

Abstract
Background
Glanzmann thrombasthenia (GT) is an autosomal recessive bleeding disorder characterized by lack of platelet aggregation in response to most physiological agonists and caused by either a lack or dysfunction of the platelet integrin αIIbβ3 (glycoprotein IIb/IIIa).
Objectives
To determine the molecular basis of GT and characterize the mutations by in vitro expression studies.
Patients
We studied three unrelated patients from southern India whose diagnosis was consistent with GT.
Results
Immunoprecipitation of the cell lysates and immunoblotting showed no detectable mature αIIb in the G128S mutant, in contrast to 6% and 33% of the normal amount of mature αIIb in the S287L and G357S mutants, respectively. Pulse-chase analysis demonstrated pro-αIIb in the mutants comparable to the normal pro-αIIb, but no conversion to mature αIIb in the G128S mutant, and only trace conversion to mature αIIb in the S287L and G357S mutants. The disappearance of pro-αIIb in the three mutants was similar to that in cells expressing normal αIIbβ3 or αIIb only. All three mutants demonstrated pro-αIIbβ3 complexes and co-localized with an ER marker by immunofluorescence. The G128S mutant showed no co-localization with a Golgi marker, and the other two mutants showed minimal and moderate co-localization with the Golgi marker.
Conclusions
These three β-propeller mutations do not affect the production of pro-αIIb, its ability to complex with β3, or its stability, but do cause variable defects in transport of pro-αIIbβ3 complexes from the ER to the Golgi.
Keywords: β-propeller mutations, αIIbβ3 biogenesis, Glanzmann thrombasthenia
Introduction
Glanzmann thrombasthenia (GT) is a rare, autosomal recessive bleeding disorder characterized by a lifelong mucocutaneous bleeding tendency and absent or severely reduced platelet aggregation in response to the physiological agonists ADP, epinephrine, and collagen, but relatively normal aggregation in response to ristocetin [1–3]. The disease is caused by either a lack or dysfunction of the platelet integrin αIIbβ3 (glycoprotein IIb/IIIa), which serves as a receptor for fibrinogen, von Willebrand factor, and perhaps other adhesive glycoproteins [4]. Mutations in either the αIIb or β3 genes have been identified in more than 100 patients with GT, and include minor or major deletions, insertions, inversions, and point mutations [5–8]. αIIb and β3 are synthesized as two independent polypeptide chains and enter the endoplasmic reticulum (ER) where they form a complex, undergo N-linked glycosylation and form intrachain disulphide bonds [9]. The complexes are then transported to the Golgi apparatus for final oligosaccharide processing and cleavage of αIIb into heavy and light chains, before being transported to the membranes of α granules and the plasma membrane [10]. The assembly of the αIIbβ3 complex appears to be a prerequisite for transport out of the ER and thus cell surface expression [11,12]. αIIb that is not complexed to β3 is likely retained in the ER and degraded, while the uncomplexed β3 can either be degraded or combine with an αV subunit to form the vitronectin receptor, αVβ3 [13].
Mutations that impair the synthesis of either αIIb or β3 prevent the export of sufficient numbers of αIIbβ3 complexes to the platelet surface resulting in Glanzmann thrombasthenia [1]. However, αIIb or β3 mutations that have no apparent effect on the synthesis of either subunit can also give rise to thrombasthenia, either by perturbing the conformation of pro-αIIbβ3 complexes so they fail to be exported out of the ER [14–21] or by impairing αIIbβ3 function [22–28]. So far, twenty missense mutations causing GT have been reported within the β-propeller domain of αIIb, the region that both complexes with β3 and contributes to the ligand binding site. Thirteen of these mutations were associated with reduced surface expression, out of which six mutations namely, G242D [15], V298F [20], E324K [19], R327H [16], I374T [20], and G418D [14] were directly demonstrated to disrupt biogenesis by preventing transport of pro-αIIbβ3 from the ER to the Golgi, leading to intracellular retention. In this paper, we describe three new missense mutations, G128S, S287L and G357S within αIIb β-propeller blades 2, 5 and 6; all three are located on the upper face of propeller in the area involved in interaction with β3. We show that all three mutant pro-αIIb were synthesized, stable and able to form a complex with β3. The G128S mutation completely prevents the transport of the pro-αIIbβ3 complex from the ER to the Golgi, whereas the S287L and G357S mutations variably impair transport from the ER to the Golgi and subsequent surface expression.
Patients
Patient 1, from Andhra Pradesh, was diagnosed as having GT at 4 years of age based on a history of easy bruising, epistaxis, prolonged bleeding time (more than 15 min), absent clot retraction, and absent in vitro platelet aggregation in response to ADP, epinephrine, and collagen. His parents were first degree relatives (uncle-niece), but had no bleeding symptoms. Patient 2, also from Andhra Pradesh, was diagnosed at 5 years of age based on a history of epistaxis, gum bleeding, hematuria, reduced clot retraction, and absent in vitro platelet aggregation in response to ADP, epinephrine, and collagen, but a normal initial phase response to ristocetin. His parents were also first degree relatives (uncle-niece) and had no bleeding symptoms. Patient 3, from Tamil Nadu, had more mild symptoms (gingival bleeding) and was diagnosed at 21 years of age based on prolonged bleeding time (more than 15 min), absent in vitro platelet aggregation in response to ADP, epinephrine, and collagen, but a normal initial phase response to ristocetin. His parents had no significant bleeding symptoms, but two of his paternal aunts died of bleeding complications before 10 years of age. There was no known consanguinity.
Materials and methods
Immunoblot analysis of platelet lysates
Immunoblot analysis of washed platelets was performed essentially as previously described [29] using murine mAbs specific for αIIb (CD41, SZ22) and β3 (CD61, SZ21) [Immunotech, Marseille, Cedex, France]. Samples for αIIb were tested reduced to display both pro-αIIb and the heavy chain of mature (cleaved) αIIb, and samples for β3 were tested non-reduced.
Polymerase chain reaction (PCR), single strand conformation polymorphism (SSCP) analysis, and DNA sequencing
A total of 38 PCR reactions were performed on genomic DNA extracted from buffy coat cells using intragenic primers and conditions that were previously described [30,31] and additional primers and conditions that can be obtained by contacting one of us (B.S.C). SSCP analysis was performed by either an automated (PhastGel System; Pharmacia Biotech, Uppsala, Sweden) or manual method (Protean II Cell; Bio-Rad, Hercules, CA). DNA sequencing was performed on amplicons that showed mobility shifts by SSCP analysis (Big Dye terminator kit; PE Biosystems, Foster City, CA).
Site-directed mutagenesis
Mutant αIIb cDNA constructs were generated by site-directed mutagenesis (QuikChange XL Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA). Wild-type αIIb cDNA in the pEF1/V5-His mammalian expression vector was used as the template (kindly provided by Dr. Junichi Takagi, Harvard University, Boston, MA). After mutagenesis, the mutant cDNA was used to transform XL10-Gold ultracompetent cells as per the manufacturer’s instructions (Stratagene).
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells were grown in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum, 1% nonessential amino acids (GIBCO, Carlsbad, CA), and 1% each of penicillin and streptomycin. Cells were grown to 70–80% confluency and then transfected with vector alone (mock), or normal αIIb cDNA alone (about 7.5 μg), or normal or mutant αIIb cDNA (about 7.5 μg) co-transfected with an equal amount of normal β3 cDNA in pcDNA 3.1/Myc-His vector (also provided by Dr. Takagi) [Perfectin Transfection Reagent; Gene Therapy Systems, San Diego, CA]. 48 h after transfection, stable cell lines were generated by G418 selection (0.5 mg/ml, GIBCO) for 14 days. To ensure comparability among cell lines, stable cell lines were not selected for high surface αIIbβ3 expression.
