

Summary
The functional consequences of Connexin43 (Cx43) phosphorylation remain largely unexplored. Using an antibody that specifically recognizes Cx43 phosphorylated at serines 325/328/330 (pS325/328/330), we show that this form of Cx43 as well as total Cx43 labeling was restricted to the intercalated disk region of normal ventricular tissue. In ischemic heart, significant relocalization of total Cx43 to the lateral edges of myocytes was evident; however pS325/328/330-Cx43 remained predominately at the intercalated disk. Western immunoblots indicated a 8-fold decrease in pS325/328/330-Cx43 in ischemic tissue. Peptide binding and competition experiments indicated that our antibody mainly detected Cx43 phosphorylated at S328 and/or S330 in heart tissue. To evaluate how this change in Cx43 phosphorylation might contribute to ischemia-induced downregulation of intercellular communication, we stably transfected Cx43−/− cells with Cx43-S325/328/330A (Cx43-TM). Cx43-TM was not efficiently processed to isoforms that have been correlated with gap junction assembly. Nevertheless, Cx43-TM cells were electrically coupled, although coupling developed over a slowed time course. Fully open channels were only rarely observed in the Cx43-TM cells, and Lucifer Yellow dye coupling was significantly reduced compared to wild-type. These data suggest that phosphorylation of Cx43 at serines 325, 328 and/or 330 may influence channel permselectivity and regulate the efficiency of gap junction assembly.
Keywords: Connexin43, Gap junction, heart, ischemia, phosphorylation
Introduction
Gap junction mediated intercellular communication facilitates direct communication among adjacent cells by allowing passage of ions and small metabolites (White and Paul, 1999; Saez et al., 2003; Sohl and Willecke, 2004). Vertebrate gap junctions, composed of integral membrane proteins from the Connexin gene family, are critically important in regulating embryonic development, coordinated contraction of excitable cells, tissue homeostasis, normal cell growth and differentiation (Saez et al., 2003; Sohl and Willecke, 2004). Furthermore, connexin mutations have been linked to several diseases (Bergoffen et al., 1993; Gong et al., 1997; Kelsell et al., 1997) including oculodentodigital dysplasia, a disease linked to Connexin43 (Cx43) mutations that can cause atrioseptal defects and arrhythmias (Paznekas et al., 2003). Twenty-one connexin genes have been identified in humans (Sohl and Willecke, 2004). During intercellular channel formation, six connexin proteins oligomerize into a hemi-channel or connexon; connexons are then transported to the plasma membrane by as yet unknown mechanisms. The intact channel is formed when one hemi-channel docks with a second in an opposing cell. Once assembled, groups of these intercellular channels (termed gap junctional plaques) mediate the diffusion of ions, amino acids, second messengers and other metabolites between the cytoplasms of the two cells (White and Paul, 1999; Sohl and Willecke, 2004). The channels can be gated in response to various stimuli, including changes in voltage, pH and connexin phosphorylation. Regulation of gap junctional communication could occur by controlling any one of the steps mentioned above, however, many of the regulatory mechanisms underlying these events remain elusive.
Cx43, the most ubiquitously expressed connexin, is differentially phosphorylated at a dozen or more serine residues throughout its life cycle (Lampe and Lau, 2004). Cx43 from cultured cells commonly demonstrates multiple electrophoretic isoforms when analyzed by SDS-PAGE: a faster migrating form (sometimes referred to as P0 or NP) that includes the non-phosphorylated isoform, and multiple slower migrating forms (sometimes termed P1 and P2 (Musil and Goodenough, 1991)). Following alkaline phosphatase treatment, the phosphorylated species collapse to the fastest migrating form, suggesting that phosphorylation is the primary covalent modification detected in SDS-PAGE analysis although no assignment of specific phosphorylation sites to a change in Cx43 migration has been made. Pulse-chase studies using Brefeldin A indicate some Cx43 phosphorylation occurs prior to reaching the plasma membrane (Laird et al., 1995). This phosphorylation event may be necessary for maintaining hemichannels in their closed state until docking occurs (Bao et al., 2004). In addition, studies investigating phosphorylation in normal rat kidney (NRK) cells show that Cx43 acquires resistance to Triton X-100 once it has been phosphorylated to the slower migrating isoforms and assembled into gap junction plaques (Musil and Goodenough, 1991). Thus, uncharacterized phosphorylation events have been correlated with changes in assembly, acquisition of Triton X-100 insolubility and, potentially, degradation of Cx43 gap junction channels.
In the normally functioning ventricle, Cx43 is localized to intercalated disks where it supports the longitudinal and transverse spread of the action potential resulting in coordinated contractile activation. Myocardial ischemia leads to Cx43 “dephosphorylation” and loss of localization to the intercalated disk, which likely contributes to contractile failure and arrhythmias (Beardslee et al., 2000; Schulz et al., 2003). Casein kinase 1 (CK1) mediates phosphorylation of Cx43 at S325/328/330 in vitro. In cultured cells these sites are routinely phosphorylated; inhibition of CK1 reduces phosphorylation at these sites and reduces gap junction assembly (Cooper and Lampe, 2002). We sought to determine here whether phosphorylation at these sites occurs in heart tissue and whether this phosphorylation event is affected during ischemia. Using an antibody specific for Cx43 when it is phosphorylated at S325/328/330, we show that Cx43 localized to intercalated disks is phosphorylated at one or more of these residues (likely S328 and S330) and that ischemia leads to loss of this phosphorylation and relocalization of the protein. Furthermore, we show that mutation of these sites increases the migration of the Cx43 phospho-isoforms on SDS-PAGE, reduces the extent of gap junction formation and reduces gap junctional communication.
