in the population. For the parental genotype (
can be any haplotype in the population. For the parental genotype (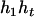 ,
, 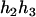 ), the denominator of Equation 5 can be expressed as
), the denominator of Equation 5 can be expressed as
The numerator of Equation 5 can be expressed as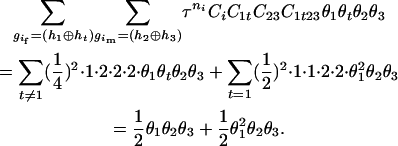
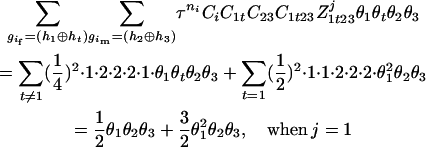
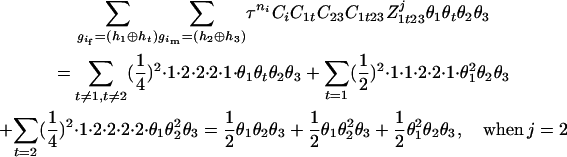
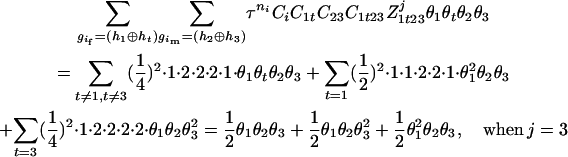
For the parental genotype (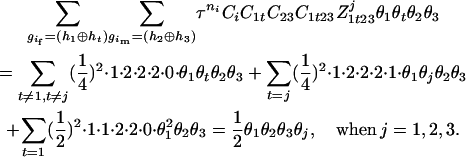
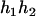 ,
, 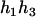 ), the denominator of Equation 5 can be expressed as
), the denominator of Equation 5 can be expressed as
The numerator of Equation 5 can be expressed as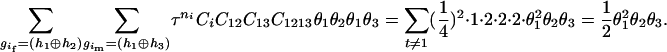
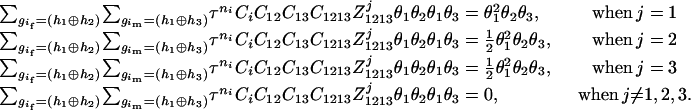
- Suppose that the two siblings take two haplotypes and their possible haplotype configurations are (
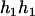 ,
, 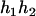 ), (
), (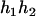 ,
, 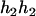 ), (
), (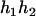 ,
, 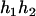 ), and (
), and (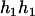 ,
, 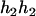 ). Accordingly, their compatible parental genotypes should be (
). Accordingly, their compatible parental genotypes should be (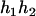 ,
, 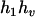 ), (
), (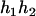 ,
, 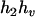 ), (
), (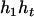 ,
, 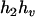 ), and (
), and (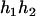 ,
, 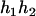 ), respectively, where
), respectively, where 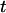 and
and  can be any haplotypes in the population. For the parental genotype (
can be any haplotypes in the population. For the parental genotype (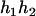 ,
, 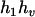 ), the denominator of Equation 5 can be expressed as
), the denominator of Equation 5 can be expressed as
The numerator of Equation 5 can be expressed as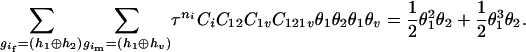
The same results can be obtained from the parental genotype (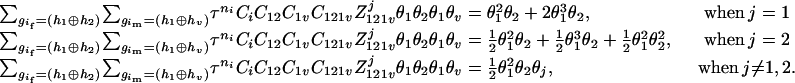
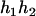 ,
, 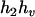 ) by exchanging the subscripts 1 and 2 in the above derivations. For the parental genotype (
) by exchanging the subscripts 1 and 2 in the above derivations. For the parental genotype (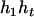 ,
, 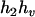 ), the denominator of Equation 5 can be expressed as
), the denominator of Equation 5 can be expressed as
The numerator of Equation 5 can be expressed as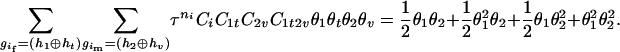
For the parental genotype (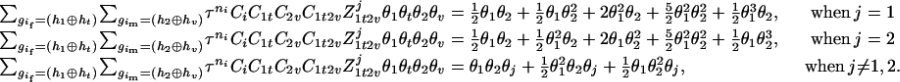
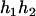 ,
, 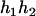 ), the denominator of Equation 5 can be expressed as
), the denominator of Equation 5 can be expressed as
The numerator of Equation 5 can be expressed as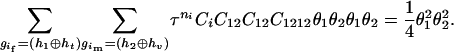
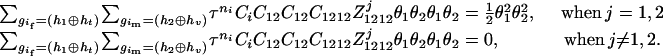
- Suppose that the two siblings take one haplotype and their haplotype configuration is (
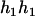 ,
, 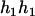 ). Accordingly, their compatible parental genotype should be (
). Accordingly, their compatible parental genotype should be (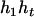 ,
, 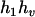 ). The denominator of Equation 5 can be expressed as
). The denominator of Equation 5 can be expressed as
The numerator of Equation 5 can be expressed as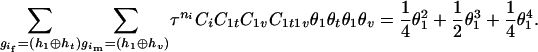
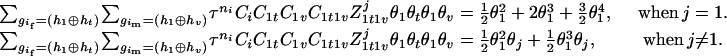
This collapse technique can be also implemented similarly in other sibship data structures. As shown in the above derivations, the haplotype inference using sibship data does not dramatically increase the EM computation compared with that using unrelated parental data. Also, increasing sibship size does not result in a larger computational burden since the number of compatible parental genotypes is reduced and more easily resolved given the larger sibship size.