Flow cytometry
Transfected cells were harvested (0.05% trypsin, 1 mM EDTA), washed, and incubated with Alexa 488-labeled mouse mAbs 10E5 (αIIbβ3) or 7E3 (αIIbβ3 + αVβ3) for 30 min at 22° C before dilution with PBS and analysis by flow cytometry (FACS Calibur; Becton Dickinson, San Jose, CA).
Immunoprecipitation and immunoblotting
Lysates of transfected cells [50 mM Tris HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 5mM N-ethyl maleimide, 0.2% protease inhibitor mixture (Protease Inhibitor Cocktail set III, Calbiochem, San Diego, CA)] were precleared with protein G-Sepharose beads (Amersham Pharmacia Biotech), and then incubated with 2 μg of mouse anti-V5 tag (anti-αIIb) mAb (Invitrogen Corporation, Carlsbad, CA) at 4° C overnight followed by protein G beads. Immunoprecipitates were eluted from the beads by incubating in SDS sample buffer, with (for αIIb), or without (for β3) 10% β-mercaptoethanol at 100° C for 5 min. SDS-PAGE and immunoblotting was performed with mouse mAbs CA3 (anti-αIIb; Chemicon International, Temecula, CA) or CD61 (anti-β3; Dako Corporation, Carpinteria, CA).
Pulse-chase analysis
Stably transfected cells were incubated in methionine- and cysteine-free DMEM for 30 min and then incubated with 480 μCi of 35S protein labeling mix (Perkin Elmer Life Sciences, Boston, MA) at 37° C for 15 min. Cells were incubated with DMEM containing 1 mg/ml each of cold methionine and cysteine for 0, 1, 2, 4, 8 and 24 h at 37° C. Cells were then harvested and solubilized; after preclearing the lysates with protein G beads, 2 μg of anti-V5 tag antibody was added, followed by protein G beads. The beads were washed twice and immunoprecipitates were eluted in SDS sample buffer containing 10% β-mercaptomethanol. After SDS-PAGE, gels were fixed, washed, soaked in an autoradiographic image intensifying reagent (Autoflour; National Diagnostics, Atlanta, GA), dried, and subjected to autoradiography.
Immunofluorescence microscopy
Transfected cells were grown overnight in DMEM on poly-l-lysine-coated glass cover slips, washed twice, and fixed in ice-cold acetone-methanol (1:1). The cells were washed, incubated in blocking solution (2.5% bovine serum albumin (BSA) BSA/0.05% NP-40 in PBS) and reacted with either rabbit anti-calnexin polyclonal antibody (ER marker, 1:200 dilution, Stressgen Bioreagents, Victoria, BC, Canada) or rabbit anti-mannosidase II polyclonal antibody (Golgi marker, 1:100 dilution, US Biological, Swampscott, MA), followed by labeling with TRITC-conjugated F(ab’)2 fragment of goat anti-rabbit IgG (H+L) (Jackson Immunoresearch Laboratories, West Grove, PA). The cells were washed thrice and then incubated with a 1:100 dilution of FITC-conjugated anti-αIIb antibody (SZ22). The cover slips were then washed, air-dried, mounted onto a glass slide with antifade solution (Molecular Probes, Eugene, OR), and analyzed using an inverted microscope (Olympus 1X70, Melville, NY). Images were deconvolved using Deltavision software (Applied Precision, Issaquah, WA).
Results
Platelet content of αIIbβ3
Immunoblot analysis of platelet lysates showed no detectable pro-αIIb or mature αIIb in any of the patient samples; trace amounts of β3 were detected only in samples from patients 2 and 3, but no β3 was detectable in the sample from patient 1 (data not shown).
Mutation detection
SSCP analysis of the genomic DNA of patient 1 revealed a mobility shift in the PCR product of exon 4 of αIIb and DNA sequencing confirmed a G→A nucleotide substitution at position 475 of the pro-αIIb sequence. This predicts a substitution of glycine to serine at amino acid position 128 within the β-propeller domain at the end of the S3–S4 loop of blade 2, just before the third β strand (W2S3) [32,33]. In patient 2, a mobility shift was observed in the PCR product of exon 11 of αIIb, and this was found to be due to a C→T nucleotide substitution at position 953 of the pro-αIIb sequence. This predicts a substitution of serine to leucine at amino acid position 287, which is located in the β-propeller domain just before the beginning of the first β strand in blade 5 (W5S1). In patient 3, a mobility shift was observed in the PCR product of exon 12 of αIIb and a G→A nucleotide substitution at position 1162 of the pro-αIIb sequence was subsequently identified. This predicts a substitution of glycine to serine at amino acid position 357, which is also located in the β-propeller domain, in the highly conserved region just before the first β strand in blade 6 (W6S1).
Expression of mutant αIIbβ3 receptors in HEK 293 cells
52% of cells transfected with normal αIIb and β3 bound 10E5 (MFI - 43), whereas mock transfected cells (vector alone) showed minimal 10E5 binding (5%). In sharp contrast, only 0, 2, and 8% of the G128S, S287L and G357S mutant cells bound 10E5 above background, and among the positive cells the extent of binding was lower (MFIs 14, 14, and 35). Immunoprecipitation of the cell lysates with an antibody to αIIb, followed by immunoblotting with an antibody to αIIb showed the presence of pro-αIIb in the normal and all three mutant cells (Figure 1, top panel). While mature αIIb was seen in the normal αIIbβ3 cells, there was no detectable mature αIIb in the G128S mutant, 6% of the normal value of mature αIIb in the S287L mutant, and 33% of the normal value in the G357S mutant. Immunoprecipitation with an antibody to αIIb also pulled down β3 as part of the αIIbβ3 complex (Figure 1, bottom panel). β3 was not detected in cells expressing the G128S mutant, while 6% and 35% of the normal amount of β3 was pulled down in complex with the S287L and G357S αIIb mutants respectively.
Figure 1. Immunoprecipitation and immunoblotting of the transfected HEK 293 cells.
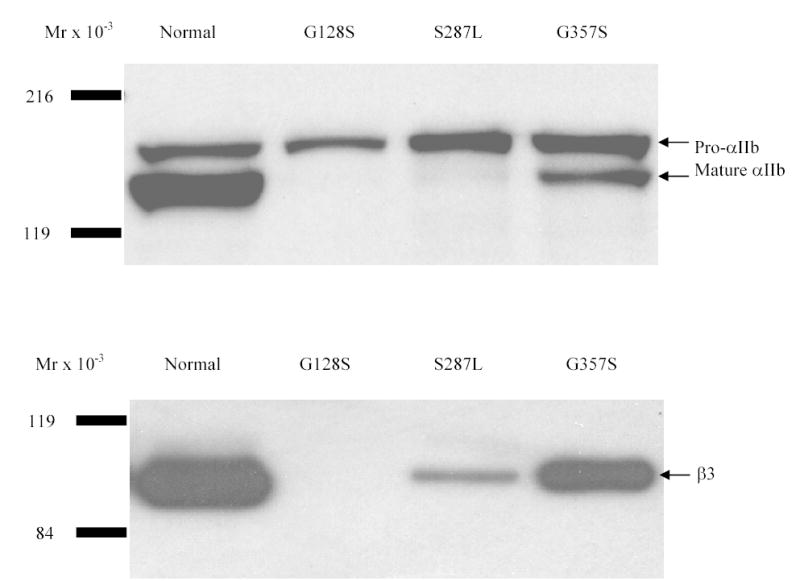
Cell lines were solubilized in lysis buffer and immunoprecipitated with an antibody to the V5 epitope tag of the αIIb subunit. Immunocomplexes were analyzed by immunoblotting under reducing (αIIb) or non-reducing (β3) conditions using an antibody to αIIb (CA3, top) or β3 (CD61, bottom). Pro-αIIb, mature αIIb and β3 are indicated by arrows.