Methods
Antibodies and Reagents
All general chemicals, unless otherwise noted, were purchased from Fisher Scientific. Mouse anti-Cx43 antibodies, Cx43CT1, Cx43CT2, and Cx43IF1 were prepared against amino acids 360–382 of Cx43 and antibody Cx43NT1 against amino acids 1–20 (Cx43NT1) of Cx43 at the Fred Hutchinson Cancer Research Center Hybridoma Development Facility (Seattle, WA). A rabbit antibody against Cx43 was purchased from Sigma (St. Louis, MO, C6219). We made a rabbit anti-pS325/328/330-Cx43 antibody by custom commercial preparation (ProSci Inc., Poway, CA; 13 week schedule) against a synthetic peptide that was phosphorylated at all 3 serines (CQAG(pS)TI(pS)N(pS)HAQ-amide) that had been linked via the N-terminal cysteine to maleimide-activated KLH (Pierce Biotechnology, Rockford, IL) according to manufacturer’s instructions. Phosphospecific antibody was affinity purified by linking to the phosphorylated peptide and the corresponding nonphosphorylated version to SulfoLink Coupling Gel (Pierce) and passing the serum first over a 3 ml column prepared with the nonphosphorylated peptide gel followed by a phosphopeptide-SulfoLink column according to the manufacturer’s instructions. After washing, phosphospecific antibody was eluted from the column with 5% acetic acid and fractions were neutralized with ammonium acetate. Antibody specific for phosphorylation at S325 and S328 was affinity purified by linking peptides phosphorylated at S325/328 and S328/330 (CGQAG(pS)TI(pS)NSH-amide and CGSTI(pS)N(pS)HAQ-amide) to SulfoLink beads to make 1.5 ml columns of each. The polyclonal antibody was then run first over the pS328/330 column and the unbound antibody was run over the pS325/328 column. The pS325/328 specific antibody that bound was then eluted as described above. All antibodies were aliquoted and stored frozen at −80°C.
Cell culture
Cx43 knockout fibroblasts lacking Cx43 (clone 23-3) or exogenously expressing wild-type Cx43 (Cx43-WT; clone 22C-3) were provided by Dr. Erica TenBroek (University of Minnesota). Normal Rat Kidney cells (E51) were obtained from American Tissue Type Collection (Manassas, VA). Cells were cultured in Dulbeccos Minimal Essential Medium (DMEM, Fisher Scientific, Pittsburgh PA) supplemented with 5–10% fetal calf serum and antibiotics (100 U/mL penicillin G and 100μg/mL streptomycin) in a humidified 5% CO2 environment. Cx43 with serines at 325, 328 and 330 mutated to alanine (Cx43-TM), kindly provided in pcDNA3.1 by Dr. Steven Taffet (SUNY Health Science Center, Syracuse NY), was subcloned into pIREShygro. To make Cx43-TM expressing fibroblasts, pIREShygro Cx43-TM was transfected into Cx43 knockout fibroblasts (clone 23-3) using lipofectAMINE Plus transfection reagent (GibcoBRL/Invitrogen, Carlsbad CA) according to the manufacturer’s instructions. Cells were selected at 500μg/ml Hygromycin B and were dilution cloned in DMEM supplemented with hygromycin and endothelial cell growth supplement (ECGS, 5mg/mL) to facilitate selection and growth of cells. Three separate clones that stably expressed Cx43-TM were isolated (TM-A, TM-B, TM-C).
Immunodetection of Cx43 in Heart
Mouse studies were conducted under FHCRC Institutional Animal Care and Use Committee approval. Inbred mice (11 months of age in a FVB/N:C57BL6 background) were anaesthetized (avertin, 0.1mL/3g body weight) and hearts were excised and placed either in ice cold PBS (calcium and glucose-free) for 30 seconds (control group) or incubated without coronary perfusion in non-oxygenated, glucose-free PBS for 30 minutes at 37°C. After 30 seconds (control) or 30 minutes (“ischemic”) of incubation, hearts were longitudinally bisected and either sonicated in Laemmli sample buffer (for Western analysis) or fixed overnight at 4°C in 10% neutral-buffered formalin (for immunohistochemistry). Although not thoroughly characterized, the 30-minute “ischemic” period in the presence or absence of 1 mM calcium reproduces the effects of ischemia on Cx43 electrophoretic mobility and gap junction remodeling (see results) demonstrated in better characterized models of ischemia.
Formalin-fixed tissue was processed, sectioned, immunostained and microscopically analyzed as previously described (King and Lampe, 2004). Briefly, tissue sections were deparaffinized, antigen retrieved, blocked and detected utilizing rabbit anti-Cx43 (1:250, Sigma) or rabbit anti-pS325/328/330 (1:1000). Slides were washed and incubated with a biotinylated anti-rabbit secondary antibody (1:250) and detected with ABC-avidin/biotin conjugate (Vectastain, Vector Labs, Burlingame, CA).
Immunoblotting and Immunofluorescence
Approximately the same amount of cardiac tissue for each treatment was sonicated in sample buffer supplemented with 50mM NaF, 1mM Na3VO4, 5% β-mercaptoethanol, 1 mM PMSF and 1x Complete protease inhibitors (Roche Molecular Biochemicals, Alameda, CA). Insoluble material was removed by centrifugation, and the soluble fraction was separated on SDS-10% PAGE. After immunoblotting, protein was detected with a rabbit antibody against pS325/328/330-Cx43 and a mouse anti-Cx43 (Cx43NT). The blots were also probed with anti-GAPDH (Ambion, Austin, TX) and anti-Vinculin (Sigma) to confirm consistent loading. Primary antibodies were visualized with fluorescent dye-labeled secondary antibodies [AlexaFluor 680 goat anti-rabbit (Molecular Probes, Eugene, OR) and IRDye800-conjugated donkey anti-mouse IgG (Rockland Immunochemicals)] and directly quantified using the LI-COR Biosciences Odyssey infrared imaging system (Lincoln, NE) and associated software (inverted images are shown).