References
- Abecasis, G. R., S. S. Cherny, W. O. Cookson and L. R. Cardon, 2002. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30: 97–101. [DOI] [PubMed] [Google Scholar]
- Becker, T., and M. Knapp, 2004. Maximum-likelihood estimation of haplotype frequencies in nuclear families. Genet. Epidemiol. 27: 21–32. [DOI] [PubMed] [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via EM algorithm. J. R. Stat. Soc. Ser. B 39: 1–38. [Google Scholar]
- Drysdale, C. M., D. W. McGraw, C. B. Stack, J. C. Stephens, R. S. Judson et al., 2000. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc. Natl. Acad. Sci. USA 97: 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L., and M. Slatkin, 1995. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 12: 921–927. [DOI] [PubMed] [Google Scholar]
- Fallin, D., and N. J. Schork, 2000. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am. J. Hum. Genet. 67: 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimer, N., and C. Sabatti, 2004. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat. Genet. 36: 1045–1051. [DOI] [PubMed] [Google Scholar]
- Gabriel, S. B., S. F. Schaffner, H. Nguyen, J. M. Moore, J. Roy et al., 2002. The structure of haplotype blocks in the human genome. Science 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- Gao, G., I. Hoeschele, P. Sorensen and F. Du, 2004. Conditional probability methods for haplotyping in pedigrees. Genetics 167: 2055–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, R. A., J. W. Belmont, P. Hardenbol, T. D. Willis, F. Yu et al., 2003. The International HapMap Project. Nature 426: 789–796. [DOI] [PubMed] [Google Scholar]
- Horan, M., D. S. Millar, J. Hedderich, G. Lewis, V. Newsway et al., 2003. Human growth hormone 1 (GH1) gene expression: complex haplotype-dependent influence of polymorphic variation in the proximal promoter and locus control region. Hum. Mutat. 21: 408–423. [DOI] [PubMed] [Google Scholar]
- Horvath, S., X. Xu, S. L. Lake, E. K. Silverman, S. T. Weiss et al., 2004. Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet. Epidemiol. 26: 61–69. [DOI] [PubMed] [Google Scholar]
- Johnson, G. C., L. Esposito, B. J. Barratt, A. N. Smith, J. Heward et al., 2001. Haplotype tagging for the identification of common disease genes. Nat. Genet. 29: 233–237. [DOI] [PubMed] [Google Scholar]
- Kruglyak, L., M. J. Daly, M. P. Reeve-Daly and E. S. Lander, 1996. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 58: 1347–1363. [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and P. Green, 1987. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 84: 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. C., 1964. The interaction of selection and linkage: II. Heterotic models. Genetics 50: 757–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., and T. Jiang, 2003. Efficient inference of haplotypes from genotypes on a pedigree. J. Bioinform. Comput. Biol. 1: 41–69. [DOI] [PubMed] [Google Scholar]
- Lin, S., and T. P. Speed, 1997. An algorithm for haplotype analysis. J. Comput. Biol. 4: 535–546. [DOI] [PubMed] [Google Scholar]
- Long, J. R., L. J. Zhao, P. Y. Liu, Y. Lu, V. Dvornyk et al., 2004. Patterns of linkage disequilibrium and haplotype distribution in disease candidate genes. BMC Genet. 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, T., 1982. Finding the observed information matrix when using the EM algorithm. J. R. Stat. Soc. Ser. B 44: 226–233. [Google Scholar]
- Martin, E. R., E. H. Lai, J. R. Gilbert, A. R. Rogala, A. J. Afshari et al., 2000. SNPing away at complex diseases: analysis of single-nucleotide polymorphisms around APOE in Alzheimer disease. Am. J. Hum. Genet. 67: 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, T., Z. S. Qin, X. Xu and J. S. Liu, 2002. Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am. J. Hum. Genet. 70: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, J. R., 2000. Zero-recombinant haplotyping: applications to fine mapping using SNPs. Genet. Epidemiol. 19(Suppl. 1): S64–S70. [DOI] [PubMed] [Google Scholar]
- Patil, N., A. J. Berno, D. A. Hinds, W. A. Barrett, J. M. Doshi et al., 2001. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294: 1719–1723. [DOI] [PubMed] [Google Scholar]
- Qian, D., and L. Beckmann, 2002. Minimum-recombinant haplotyping in pedigrees. Am. J. Hum. Genet. 70: 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde, K., and R. Fuerst, 2001. Haplotyping and estimation of haplotype frequencies for closely linked biallelic multilocus genetic phenotypes including nuclear family information. Hum. Mutat. 17: 289–295. [DOI] [PubMed] [Google Scholar]
- Schaid, D. J., S. K. McDonnell, L. Wang, J. M. Cunningham and S. N. Thibodeau, 2002. Caution on pedigree haplotype inference with software that assumes linkage equilibrium. Am. J. Hum. Genet. 71: 992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel, E., and K. Lange, 1996. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 58: 1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Tapadar, P., S. Ghosh and P. P. Majumder, 2000. Haplotyping in pedigrees via a genetic algorithm. Hum. Hered. 50: 43–56. [DOI] [PubMed] [Google Scholar]
- Wijsman, E. M., 1987. A deductive method of haplotype analysis in pedigrees. Am. J. Hum. Genet. 41: 356–373. [PMC free article] [PubMed] [Google Scholar]
- Zhang, K., P. Calabrese, M. Nordborg and F. Sun, 2002. Haplotype block structure and its applications to association studies: power and study designs. Am. J. Hum. Genet. 71: 1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S., Q. Sha, H. S. Chen, J. Dong and R. Jiang, 2003. Transmission/disequilibrium test based on haplotype sharing for tightly linked markers. Am. J. Hum. Genet. 73: 566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]