Pro-αIIb subunits of the G128S, S287L and G357S mutants are synthesized and degraded at rates similar to normal pro-αIIb, but show little or no progression to mature αIIb
Cells expressing normal αIIbβ3 demonstrated maximum synthesis of pro-αIIb within 1–2 h, progressive maturation of a portion of the pro-αIIb to mature, cleaved αIIb over approximately 8 h, and disappearance of pro-αIIb between 8 and 24 h (Figure 2A). Cells transfected with normal αIIb alone gave a pattern of pro-αIIb synthesis and disappearance very similar to the cells co-transfected with both normal αIIb and β3, with no obvious evidence of more rapid degradation. Pro-αIIb molecules were also synthesized at the normal rate by the three mutant αIIbβ3 cells, but there was no progression to mature αIIb in the G128S mutant, only trace progression to mature αIIb in the S287L mutant, and slightly more progression to mature αIIb in the G357S mutant (Figure 2A). Of note, the small amounts of mature αIIb observed in the S287L and G357S mutants appeared very early in the time course and did not increase thereafter in density. The rates of disappearance of pro-αIIb in the three mutant cells were very similar to the rates in the cells expressing αIIb alone and normal αIIbβ3, with a half-disappearance time of approximately 4 h (Figure 2B). In contrast to cells transfected with normal αIIbβ3, however, all three mutant cells showed some increase in relative pro-αIIb values over the first 2 hours. Thus, despite their defects in progression to mature αIIb, the G128S, S287L and G357S mutations did not affect either the production of pro-αIIb or its rate of degradation.
Figure 2. Pulse-chase analysis of transfected HEK 293 cells expressing normal or mutant αIIbβ3 receptors.
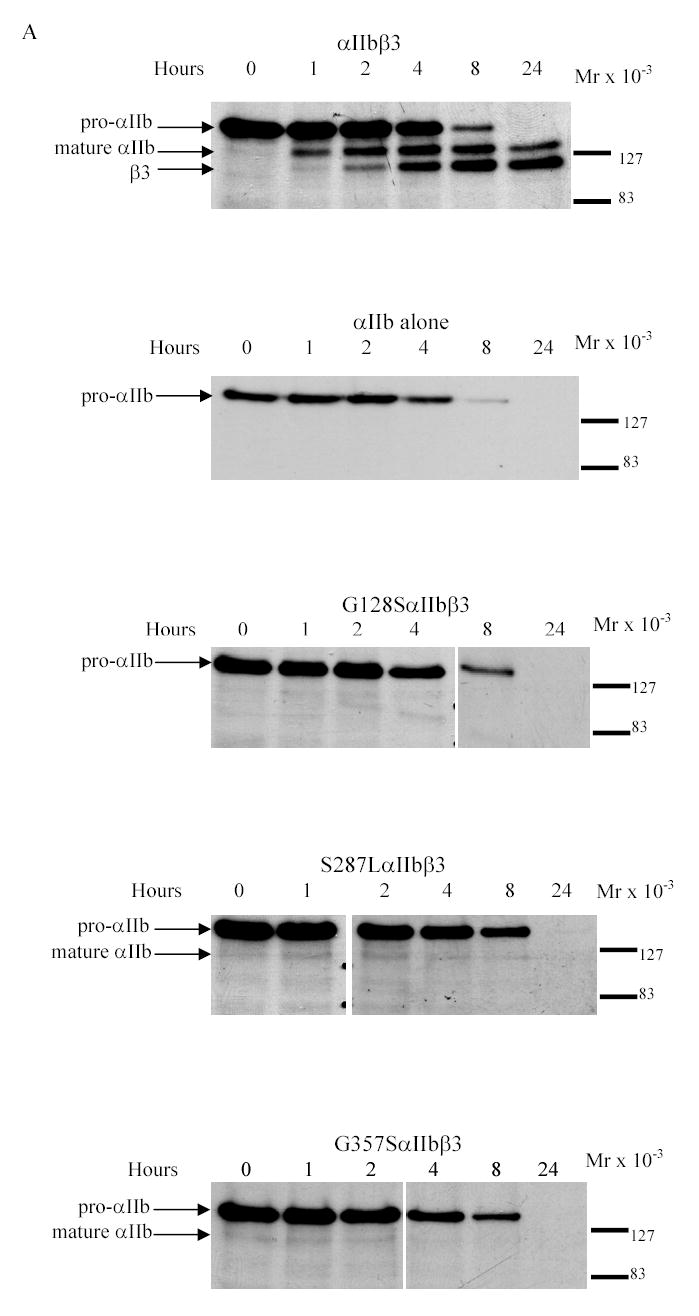
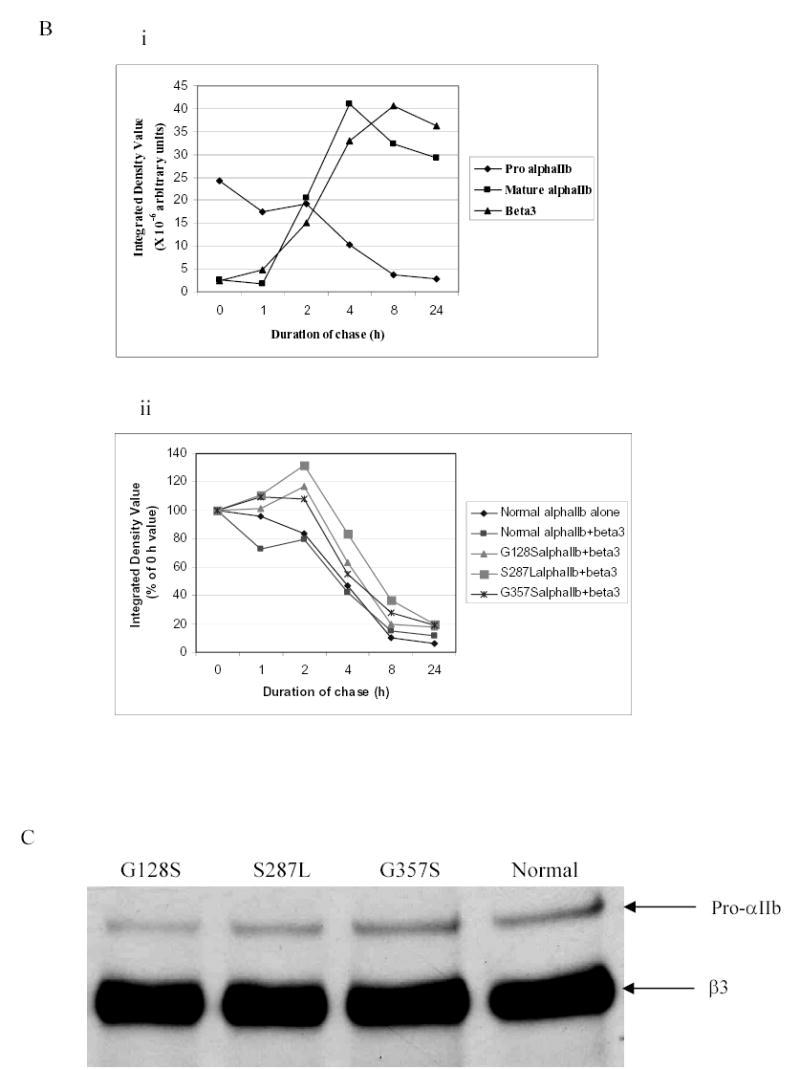
(A) Stable cell lines expressing normal or mutant αIIbβ3 were pulsed with 35S-methionine- and cysteine-containing medium for 15 min and chased in medium containing unlabeled methionine and cysteine for 0, 1, 2, 4, 8, and 24 h. The cell lysates were immunoprecipitated with an antibody to the V5-epitope tag on αIIb. Samples were electrophoresed under reduced conditions. Bands representing pro-αIIb, mature αIIb, and β3 are indicated by arrows. (B) (i) Kinetics of pro-αIIb, mature αIIb, and β3 in cell lines transfected with normal αIIb and β3 subunits as judged by band densities. There was a gradual decrease in pro-αIIb levels and this was mirrored by both an increase in mature αIIb over 24 h and an increase in β3 associated with αIIb. (ii) Kinetics of pro-αIIb in cell lines transfected with αIIb only or normal or mutant αIIbβ3 receptors. All three mutant αIIb subunits demonstrated similar rates of degradation, which were similar to the rates in the normal αIIbβ3 and αIIb only cell lines. (C) Immunoprecipitation with an antibody to β3 (7H2) 1 h after pulse-chase. Bands representing pro-αIIb and β3 are indicated by arrows.