To test the phosphospecificity of the p325/328/330 antibody, we treated with alkaline phosphatase or left untreated equal amounts of protein lysates from fibroblasts expressing Cx43-WT and Cx43-TM (clone A) as previously reported (Lampe et al., 1998), ran the lysates in SDS-PAGE and performed a Western blot with Cx43NT1 and the pS325/328/330 antibodies on the same blot as described above. To determine the specificity of binding of the antibody, peptides representing singly phosphorylated S325, 328 and 330 (CGQAG(pS)TISN-amide, CAGSTI(pS)NSHA-amide, CGSTISN(pS)HAQP-amide, respectively), doubly phosphorylated S325/328 and S328/330 (CGQAG(pS)TI(pS)NSH-amide and CGSTI(pS)N(pS)HAQ-amide) and the triply phosphorylated immunizing peptide were synthesized and individually incubated (at 200 pg/ml) with pS325/328/330 antibody for 30 minutes prior to probing Western blots of whole cell lysates from mouse heart (prepared and immunodetected as described above) using a Surf-Blot apparatus (Idea Scientific, Minneapolis, MN) for the primary antibody incubation step. The level of antibody binding was determined as described above by LI-COR densitometry and normalized to the signal in the absence of peptide. The amount of antibody that can bind to the different peptides was determined by linking the 6 peptides to Reacti-Bind Maleimide-activated 96-well plates (Pierce) at saturating concentrations. The plates were washed, blocked and incubated with pS325/328/330 antibody or the further purified pS325/328 specific antibody in quadruplicate according to manufacturer’s instructions and with peroxidase labeled donkey anti-rabbit secondary antibody (Chemicon, Temecula, CA). Peroxidase levels were detected using ABTS Peroxidase substrate (KPL, Gaithersburg, MD). The amount of antibody bound was determined by normalizing to the total Abs410nm for the immunizing peptide and mean and standard deviation were calculated.
To test the effect of mutation at S325/328/330, fibroblasts exogenously expressing wild-type Cx43 or Cx43-TM were grown to 90–95% confluency and cells were harvested in 1% Triton X-100 in PBS supplemented with 50mM NaF, 1mM Na3VO4 and protease inhibitors. These samples were separated into Triton soluble and insoluble fractions by centrifugation at 13,000g at 4°C for 10 minutes. Triton insoluble fractions (pellets) were resuspended in Laemmli sample buffer and sonicated. Duplicate parallel cultures were lysed in 1X Laemmli SDS-sample buffer supplemented with phosphatase/protease inhibitors and 5% β-mercaptoethanol (whole cell lysate), followed by brief sonication. Protein assays for equal loading, electrophoresis and immunodetection using Cx43CT2 antibody was performed as previously described (Cooper and Lampe, 2002).
Immunofluorescence was performed as previously described (Solan et al., 2003). Cx43 was detected with anti-Cx43 antibody (C6219) and Alexa 594 anti-rabbit secondary antibody. DAPI was added to visualize cellular nuclei. The coverslips were mounted onto slides with DABCO antifade medium [25mg/ml of 1,4-Diazobicyclo-(2,2,2)octane (Sigma) diluted in Spectroglycerol (Eastman Kodak Co.) and 10% PBS, pH 8.6] and viewed with a Nikon Diaphot TE300 fluorescence microscope, equipped with a 40x (1.3 n.a.) oil objective and a Princeton Instruments digital camera driven by an attached PC and Metamorph imaging software.
Dye Injection
Cx43-WT or Cx43-TM expressing cells were grown in 35mm dishes to 70–80% confluency. Donor cells were microinjected with Lucifer yellow (1mg/ml dissolved in 0.15M LiCl) and allowed to transfer dye for three minutes. Digital images were collected at identical camera settings on the Nikon Diaphot TE300 fluorescence microscope described above. Following imaging, the number of recipient cells for both conditions was quantified in a blind manner.
Junctional Conductance and Single Channel Activity
Confluent cells were trypsinized and replated at low density on glass coverslips. After 2–5h coverslips were mounted in a custom made chamber and cells were visualized on an inverted (Olympus IMT2) microscope equipped for phase contrast observation. Cells were bathed in solution containing (in mM): 142.5 NaCl, 4 KCl, 1 MgCl2, 5 glucose, 2 sodium pyruvate, 10 HEPES, 1 BaCl2, 1 CaCl2, 15 CsCl, and 10 TEACl, pH 7.2 and osmolarity 315 mOsM. Junctional conductance was determined using dual whole-cell voltage clamp techniques as previously described (Cottrell et al., 2003). The pipette solution contained (in mM): 120 KCl, 14 CsCl, 3 MgCl2, 5 glucose, 9 HEPES, 9 EGTA, 9 TEACl, 5 Na2ATP, 30 KOH to adjust pH to 7.2, 315 mOsM. Because Cx45 is sometimes detected at low levels in the parental Cx43−/− cell line (Martyn et al, 1997), we first evaluated each cell pair for voltage dependent loss of junctional conductance. Junctions displaying significant voltage dependent gating at transjunctional voltages of 40mV (>10–15% decrease in conductance in 5–10s) were not studied further. For voltage insensitive junctions, junctional conductance was evaluated with 10mV transjunctional pulses and single channel events were visualized with a transjunctional driving force of 40mV following superfusion with 4mM halothane (Burt and Spray, 1989). Event amplitudes were measured by hand for each cell pair, binned into 6.25pS bins, the binned data from all pairs pooled, and the relative frequencies of each bin calculated and plotted. Data were fit using Origin software as previously described (Cottrell et al., 2003).
RESULTS
Ischemia-induced “dephosphorylation” of Cx43
Previous studies have shown that the electrophoretic mobility of Cx43 isolated from ischemic heart is increased, such that most of the protein migrates at a position that includes dephosphorylated Cx43 so the assumption has been made that these changes in electrophoretic mobility represent a reduction in phosphorylation at undefined sites (Beardslee et al., 2000; Jain et al., 2003). In addition, inhibition of CK1 has been shown to reduce gap junction assembly and to phosphorylate Cx43 in vitro at one or more of serines 325/328/330 (Cooper and Lampe, 2002). We hypothesized that phosphorylation at sites necessary for efficient assembly of channels might be compromised in the ischemic setting. To evaluate whether these phosphorylation events occur in control and ischemic heart tissue, we generated a polyclonal antibody that uniquely detects Cx43 phosphorylated at S325/328/330 (pS325/328/330–the specificity of which is described in the next section).
In agreement with previous studies, the electrophoretic mobility of total Cx43 from ischemic heart was increased compared to control possibly indicating unspecified “dephosphorylation” event(s) had occurred (Fig. 1, each lane represents a distinct heart). When detected with the phosphospecific antibody, we found that the pS325/328/330-Cx43 content decreased ~8 fold (p<0.03) in ischemic hearts (6-fold when the loss of total Cx43 was taken into account).
Fig. 1.
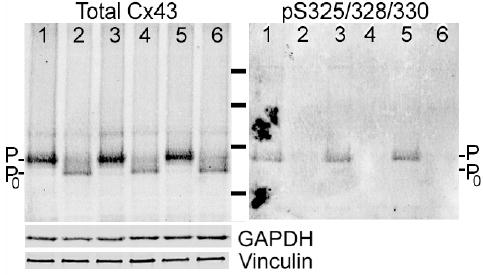
Increased electrophoretic mobility of Cx43 in the ischemic heart involves dephosphorylation at serines 325/328/330. Western blot of total protein isolated from 3 control (Lanes 1,3,5, respectively) or 3 globally ischemic (30’ at 37°C; Lanes 2,4,6, respectively) hearts simultaneously probed for total Cx43 (Cx43NT-1) and Cx43 phosphorylated at S325/328/330 followed by GAPDH, and vinculin antibodies. Note the increased mobility of Cx43 and decreased pS325/328/330 content in the ischemic samples. Molecular weight standards are 28, 49, 62 and 98 kDa.
The cellular distribution of Cx43 detected with the phosphospecific antibody and one to total Cx43 varied in ischemic vs. normal tissue. Fig. 2 shows that in normal hearts virtually all total Cx43 and pS325/328/330-Cx43 were localized to intercalated disks. As shown previously (Severs et al., 2004), ischemia caused a redistribution of total Cx43 from the intercalated disk to the lateral borders of myocytes. The pS325/328/330 signal was reduced at the intercalated disk of ischemic heart tissue, consistent with the total Cx43 reduction, but, surprisingly, pS325/328/330-Cx43 was undetectable at the lateral borders. If phosphorylation at these sites promotes gap junctional functionality, then transverse conduction in the ischemic heart would not be expected to increase (dramatically) despite increased Cx43 at lateral borders because of the reduced phosphorylation at the S325/328/330 sites and consequent poor gap junction functionality.
Fig. 2.
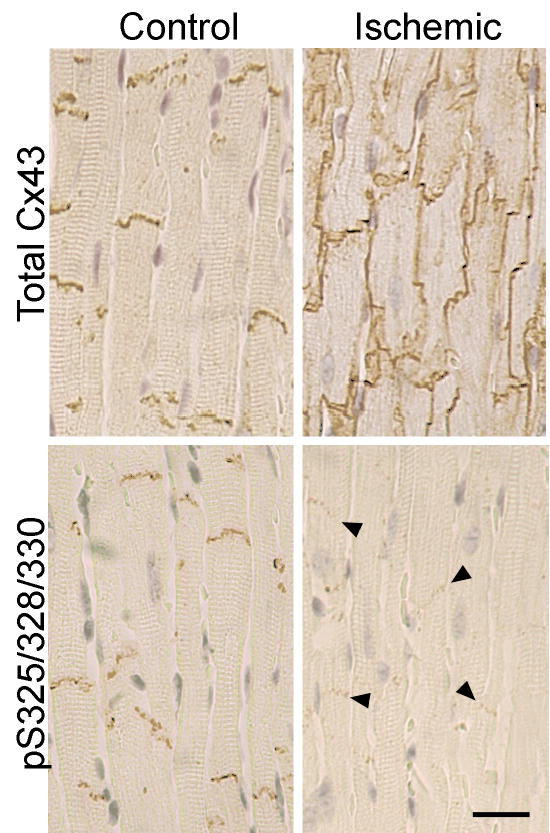
Differential phosphorylation of Cx43 localized at intercalated disks vs. lateral edges of myocytes in the normal and ischemic heart. The localization of total Cx43 (Sigma C6219) and pS325/328/330-Cx43 in normal and ischemic heart is shown. Note the increase in Cx43 at the lateral edges of the myocytes of the ischemic heart. Lateral edge Cx43 was not detectably phosphorylated at S325/328/330. In contrast, Cx43 at the intercalated disks of ischemic heart retained phosphorylation at S325/328/330 (bar=20μm).
Analysis of the specificity of the pS325/328/330 antibody
To test the specificity of the pS325/328/330 antibody and to determine whether phosphorylation at S325/328/330 directly affects protein localization and gap junction formation, we transfected constructs encoding either wild-type Cx43 (WT) or Cx43 with serine to alanine mutations at residues 325, 328 and 330 (Cx43-TM) into fibroblasts derived from Cx43−/− mice and examined the properties of the gap junctions formed by these cells. We first validated the phosphospecific nature of the antibody by examining its binding to extracts from cells expressing Cx43-WT or Cx43-TM both before and after treatment with alkaline phosphatase. As shown in Fig. 3A, the antibody to total Cx43 bound to 41–44 kDa bands in all cases and revealed the classic increase in mobility to the fastest migrating position upon alkaline phosphatase treatment. In contrast, pS325/328/330-specific antibody bound to wild-type Cx43 only in the absence of alkaline phosphatase treatment and did not bind to the Cx43-TM under any conditions. Furthermore, pS325/328/330 antibody appeared to bind only to the slowest migrating isoform of Cx43; the Cx43-TM expressing cells also did not efficiently convert Cx43 to the slower migrating phosphorylated isoforms.
Fig. 3.