Detection of pro-αIIbβ3 complexes in normal and mutant receptors
To assess whether the mutant pro-αIIb subunits formed complexes with β3, immunoprecipitations of pulse-labeled cells 1 h after the chase were performed with antibodies to β3. Cells transfected with normal αIIbβ3 demonstrated pro-αIIb immunoprecipitating with antibody to β3 whereas cells transfected with αIIb alone did not (data not shown). All three mutant cells demonstrated pro-αIIb immunoprecipitating with antibodies to β3 (Figure 2C). The pro-αIIbβ3 complexes of all three mutants were also detected up to 8 h post-chase in a separate experiment (data not shown).
Progression of αIIbβ3 from the ER to Golgi to surface membrane in normal and mutant cells
Fluorescent labeling of αIIb in cells transfected with normal αIIbβ3 demonstrated strong staining at the periphery of the cell consistent with αIIbβ3 surface expression, and diffuse staining throughout the remainder of the cell (Figure 3A, left top and bottom panels). The ER (calnexin) stain was distributed diffusely throughout the cell in clusters that were more accentuated near the periphery (Figure 3A, top center panel), while the Golgi (mannosidase II) stain was present in more discrete clusters, primarily in the periphery (Figure 3A, bottom center panel). The merged image of αIIb and calnexin demonstrated extensive co-localization, seen as yellow, indicating the presence of αIIb in the ER (Figure 3A, top right panel). The cell surface αIIbβ3, however, appeared green, indicating that it did not co-localize with the calnexin stain, and thus served as a control. In the normal αIIbβ3 cells, αIIb stain also co-localized with the mannosidase-II stain (Figure 3A, bottom right panel), indicating the presence of αIIb in the Golgi. The insert figures in Figure 3A are from mock transfected cells demonstrating the absence of αIIb, but the presence of calnexin- and mannosidase-II positive organelles. The G128S mutant showed enhanced staining for αIIb in a ring-like configuration within the cell, but no surface αIIb staining (Figure 3B, left top and bottom panels). Co-localization with calnexin (Figure 3B, top center and right panels) indicated that the vast majority of the αIIb was in the ER. In the αIIb and mannosidase-II merged image, the mannosidase-II-positive organelles stained red rather than yellow, indicating little or no presence of G128SαIIbβ3 in the Golgi (Figure 3B, bottom center and right panels). The S287L mutant cells demonstrated barely detectable surface labeling for αIIb, strong ring-like staining for αIIb inside the cell that co-localized with calnexin, indicating an ER localization, and some co-localization with mannosidase-II (Figure 3C). In contrast, a small amount of surface αIIb was detectable in the G357S mutant cells and there was stronger co-localization with mannosidase-II, indicating that more G357SαIIbβ3 was transported to the Golgi. The strong ring-like staining for αIIb, which co-localized with calnexin was also observed in these cells, indicating the presence of G357S αIIb in the ER (Figure 3D).
Figure 3. Co-localization of αIIb in the ER and Golgi apparatus in the transfected HEK 293 cells.
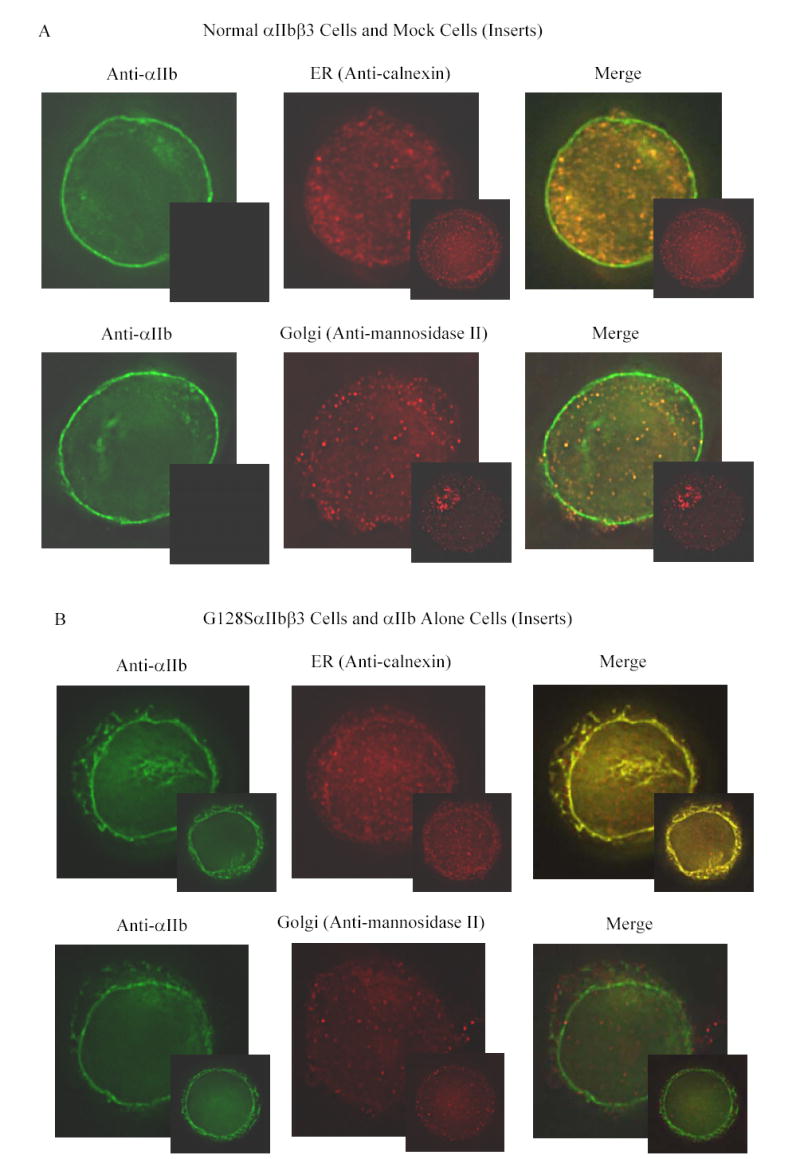
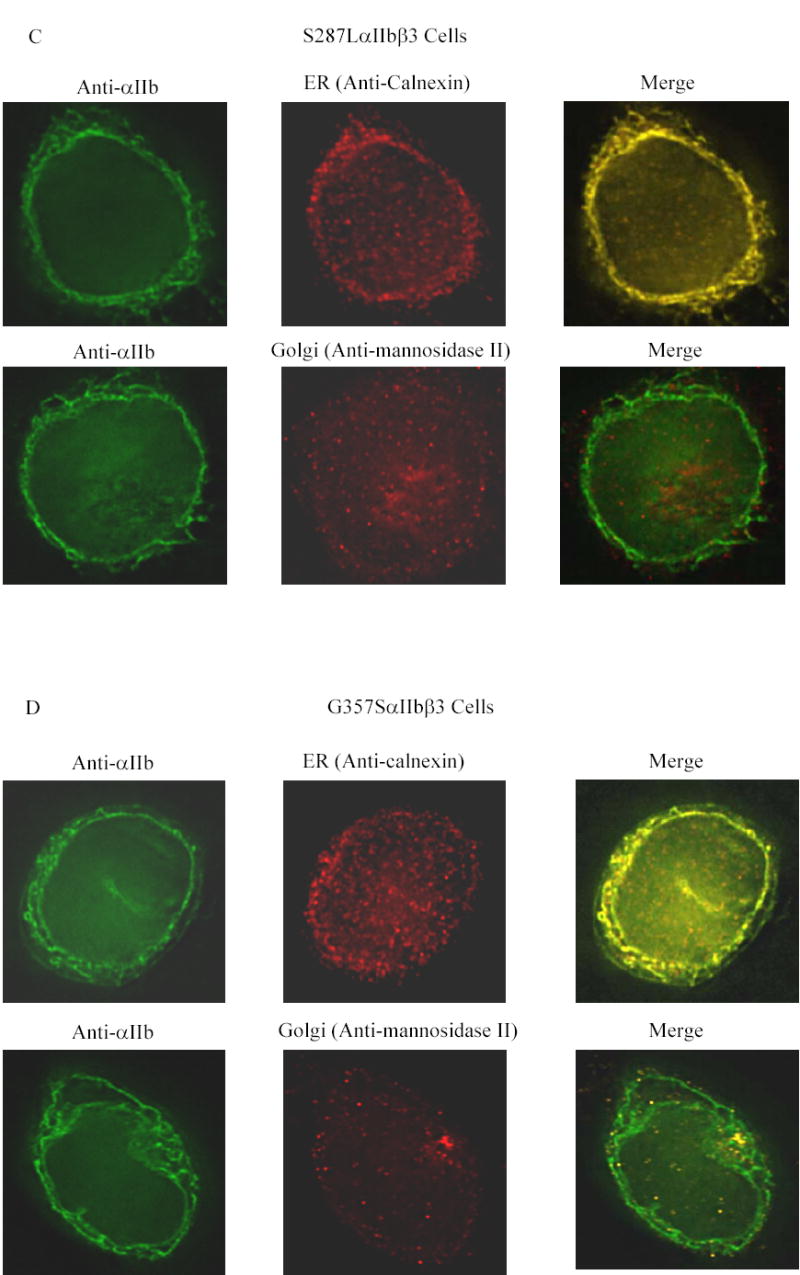
Transfected cells were labeled with an anti-αIIb antibody (green), and either an antibody to an ER component (calnexin) or a Golgi component (mannosidase II) (red). (A) Cells transfected with normal αIIbβ3 showed strong labeling of the cell surface, evident as a green outline, indicating normal αIIbβ3 expression. In addition, there is αIIb staining throughout the cell, some of which co-localizes with the ER marker and some with the Golgi marker, as indicated by the yellow color of the merged images. In contrast, mock transfected cells, shown here as inserts adjacent to the corresponding images of the normal cells, showed no αIIb staining either outside or inside the cell, but did show staining of the ER and Golgi. (B) Surface staining of αIIb was not observed in cells transfected with the G128SαIIbβ3 mutant. αIIb staining of the G128SαIIbβ3 mutant co-localized with the ER marker, indicated by the yellow color of the merged image, but not with the Golgi marker. The intensity of ER staining by anti-calnexin antibody in the G128SαIIbβ3 mutant cells was greater than in the normal αIIbβ3 cells. These results are comparable to cells transfected with αIIb alone (inserts). (C) S287LαIIbβ3 mutant cells demonstrated minimal amounts of αIIb on the surface. This mutant αIIb co-localized with the ER marker, but showed only faint co-localization with the Golgi marker, implying egress of only a small amount of S287LαIIbβ3 from the ER to Golgi. (D) In contrast, the G357SαIIbβ3 mutant cells demonstrated faint, but detectable surface labeling, and showed αIIb staining co-localizing with both the ER and the Golgi markers.
Locations of the three mutations in the crystal structure of αIIbβ3 and their potential structural implications
The locations of the αIIb β-propeller mutations, G128S, S287L, and G357S were identified on the crystal structure of αIIbβ3 [33] using the MOLecule analysis and MOLecule display (MOLMOL) software. The residues glycine 128 and serine 287 are at the top of the propeller and lie within the interface with the βA domain of β3, while the glycine 357 is buried deeper within the propeller structure (Figure 4A). All three mutations are located among the residues contributing to the highly conserved “cage” motif, which consists of two concentric rings of predominantly aromatic residues within the β-propeller structure [32]. This motif has been shown to contain a consensus sequence, (X17–33-{φφ GφX13–20PX2–15GX5–8})7, where X is any residue and φ is an aromatic residue (Figure 4B) [32]. It is defined by a mostly aromatic 4-residue “cup” (φφ Gφ) that precedes the first β strand in each propeller blade (A), a proline immediately following the second β strand (B), referred to as Pro-B, and an invariant glycine at the beginning of the third β strand (C), referred to as Gly-C [32]. Glycine 128 is located at the position of the invariant glycine in blade 2. The conservation of glycine at this or an adjacent relative location in all seven β-propeller blades of αIIb, αV, α1-α11, αM, αL, and αX subunits in humans indicates the importance of having the small size and/or flexibility of glycine in this region. The serine 287 is located immediately adjacent to the φφ Gφ “cup” in blade 5, and is conserved in α1, α2, α4-α11, αL, and αX subunits in humans. The glycine 357 is located directly within the “cup” of blade 6 and is conserved in αV, α1-α11, αL, αM, and αX subunits in humans. Of especial note, a previously described Glanzmann thrombasthenia mutation (G418D) is in the same position as glycine 357 in the “cup” of αIIb blade 7 [14]. The three mutated αIIb residues are also conserved across species, including αIIb of mouse, rat, rabbit, horse, pig, and dog.
Figure 4. Location of the three mutations in the αIIbβ3 crystal structure.
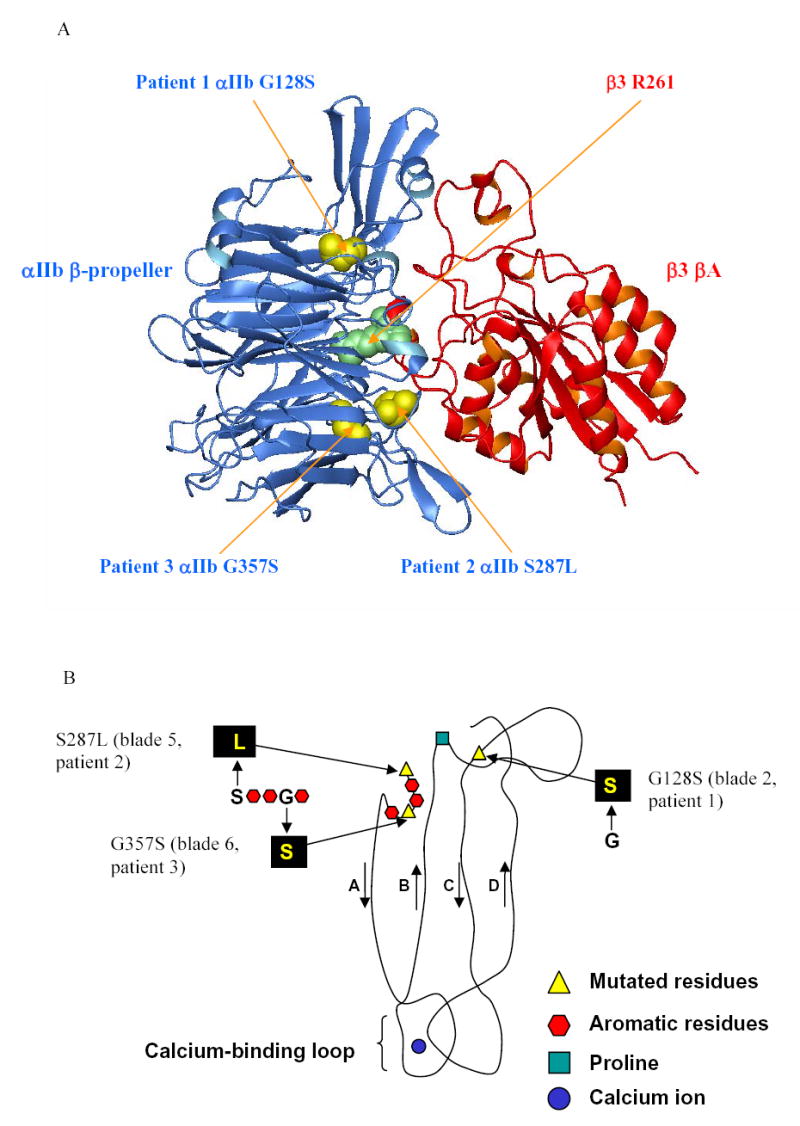
(A) The crystal structure of αIIbβ3 [33] is shown, highlighting the interface between the β-propeller domain of αIIb (blue) and the βA domain of β3 (red). The mutated residues of αIIb (yellow), and the R261 of β3 (green) are represented by space-filling models, and the remainder of the protein is shown as a ribbon diagram. (B) Schematic diagram of one blade of the αIIb β -propeller (viewed from the side), derived from the crystal structure, showing residues of the cage motif and the relative positions of the three mutations reported in this study, which lie in blades 2, 5 and 6 respectively.