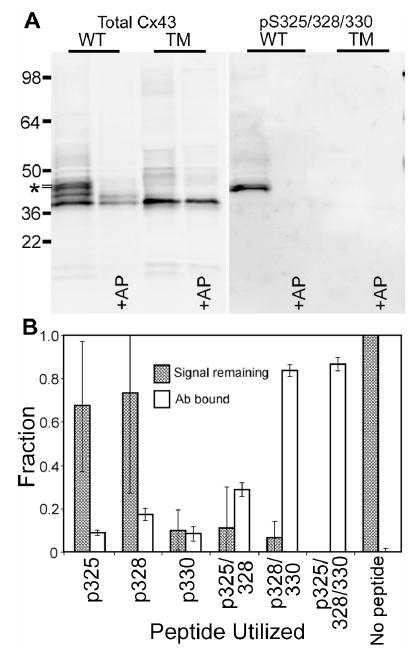
The pS325/328/330 antibody is specific for the P2 isoform of Cx43 and pS328/330. (A) Equal amounts of protein lysate from fibroblasts expressing wild-type Cx43 (WT) and Cx43-TM (TM, clone A) were either treated with alkaline phosphatase (+AP) or untreated and probed in a Western blot with Cx43NT1 (Total Cx43-left panel) and the pS325/328/330 (right panel) antibodies on the same blot. Molecular weight standards are marked on the left side and the P2 isoform is marked with an *. (B) The amount of antibody bound to the different peptides representing singly, doubly and triply phosphorylated Cx43 linked to an ELISA well (open bars) is shown along with the ability of these peptides to compete with Cx43 present in heart lysates in a Western immunoblot format (filled bars). Error bars represent standard deviation from the mean.
To examine the specificity of the pS325/328/330 antibody and determine which sites are likely phosphorylated in vivo, we synthesized 3 peptides with S325, 328 and 330 each singly phosphorylated, two peptides, S325/328 and S328/330, doubly phosphorylated, and the immunogen peptide triply phosphorylated and covalently bound these peptides to an activated 96-well plate. The p325/328/330 antibody was incubated in the well and a standard enzyme-linked immunoabsorbent assay (ELISA) was performed (Fig. 3B, open bars). The pS328/330 peptide bound almost as much total antibody as the immunizing (triply phosphorylated) peptide indicating that most of the antibody bound an epitope centered on these sites. The singly phosphorylated peptides bound only a small amount of antibody. To further answer the question of the specificity of this antibody preparation, we removed the antibody that bound to the p328/330 peptide and purified that specific for pS325/328. Analysis of the binding of this preparation to the 6 peptides in the ELISA format indicated that the pS325/328 purified antibody indeed bound to mostly to the pS325/328 peptide followed by the triply phosphorylated and the pS325 singly phosphorylated peptides (data not shown). These results indicate that the antibody preparation is phosphospecific but polyclonal with reactivity to all 3 sites. To examine the ability of the 6 peptides to block antibody binding to Cx43, we preincubated these peptides with the pS325/328/330 antibody prior to Western blot analysis of heart lysates. Neither the pS325 nor pS328 singly phosphorylated peptides interfered significantly with antibody binding to intact Cx43 leaving most of the signal remaining, whereas the pS330 singly and all of the more extensively phosphorylated peptides did reduce the Western signal to close to background levels (Fig. 3B, filled bars). Further, we tested the ability of the pS325/328 specific antibody and found that it could bind to Cx43 prepared from heart in an immunoblot but much less extensively than the antibody prior to the removal of the pS328/330 reactivity (data not shown). Combined these antibody binding and competition results indicate that this polyclonal antibody can bind to Cx43 phosphorylated at S325, S328 and S330 and that Cx43 is phosphorylated at S328 and/or S330 and to a lesser extent at S325 in heart tissue.
The role of phosphorylation at S325/328/330 in gap junction assembly
To be more quantitative about the extent of gap junction formation, we assayed the ability of three separate Cx43-TM clones to phosphorylate Cx43 to the P2 isoform and incorporate it into a Triton X-100 insoluble fraction, a hallmark of conventional gap junctions (Musil and Goodenough, 1991). Like most cell lines, fibroblasts expressing wild-type Cx43 showed several isoforms of Cx43 in the whole cell lysate and a dramatic enrichment of the P2 isoform in the Triton insoluble fraction (Fig. 4, WT lanes). Transfectants expressing Cx43 with the TM mutations (clones A, B, C) showed multiple differences in their Cx43 migration. In the whole cell fraction, Cx43-TM migrated primarily at P0 with a second band between P0 and P1 and no apparent P2. There was a significant reduction in the total amount of Triton insoluble Cx43 in clone B and C and essentially no bands that co-migrated with P2 were observed in any of the clones. Variation in total Cx43 protein levels most likely represents clonal heterogeneity among individual Cx43 expressing clones. Since the TM-A clone showed comparable levels of total Cx43 expression to the Cx43-WT expressing cells, we used it for most of the subsequent studies. We conclude that phosphorylation of Cx43 at S325, 328 and/or 330 is necessary for Cx43 to migrate as the P2 isoform.
Fig. 4.
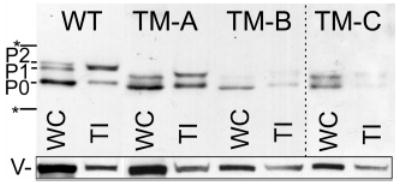
Cx43-TM expressing cells do not phosphorylate Cx43 to the P2 isoform and contain less Triton insoluble (TI) Cx43. Whole cell (WC) and Triton X-100 insoluble (TI) cellular lysates from cells expressing wild-type Cx43 (WT) or three Cx43-TM clones (TM-A, TM-B, and TM-C) were western immunoblotted and probed for total Cx43 with the Cx43NT1 antibody. A darker exposure of the TM-C clone is shown. The migration position of a 50 and 36 kDa marker are indicated with an * and the vinculin loading controls are indicated with a V. The Triton insoluble vinculin could be associated with the cytoskeleton or adhesive junctions.