Discussion
We have characterized three separate αIIb mutations in the β-propeller domain, in blades 2 (G128S), 5 (S287L), and 6 (G357S), in three unrelated patients with Glanzmann thrombasthenia from southern India. The pro-αIIb of the G128S mutant was synthesized in HEK 293 cells and formed a complex with β3 but we could not identify any mature αIIbβ3 complex either inside or on the surface of the cells. Moreover, although the G128SαIIbβ3 colocalized with the ER marker, it did not colocalize with the Golgi marker. As a result we conclude that the αIIb G128S mutation profoundly interferes with transport of the pro-αIIbβ3 complex to the Golgi apparatus and subsequent processing to mature, cleaved αIIb. Although HEK 293 cells differ from megakaryocytes, our data indicating the need for pro-αIIbβ3 transport to the Golgi as a prerequisite for processing of pro-αIIb to mature αIIb are similar to those previously reported in megakaryocytes [10].
The S287L and G357S mutations in patients 2 and 3 respectively, resulted in abnormalities similar to those in patient 1, but the defects in αIIbβ3 complex formation, transport to the Golgi, and surface expression were less severe. Of note, the pro-αIIb in all three mutations was synthesized and disappeared with kinetics that were similar to normal pro-αIIb when expressed either alone or in combination with normal β3. Similar observations have been made with other mutations affecting the αIIb β-propeller domain [14,15]. Moreover, since the overall disappearance of pro-αIIb from cells lacking β3 is not more rapid than in cells containing normal β3, these data suggest that the rate of pro-αIIb degradation is not primarily determined by the formation of the pro-αIIbβ3 complex. Immunoprecipitation with anti-β3 antibody detected only a small amount of pro-αIIb in complex with β3 in cells expressing normal and mutant αIIbβ3 receptors.
The availability of the crystal structures of αVβ3 and αIIbβ3 permits us to correlate the molecular abnormalities in the αIIb β-propeller domain in GT patients with their functional consequences [32,33]. The various mutations identified in the β-propeller domain thus far, including the ones reported in this paper, are depicted in the ribbon diagram model of the αIIbβ3 in Figures 5A and 5B. The vast majority of the missense mutations that have been characterized result in decreased or absent αIIbβ3 surface expression. The two missense mutations that affect ligand binding (Y143H [27] and P145A [26]) are located adjacent to the αIIb-specific cap domain that has been implicated in ligand binding [33]. Many of the other mutations are at the interface between the αIIb β-propeller domain and the β3 βA-domain, where one might have expected them to interfere with complex formation between pro-αIIb and β3. However, in our studies [20] and in studies conducted by others [14–17,19,24,28], immunoprecipitation experiments demonstrated the presence of pro-αIIbβ3 complexes. These observations are, in fact, consistent with the studies of Wilcox et al [16], Basani et al [17], and McKay et al [34], who demonstrated that recombinant forms of αIIb containing only the first three blades of the propeller could form a complex with β3. Even among mutations affecting the non-cap region of the first three blades of αIIb (Y143H [27], P145A [26], P145L [26], F171C [21], L183P [24], and the G128S that we now report), only the F171C mutation resulted in failure to form the pro-αIIbβ3 complex, and even in this case it is uncertain whether this resulted from it selectively altering the interface between pro-αIIb and β3 or from it causing more general disruption of pro-αIIb folding as a result of the introduction of a new unpaired cysteine residue. Several αIIb β-propeller mutations affect the regions in blades 4–7 that are near to or in the β-hairpin calcium binding loops [14–17,19]. These mutations also do not affect pro-αIIbβ3 complex formation, but dramatically affect receptor maturation and surface expression.
Figure 5. Localization of the β-propeller mutations identified thus far in the αIIbβ3 crystal structure.

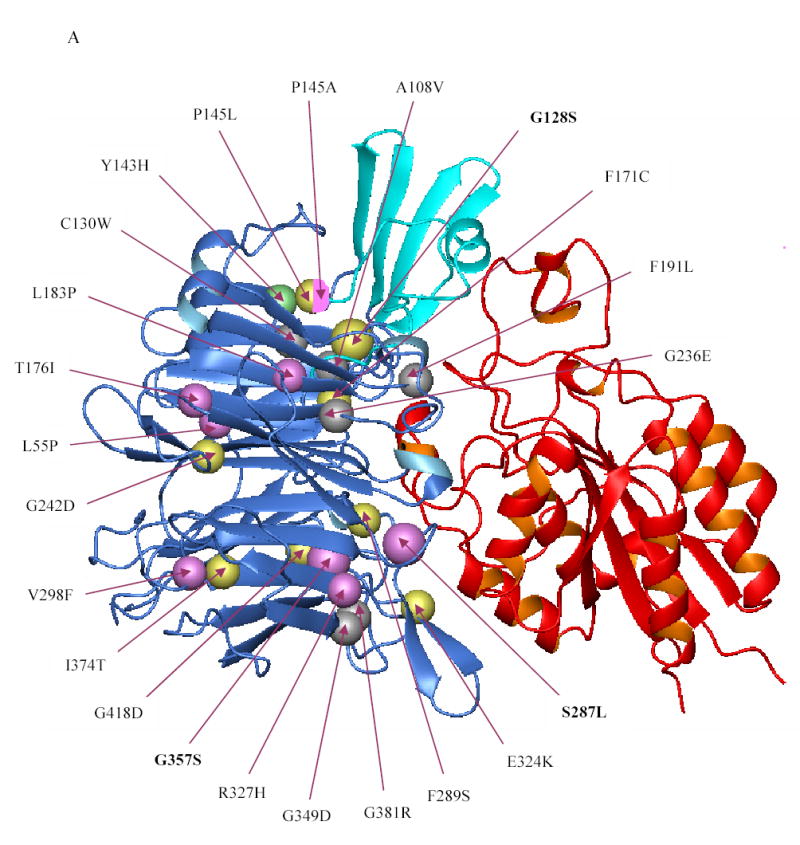
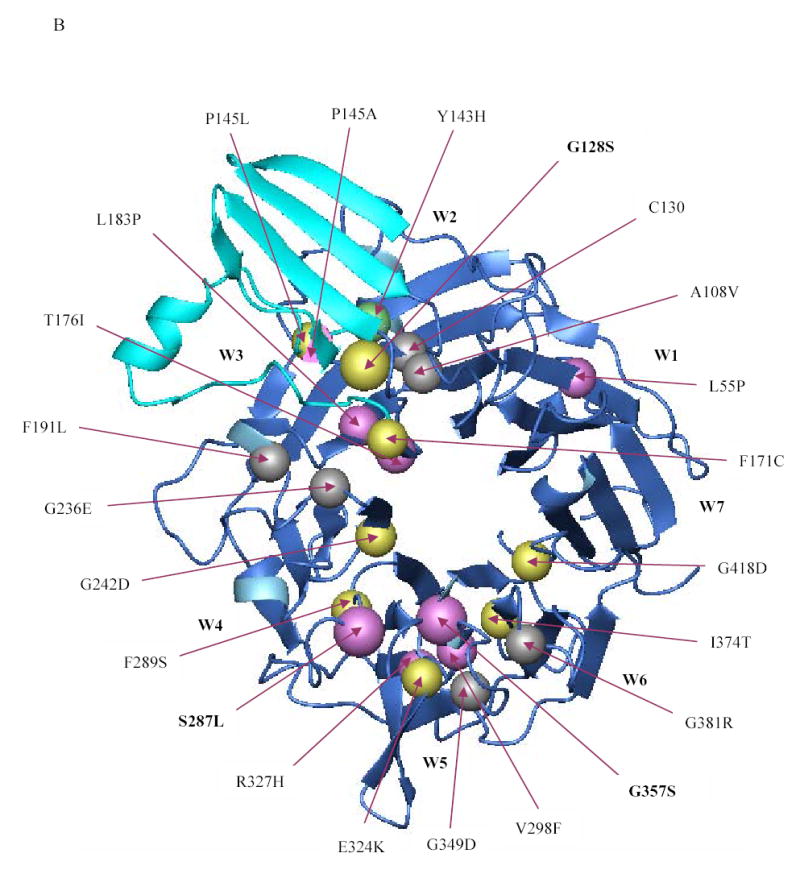
(A) Crystal structure of the interface between the β-propeller domain of αIIb (blue) and the βA domain of β3 (red) showing the location of the various β-propeller mutations reported thus far (including the ones reported in this paper). Mutations that result in no or low surface expression of αIIbβ3 are depicted in yellow and magenta, respectively. The Y143H mutation which affected ligand binding, but had little effect on surface expression, is shown in green, and lies adjacent to the cap region (cyan), implicated in ligand binding. Note that the mutations P145A, T176I, and L183P affected ligand binding as well as surface expression. The substitution of leucine or alanine for proline at position 145, respectively, resulted in no or low expression and this is depicted by a sphere that is half yellow and half magenta. Published mutations on which no data are available on either platelet surface expression or in vitro expression are depicted in gray. (B) Crystal structure of the β-propeller domain of αIIb as seen from the surface facing the β3 βA domain showing the various mutations published in the literature (including the ones in this paper). The central core formed by two concentric rings of aromatic residues is key to interacting with the βA domain of β3, which is not shown here.
The major limiting step in αIIbβ3 biogenesis in the mutations we describe and most of the previously described mutations is not, therefore, complex formation, but rather transport from the ER to the Golgi for carbohydrate modification, pro-αIIb cleavage, and transport to the cell surface. It is likely, therefore, that a molecular mechanism exists to assess the quality of the folding of pro-αIIb and/or the conformation of the pro-αIIbβ3 complex as a prelude to transport out of the ER. Direct support for alterations in the conformation of the pro-αIIbβ3 complex as a result of a number of the reported mutations (V298F, I374T, G242D, and G418D) derives from studies demonstrating lack of binding of monoclonal antibodies that are αIIbβ3 complex-specific or conformation-specific to the mutant proteins [14–16]. It is unlikely that the quality control mechanism involves a ligand-like molecule since some mutations that profoundly affect ligand binding do not result in markedly reduced receptor expression, as for example, β3 D119Y [22,35] and R214Q [36,37]. Alterations in binding to a chaperone-like protein(s) seem likely.
Acknowledgments
We thank Dr. Uri Seligsohn and Dr. Hava Peretz for their collaboration and their scientific efforts in training the first author of this manuscript in some of the techniques used in this study. We also thank Dr. Nurit Rosenberg for kindly sharing the protocol for immunofluorescence studies.
Footnotes
This research was supported in part by grant 19278 from the National Heart, Lung and Blood Institute, a General Clinical Research Center grant (M01-RR00102) from the National Center for Research Resources at the National Institutes of Health, funds from Stony Brook University (B.S.C), and grant BT/PR2614/Med/13/113/2001 from the Department of Biotechnology (A.S), Government of India.
Conflict of interest disclosure
Dr. Coller is an inventor of abciximab and in accord with Federal law and the policies of the Research Foundation of the State University of New York, shares in royalties paid to the Foundation for sales of abciximab. No other authors have conflicts of interest.
References
- 1.George JN, Caen J-P, Nurden AT. Glanzmann’s thrombasthenia: the spectrum of clinical disease. Blood. 1990;75:1383–95. [PubMed] [Google Scholar]
- 2.Caen JP, Castaldi PA, Leclerc JC, Inceman S, Larrieu MJ, Probst M, Bernard J. Congenital bleeding disorders with long bleeding time and normal platelet count: Glanzmann’s thrombasthenia. Am J Med. 1966;41:4–21. [Google Scholar]
- 3.George JN, Nurden AT, Phillips DR. Molecular defects in interactions of platelets with the vessel wall. N Engl J Med. 1984;311:1084–98. doi: 10.1056/NEJM198410253111705. [DOI] [PubMed] [Google Scholar]
- 4.Nurden AT, Caen JP. An abnormal platelet glycoprotein pattern in three cases of Glanzmann’s thrombasthenia. Br J Haematol. 1974;28:253–60. doi: 10.1111/j.1365-2141.1974.tb06660.x. [DOI] [PubMed] [Google Scholar]
- 5.French DL, Coller BS. Hematologically important mutations: Glanzmann thrombasthenia. Blood Cells Mol Dis. 1997;23:39–51. doi: 10.1006/bcmd.1997.0117. [DOI] [PubMed] [Google Scholar]
- 6.Vinciguerra C, Bordet JC, Beaune G, Grenier C, Dechavanne M, Negrier C. Description of 10 new mutations in platelet glycoprotein IIb (αIIb) and glycoprotein IIIa (β3) genes. Platelets. 2002;12:486–95. doi: 10.1080/095371001317126383. [DOI] [PubMed] [Google Scholar]
- 7.D’Andrea G, Colaizzo D, Vecchione G, Grandone E, Di Minno G, Margaglione M. Glanzmann’s thrombasthenia: Identification of 19 new mutations in 30 patients. Thromb Haemost. 2002;87:1034–42. [PubMed] [Google Scholar]
- 8.http://sinaicentral.mssm.edu/intranet/research/glanzmann/listmutations?mut=all
- 9.Bray PF, Rosa J-P, Lingappa VR, Kan YW, McEver RP, Shuman MA. Biogenesis of the platelet receptor for fibrinogen: evidence for separate precursors for glycoproteins IIb and IIIa. Proc Natl Acad Sci USA. 1986;83:1480–84. doi: 10.1073/pnas.83.5.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duperray A, Berthier R, Chagnon E, Ryckewaert JJ, Ginsberg M, Plow E, Marguerie G. Biosynthesis and processing of platelet GPIIb-IIIa in human megakaryocytes. J Cell Biol. 1987;104:1665–73. doi: 10.1083/jcb.104.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duperray A, Troesch A, Berthier R, Chagnon E, Frachet P, Uzan G, Marguerie G. Biosynthesis and assembly of platelet GPIIb-IIIa in human megakaryocytes: evidence that assembly between pro-GPIIb and GPIIIa is a prerequisite for expression of the complex on the cell surface. Blood. 1989;74:1603–11. [PubMed] [Google Scholar]
- 12.O’Toole TE, Loftus JC, Plow EF, Glass AA, Harper JR, Ginsberg MH. Efficient surface expression of platelet GPIIb-IIIa requires both subunits. Blood. 1989;74:14–18. [PubMed] [Google Scholar]
- 13.Polack B, Duperray A, Troesch A, Berthier R, Marguerie G. Biogenesis of the vitronectin receptor in human endothelial cell: evidence that the vitronectin receptor and GPIIb-IIIa are synthesized by a common mechanism. Blood. 1989;73:1519–24. [PubMed] [Google Scholar]
- 14.Wilcox DA, Wautier JL, Pidard D, Newman PJ. A Single amino acid substitution flanking the fourth calcium-binding domain of αIIb prevents maturation of the αIIbβ3 integrin complex. J Biol Chem. 1994;269:4450–57. [PubMed] [Google Scholar]
- 15.Poncz M, Rifat S, Coller BS, Newman PJ, Shattil SJ, Parrella T, Fortina P, Bennett JS. Glanzmann thrombasthenia secondary to a Gly273→Asp mutation adjacent to the first calcium-binding domain of platelet glycoprotein IIb. J Clin Invest. 1994;93:172–79. doi: 10.1172/JCI116942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilcox DA, Paddock CM, Lyman S, Gill JC, Newman PJ. Glanzmann thrombasthenia resulting from a single amino acid substitution between the second and third calcium-binding domains of GPIIb. Role of the GPIIb amino terminus in integrin subunit association. J Clin Invest. 1995;95:1553–60. doi: 10.1172/JCI117828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basani RB, Vilaire G, Shattil SJ, Kolodziej MA, Bennett JS, Poncz M. Glanzmann thrombasthenia due to a two amino acid deletion in the fourth calcium-binding domain of alphaIIb: demonstration of the importance of calcium-binding domains in the conformation of alphaIIb/beta3. Blood. 1996;88:167–73. [PubMed] [Google Scholar]