The localization of Cx43 in the Cx43-TM expressing clones was also examined via immunofluorescence. In Fig. 5, we show that fibroblasts transfected with wild-type Cx43 showed both intracellular and punctate, gap junctional plaque-associated Cx43 (compare to the lack of signal in the KO panel). In contrast, the TM mutant cell lines showed predominately intracellular Cx43 although occasional apparent appositional labeling was observed (TM-A clone is shown).
Fig. 5.
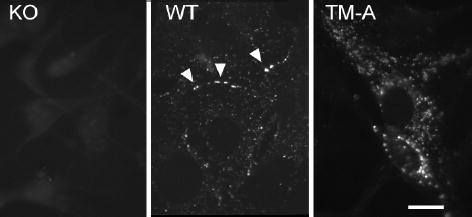
Comparison of Cx43 localization in parental knockout (KO) cells, wild-type Cx43 (WT) and Cx43-TM (TM-A) expressing cells. Apparent gap junctions are marked by arrowheads in the WT panel. Bar=10μm.
We next determined whether cells expressing Cx43-TM supported intercellular communication. Parental Cx43 knockout fibroblasts (KO), wild-type Cx43 expressing fibroblasts (WT) and the three individual Cx43-TM expressing clones A-C were microinjected with Lucifer yellow dye and evaluated for dye transfer 3 minutes later. Digital images were taken and the number of recipient cells was quantified. As shown in Fig. 6, the number of cells receiving dye from donor cells was 60–85% lower in the Cx43-TM clones than for Cx43-WT expressing cells and was comparable to the coupling observed in the parental fibroblasts that lacked Cx43. Although Cx43 protein expression levels varied between the clones (see Fig. 4), all 3 Cx43-TM clones including TM-A, the one that expressed similar levels of Cx43, showed significant reductions in communication compared to the wild-type control (p<0.02). These data suggest that phosphorylation at S325, S328 and/or S330 is critical for efficient dye transfer in mouse fibroblasts.
Fig. 6.
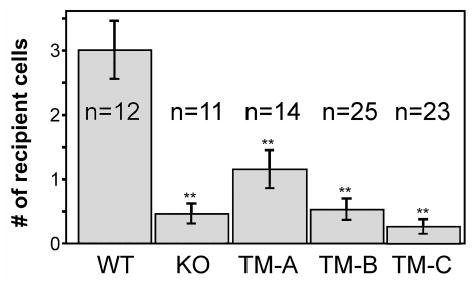
Cells expressing Cx43 S325/328/330A are inefficient at dye transfer. Wild-type (WT), parental (KO) and Cx43-TM (clones TM-A, TM-B and TM-C) expressing cells were microinjected with Lucifer yellow dye. After 3 minutes of transfer, digital images were taken and the number of recipient cells was determined (n=the number of injected cells). Mean and standard error are shown. The extent of dye transfer was significantly different (p<0.02) for WT compared to all of the other cell types as indicated by **.
We next evaluated whether junctional conductance and channel behavior differed as a consequence of the serine to alanine mutations at the S325/328/330 sites. Based on the dye-coupling data presented above, we expected junctional conductance to be significantly reduced in the Cx43-TM cell clones. This was indeed the case for Cx43-TM cells plated for an equivalent period of time as wild-type cells (2–5h) - junctional conductance in Cx43-TM cells was 3.5±0.7 nS (n=24) vs. 6.2 ±1.1 nS (n=35, p<0.05) in Cx43-WT cells. Coupling between Cx43-TM cells improved with time such that 24–30h after plating, conductance was 4.9±0.8 nS (n=28), a value not significantly different from Cx43-WT cells at 2–5h post plating. The profile of channel conductances in the Cx43-TM cells was substantially different from that observed in wild-type cells as shown in Fig. 7A and emphasized in the difference plot (Fig. 7B), with a severely decreased frequency for activity of fully-open channels (~110pS) and increased frequency of 60–85pS channels.
Fig. 7.
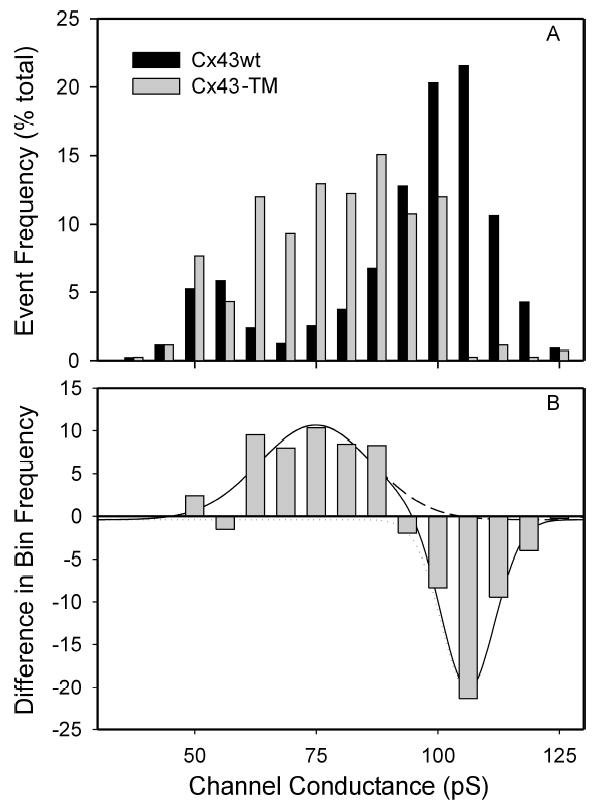
Histogram of channel events observed in Cx43-TM (gray) and Cx43-WT expressing cells. Panel A shows event frequency as a percent of total events in each bin; panel B shows the difference induced by mutation of the 325, 328, and 330 sites. Note the reduced incidence of events corresponding to fully open channels (106 ± 1 pS) and increased incidence of 75 ± 2 pS. The 54 ± 1 pS population did not differ between wild-type and mutant data sets. Difference data were fit using Origin, Chi2 and R2 for fit are 5.677 and 0.94, respectively (−WT: 1851 events, n=13; TM: 418 events, n=7).