- 18.Ambo H, Kamata T, Handa M, Kawai Y, Oda A, Murata M, Takada Y, Ikeda Y. Novel point mutations in the alphaIIb subunit (Phe289→Ser, Glu324→Lys and Gln747→Pro) causing thrombasthenic phenotypes in four Japanese patients. Br J Haematol. 1998;102:829–40. doi: 10.1046/j.1365-2141.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 19.Milet-Marsal S, Breillat C, Peyruchaud O, Nurden P, Combrie R, Nurden AT, Bourre F. Analysis of the amino acid requirement for a normal αIIbβ3 maturation at αIIbGlu324 commonly mutated in Glanzmann thrombasthenia. Thromb Haemost. 2002;88:655–62. [PubMed] [Google Scholar]
- 20.Mitchell WB, Li J, Singh F, Michelson AD, Bussel J, Coller BS, French DL. Two novel mutations in the αIIb calcium-binding domains identify hydrophobic regions essential for αIIbβ3 biogenesis. Blood. 2003;101:2268–76. doi: 10.1182/blood-2002-07-2266. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg N, Landau M, Luboshitz J, Rechavi G, Seligsohn U. A novel Phe171Cys mutation in integrin alpha causes Glanzmann thrombasthenia by abrogating alphabeta complex formation. J Thromb Haemost. 2004;2:1167–75. doi: 10.1111/j.1538-7836.2004.00758.x. [DOI] [PubMed] [Google Scholar]
- 22.Loftus JC, O’Toole TE, Plow EF, Glass A, Frelinger AL, 3rd, Ginsberg MH. A beta 3 integrin mutation abolishes ligand binding and alters divalent cation-dependent conformation. Science. 1990;249:915–18. doi: 10.1126/science.2392682. [DOI] [PubMed] [Google Scholar]
- 23.Chen YP, Djaffar I, Pidard D, Steiner B, Cieutat AM, Caen JP, Rosa JP. Ser-752→Pro mutation in the cytoplasmic domain of integrin beta 3 subunit and defective activation of platelet integrin alpha IIb beta 3 (glycoprotein IIb–IIIa) in a variant of Glanzmann thrombasthenia. Proc Natl Acad Sci U S A. 1992;89:10169–73. doi: 10.1073/pnas.89.21.10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grimaldi CM, Chen F, Wu C, Weiss HJ, Coller BS, French DL. Glycoprotein IIb Leu214Pro mutation produces glanzmann thrombasthenia with both quantitative and qualitative abnormalities in GPIIb/IIIa. Blood. 1998;91:1562–71. [PubMed] [Google Scholar]
- 25.Honda S, Tomiyama Y, Shiraga M, Tadokoro S, Takamatsu J, Saito H, Kurata Y, Matsuzawa Y. A two-amino acid insertion in the Cys146- Cys167 loop of the alphaIIb subunit is associated with a variant of Glanzmann thrombasthenia. Critical role of Asp163 in ligand binding. J Clin Invest. 1998;102:1183–92. doi: 10.1172/JCI3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basani RB, French DL, Vilaire G, Brown DL, Chen F, Coller BS, Derrick JM, Gartner TK, Bennett JS, Poncz M. A naturally occurring mutation near the amino terminus of alphaIIb defines a new region involved in ligand binding to alphaIIbbeta3. Blood. 2000;95:180–88. [PubMed] [Google Scholar]
- 27.Kiyoi T, Tomiyama Y, Honda S, Tadokoro S, Arai M, Kashiwagi H, Kosugi S, Kato H, Kurata Y, Matsuzawa Y. A naturally occurring Tyr143His alpha IIb mutation abolishes alphaIIb beta3 function for soluble ligands but retains its ability for mediating cell adhesion and clot retraction: comparison with other mutations causing ligand-binding defects. Blood. 2003;101:3485–91. doi: 10.1182/blood-2002-07-2144. [DOI] [PubMed] [Google Scholar]
- 28.Westrup D, Santoso S, Follert-Hagendorff K, Bassus S, Just M, Jablonka B, Kirchmaier CM. Glanzmann thrombasthenia Frankfurt I is associated with a point mutation Thr176Ile in the N-terminal region of αIIb subunit integrin. Thromb Haemost. 2004;92:1040–51. doi: 10.1160/TH04-03-0170. [DOI] [PubMed] [Google Scholar]
- 29.Coller BS, Seligsohn U, Zivelin A, Zwang E, Lusky A, Modan M. Immunologic and biochemical characterization of homozygous and heterozygous Glanzmann thrombasthenia in the Iraqi-Jewish and Arab populations of Israel: comparison of techniques for carrier detection. Br J Haematol. 1986;62:723–35. doi: 10.1111/j.1365-2141.1986.tb04096.x. [DOI] [PubMed] [Google Scholar]
- 30.Yatuv R, Rosenberg N, Dardik R, Brenner B, Seligsohn U. Glanzmann thrombasthenia in two Iraqi-Jewish siblings is caused by a novel splice junction mutation in the glycoprotein IIb. Blood Coagul Fibrinolysis. 1998;9:285–88. doi: 10.1097/00001721-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 31.Jin Y, Dietz HC, Nurden A, Bray PF. Single-strand conformation polymorphism analysis is a rapid and effective method for the identification of mutations and polymorphisms in the gene for glycoprotein IIIa. Blood. 1993;82:2281–88. [PubMed] [Google Scholar]
- 32.Xiong J-P, Stehle T, Diefenbach B, et al. Crystal structure of the extracellular segment of integrin alphaVbeta3. Science. 2001;294:339–45. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKay BS, Annis DS, Honda S, Christie D, Kunicki TJ. Molecular requirements for assembly and function of a minimized human integrin αIIbβ3. J Biol Chem. 1996;271:30544–547. doi: 10.1074/jbc.271.48.30544. [DOI] [PubMed] [Google Scholar]
- 35.Ginsberg MH, Lightsey A, Kunicki TJ, Kaufmann A, Marguerie G, Plow EF. Divalent cation regulation of the surface orientation of platelet membrane glycoprotein IIb. Correlation with fibrinogen binding function and definition of a novel variant of Glanzmann’s thrombasthenia. J Clin Invest. 1986;78:1103–11. doi: 10.1172/JCI112667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fournier DJ, Kabral A, Castaldi PA, Berndt MC. A variant of Glanzmann’s thrombasthenia characterized by abnormal glycoprotein IIb/IIIa complex formation. Thromb Haemost. 1989;62:977–83. [PubMed] [Google Scholar]
- 37.Bajt ML, Ginsberg MH, Frelinger AL, III, Berndt MC, Loftus JC. A spontaneous mutation of integrin αIIbβ3 (platelet glycoprotein IIb–IIIa) helps define a ligand binding site. J Biol Chem. 1992;267:3789–94. [PubMed] [Google Scholar]