Discussion
Previous work has established that myocardial ischemia rapidly induces uncharacterized changes in Cx43 phosphorylation and localization, however no mechanistic connections between specific phosphorylation events and changes in conduction or localization have been identified. Here, we show that phosphorylation of Cx43 at serines 325, 328, and/or 330 was drastically reduced in the ischemic heart. Further, we demonstrate that Cx43 localized to the lateral borders of myocytes was not phosphorylated at these sites, whereas the Cx43 that remained at the intercalated disk retained these modifications. Finally, we show that when these sites were not phosphorylated, the event frequency for fully open channels, the overall junctional conductance and the extent of Lucifer Yellow dye coupling were all significantly reduced.
Although direct Cx43 phosphorylation on serines has long been correlated with gap junction formation and function under basal conditions (Musil and Goodenough, 1991), few mechanistic connections to specific phosphorylation events have been drawn. For instance, phosphorylation at S364 was shown to be important for cAMP induced up-regulation of gap junction assembly. However, cells expressing Cx43 with a S364A mutation assembled Cx43 into junctions nearly as well as wild-type expressing cells, suggesting S364 was not necessary for assembly in homeostatic cells (TenBroek et al., 2001). Similarly, several serines are known to be involved in mitogen-induced downregulation of communication including serines 255, 279 and 282 (MAPK) and serines 262 and 368 (protein kinase C), but these phosphorylation events have been reported to negatively affect channel gating properties (Lampe et al., 2000; Cottrell et al., 2003). S325, 328 and/or 330 were previously shown to be substrates for CK1 phosphorylation, a kinase implicated in the regulation of gap junction assembly (Cooper and Lampe, 2002). Our phosphospecific antibody could react with Cx43 phosphorylated at all 3 sites in heart tissue. We probed heart lysates with 7 other Cx43 phosphospecific antibodies that we created or are available from commercial sources and that we find to be specific for phosphorylation at S255, S262, S279/282, S364, S368, or S372. S325/328/330 was the only one that gave a significant decrease in phosphorylation (results not shown, several showed little binding to heart lysate under either condition). The antibody to pS368 showed increased binding upon hypoxia that we have investigated separately (Ek-Vitorin et al., 2006). Similar to a previous report (Jeyaraman et al., 2003), we found (data not shown) that ischemia led to increased binding of an anti-Cx43 antibody we prepared to a nonphosphorylated peptide corresponding to residues 360–382 of Cx43 that appears to be identical to Zymed 13800, an antibody prepared to a similar epitope that has been reported to bind to “dephosphorylated Cx43” (Nagy et al., 1997).
We probed the functional consequences of these phosphorylation events by expressing Cx43 with serine to alanine conversions at all 3 sites (i.e., the TM mutant). This mutant, rather than double or single site mutants, was selected for study for two reasons: 1) all 3 sites appear to be phosphorylated in heart tissue and 2) the functional consequences of phosphorylation at clusters of (phospho)serines can be misinterpreted from single site mutants because either the remaining sites partially or fully compensate for the missing site or the remaining sites are not phosphorylated because phosphorylation is sequential with the mutated site necessary for subsequent phosphorylation events (as can be the case for CK1 (Flotow et al., 1990)). Microinjected Cx43-TM cells and parental (Cx43−/−) fibroblasts were occasionally dye coupled to 1 or 2 cells. In contrast, although electrical coupling between Cx43-TM cells was reduced compared to wild-type in the early hours after plating, significant coupling was routinely observed at 24–30 hours. This coupling was mediated by channels with conductances of 70–90pS, a value intermediate to the fully open channel (~110pS) and the previously reported, phosphorylation-induced 50–60pS substate (Lampe et al., 2000). These results are consistent with at least two explanations. First, phosphorylation at S325/328/330 is necessary for Cx43 channels to open to their fully open state. If correct, the intermediate conductance substate of Cx43 observed in the Cx43-TM cells would have to be poorly permeated by LY to accommodate the low levels of dye coupling observed for these cells. Interestingly, this intermediate conductance state is commonly observed in Cx43-WT expressing rat insulinoma cells that do not express Cx45 (Ek-Vitorin, personal communication), and the junctions formed by these cells are poorly permeated by dye. Second, since Cx45 is expressed at low levels in the parental fibroblasts (Martyn et al., 1997), these intermediate channels could reflect the activity of heteromeric Cx43–Cx45 channels (Martinez et al., 2002). This seems unlikely for two reasons, first both single channel data sets (Cx43-TM and Cx43-WT) were obtained from cells whose macroscopic levels of coupling were comparable, and second heteromeric Cx43–Cx45 channels are not permeated by Lucifer Yellow (Martinez et al., 2002). Regardless of which of these possibilities is correct, the data indicate that loss of the capacity for phosphorylation at S325, S328 and/or S330 renders the junctions inefficient at dye transfer and less conductive.
What mechanisms might control the level of phosphorylation at S328 and/or 330 during hypoxia? Several groups have shown that phosphatase inhibitors block the change in Cx43 electrophoretic mobility (Duthe et al., 2001; Jeyaraman et al., 2003; Turner et al., 2004). PP1 has been reported to be the main phosphatase that is active against Cx43 in cardiomyocytes and it appears to be constitutively active (Duthe et al., 2001; Jeyaraman et al., 2003; Turner et al., 2004). We have shown that CK1 can phosphorylate Cx43 at S325, 328, and/or 330, and CK1 inhibition leads to loss of phosphorylated Cx43 supporting the role of this kinase in the phosphorylation of these sites (Cooper and Lampe, 2002). However, a constant kinase/phosphatase cycling process has been reported to occur during hypoxia with Cx43 dephosphorylation resulting from reduced cellular ATP levels without kinase or phosphatase activity changes or Cx43 synthesis (Duthe et al., 2001; Turner et al., 2004). Given that kinase or phosphatase localization could also be critical, the roles that kinase activity, phosphatase activity and ATP concentration play on the control of the level of phosphorylation at S325/328/330 during hypoxia remain to be determined.
In summary, data presented here indicate that Cx43 present in cardiac tissue is phosphorylated at serines 325, 328 and/or 330. During ischemia, the amount of total Cx43 is reduced at the intercalated disk and Cx43 relocates to the myocytes’ lateral edges. Cx43 at the intercalated disk retained phosphorylation at S325/328/330 whereas Cx43 present at the lateral edges did not. We have shown that phosphorylation at these residues was necessary to form the P2 isoform, and these residues are important in gap junction assembly/stability. Thus, the expected net effect of ischemia would be a measured reduction in longitudinal conduction velocity, due to decreased localization at intercalated disks (Poelzing and Rosenbaum, 2004), without a concomitant increase in transverse conduction velocity, despite increased localization at lateral borders. Whether reduced phosphorylation at S325/328/330 triggers the relocalization of Cx43 to the lateral edges is not known. However, the spatially specific retention of phosphorylation at these sites at the intercalated disk and loss at the lateral edges would be expected to help retain the anisotropy of conduction in the heart during ischemia.
Acknowledgments
These studies were supported by Grants from the National Institutes of Health: GM055632 (PDL) and HL058732 (JMB).
References
- Bao X, Reuss L, Altenberg GA. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of Serine 368. J Biol Chem. 2004;279:20058–20066. doi: 10.1074/jbc.M311137200. [DOI] [PubMed] [Google Scholar]
- Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circulation Research. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- Bergoffen J, Scherer SS, Wang S, Oronzi Scott M, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fishbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- Burt JM, Spray DC. Volatile anesthetics block intercellular communication between neonatal rat myocardial cells. Circ Res. 1989;65:829–837. doi: 10.1161/01.res.65.3.829. [DOI] [PubMed] [Google Scholar]
- Cooper CD, Lampe PD. Casein kinase 1 regulates connexin43 gap junction assembly. J Biol Chem. 2002;277:44962–44968. doi: 10.1074/jbc.M209427200. [DOI] [PubMed] [Google Scholar]
- Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF, Burt JM. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol. 2003;284:C511–520. doi: 10.1152/ajpcell.00214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthe F, Plaisance I, Sarrouilhe D, Herve JC. Endogenous protein phosphatase 1 runs down gap junctional communication of rat ventricular myocytes. American Journal of Physiology - Cell Physiology. 2001;281:C1648–1656. doi: 10.1152/ajpcell.2001.281.5.C1648. [DOI] [PubMed] [Google Scholar]
- Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of Connexin 43 Channels Is Regulated Through Protein Kinase C-Dependent Phosphorylation. Circ Res. 2006 doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem. 1990;265:14264–14269. [PubMed] [Google Scholar]
- Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB. Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91:833–843. doi: 10.1016/s0092-8674(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Jain SK, Schuessler RB, Saffitz JE. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ Res. 2003;92:1138–1144. doi: 10.1161/01.RES.0000074883.66422.C5. [DOI] [PubMed] [Google Scholar]
- Jeyaraman M, Tanguy S, Fandrich RR, Lukas A, Kardami E. Ischemia-induced dephosphorylation of cardiomyocyte connexin-43 is reduced by okadaic acid and calyculin A but not fostriecin. Mol Cell Biochem. 2003;242:129–134. [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Laing JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- King TJ, Lampe PD. The gap junction protein connexin32 is a mouse lung tumor suppressor. Cancer Res. 2004;64:7191–7196. doi: 10.1158/0008-5472.CAN-04-0624. [DOI] [PubMed] [Google Scholar]
- Laird DL, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in Brefeldin A-treated rat mammary tumor cells. J Cell Biol. 1995;131:1193–1203. doi: 10.1083/jcb.131.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. Int J of Biochem Cell Biol. Vol. 36. 2004. The effects of connexin phosphorylation on gap junctional communication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe PD, Kurata WE, Warn-Cramer B, Lau AF. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci. 1998;111:833–841. doi: 10.1242/jcs.111.6.833. [DOI] [PubMed] [Google Scholar]
- Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;126:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez AD, Hayrapetyan V, Moreno AP, Beyer EC. Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circ Res. 2002;90:1100–1107. doi: 10.1161/01.res.0000019580.64013.31. [DOI] [PubMed] [Google Scholar]
- Martyn KD, Kurata WE, Warn-Cramer BJ, Burt JM, TenBroek E, Lau AF. Immortalized connexin43 knockout cell lines display a subset of biological properties associated with the transformed phenotype. Cell Growth & Differentiation. 1997;8:1015–1027. [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Li WEI, Roy C, Doble BW, Gilchrist JS, Kardami E, Hertzberg EL. Selective monoclonal antibody recognition and cellular localization of an unphosporylated form of connexin43. Exper Cell Res. 1997;236:127–136. doi: 10.1006/excr.1997.3716. [DOI] [PubMed] [Google Scholar]
- Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelzing S, Rosenbaum DS. Nature, significance, and mechanisms of electrical heterogeneities in ventricle. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:1010–1017. doi: 10.1002/ar.a.20103. [DOI] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. Faseb J. 2003;17:1355–1357. doi: 10.1096/fj.02-0975fje. [DOI] [PubMed] [Google Scholar]
- Severs NJ, Dupont E, Coppen SR, Halliday D, Inett E, Baylis D, Rothery S. Remodelling of gap junctions and connexin expression in heart disease. Biochim Biophys Acta. 2004;1662:138–148. doi: 10.1016/j.bbamem.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Sohl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62:228–232. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci. 2003;116:2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–1318. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, Webster KA, Bishopric NH. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. 2004;95:726–733. doi: 10.1161/01.RES.0000144805.11519.1e. [DOI] [PubMed] [Google Scholar]
- White T, Paul D. Genetic diseases and gene knockouts reveal diverse connexin functions. Ann Rev Physiology. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]