

Abstract
Transforming growth factor-alpha (TGF-α) is abnormally expressed in autosomal recessive polycystic kidney disease (ARPKD). Tumor necrosis factor-alpha converting enzyme (TACE), a metalloproteinase, mediates TGF-α processing. In this study, we sought to determine whether TGF-α was an absolute requirement for renal cystogenesis and whether its absence would modulate disease severity or related growth factors/receptors expression. Bpk heterozygotes were bred with TGF-α null mice to produce cystic and noncystic offspring with or without TGF-α. Assessments included kidney weight (KW), body weight (BW), blood urea nitrogen (BUN), and kidney and liver immunohistology. Western analysis assessed kidney expression of amphiregulin (AR), epidermal growth factor (EGF), heparin-binding EGF (HB-EGF), and their receptors, EGFR and ErbB4. A PCR-based methodology for genotyping bpk mice was also developed. No significant differences in KW, BW, KW/BW%, or BUN were seen in cystic mice with versus without TGF-α. Cystic kidney disease and liver disease histology were similar. AR, EGF, HB-EGF, EGFR, and ErbB4 were abnormally expressed to an equal degree in kidneys of mice with versus without TGF-α. Although previous data suggest a critical role of TGF-αin murine PKD, these data show that TGF-αis not required for renal cyst formation or kidney or liver disease progression. We speculate that the therapeutic effect of WTACE2 could have been due to effects on several TACE targets, including TGF-α, AR, and ErbB4, as well as metalloproteinases other than TACE.
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; AR, amphiregulin; ARPKD, autosomal recessive polycystic kidney disease; BUN, blood urea nitrogen; BW, body weight; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; HB-EGF, heparin-binding epidermal growth factor; KW, kidney weight; MMP, matrix metalloproteinase; PKD, polycystic kidney disease; TACE, tumor necrosis factor-alpha converting enzyme; TGF-α, transforming growth factor-alpha
PKD is characterized by abnormal renal tubular epithelial cell proliferation, which contributes to cyst formation and expansion (1). Numerous studies have implicated the EGFR and its ligand, TGF-α, in the pathogenesis of this abnormal cell proliferation. EGFR and TGF-α are abnormally expressed in murine and human PKD (2–4). Transgenic mice that overexpress TGF-α develop cystic kidneys (5). TGF-α overexpression also accelerates the progression of cystic kidney disease in the pcy murine model of PKD (6). Treatment with novel inhibitors of EGFR tyrosine kinase significantly attenuated the severity of cystic kidney disease in the bpk mouse model of ARPKD and the Han-SPRD rat model of autosomal dominant PKD (ADPKD) (7,8). Treatment of cystic bpk mice with a novel inhibitor of TACE, the metalloproteinase that mediates TGF-α ectodomain shedding, significantly reduced the severity of cystic kidney disease (2). Furthermore, when compared with single higher dose EGFR inhibitor therapy, combination therapy with a TACE inhibitor and a decreased dose of an EGFR inhibitor was as effective (9), suggesting a synergistic effect of the two. Based on these collective findings, we hypothesized that TGF-α has an essential role in cyst formation and progression of cystic kidney disease in murine ARPKD.
In the current study, we sought to determine 1) whether TGF-α is an absolute requirement for renal cystogenesis in ARPKD and 2) whether the absence of TGF-α would diminish the severity of cystic kidney or biliary disease or modulate the expression of other EGF-related growth factors and their receptors. As a secondary aim, we sought to develop a PCR-based methodology for rapidly genotyping bpk mice, based on the recent identification of bicaudal C (Bicc1) as the mutated gene in this model (10).
METHODS
Animal models
The inbred mouse strain C57BL/6J-Tgfatm1Ard, which is homozygous for a TGF-α null allele (11), was obtained from the Jackson Laboratories (Bar Harbor, ME). TGF-α null (TGF −/−) mice develop normally and are fertile. Their phenotypic abnormality is characterized by pronounced waviness in the coat and whiskers, and dramatic derangement of the hair follicles (11,12).
The bpk model of murine ARPKD arose out of a spontaneous mutation in a Balb/C colony (13). Mice homozygous for the bpk mutation (bpk −/−) develop severe cystic renal disease evident histologically at birth and typically die by 28 d of age. The disease phenotype and course have been characterized previously (13,14). Mice heterozygous for the bpk mutation have no phenotypic abnormalities.
All animal experiments were conducted in accordance with policies of the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University. Reagents were obtained from Sigma Chemical (St. Louis, MO), unless otherwise indicated.
Breeding strategy and animal genotyping
Bpk heterozygote mice were bred with TGF-α null mice and the resultant offspring (all obligate TGF-α heterozygotes) were backcrossed to known bpk heterozygotes. Double heterozygotes (bpk +/−; TGF +/−) were then intercrossed to create cystic and noncystic offspring with 0, 1, or 2 TGF-α null alleles. Before the identification of the bicaudal C (Bicc1) as the bpk causative gene, and the development of a genotyping protocol (see below), bpk heterozygotes were identified based on a normal appearance and their ability to produce cystic offspring. Cystic (bpk −/−) mice were identified by the typical findings of massively enlarged, cystic kidneys corresponding to advanced cystic disease. When Bicc1 was identified as the causative disease gene for the bpk mutation, a PCR-based genotyping protocol was developed (see below). Genomic DNA was isolated from tail samples and all offspring studied were then analyzed to confirm their bpk genotype. TGF-α heterozygote (TGF +/−) mice were identified by a normal appearance and PCR confirmation of the TGF-α null allele. The presence of the TGF-α null allele was identified by PCR for the neo cassette contained within the knockout construct, using a standard genotyping protocol, “NEOTD,” provided by the Jackson Laboratories and available at http://jaxmice.jax.org. TGF-α null mice were identified based on phenotypic changes (wavy hair, curly whiskers) and PCR confirmation of the TGF-α null allele. TGF-α wild-type (TGF +/+) mice were identified by a normal appearance and the absence of the TGF-α null allele.
Analysis of cystic kidney disease and liver disease severity
Kidneys were obtained at sacrifice from intercross progeny at d 21 of life, an age of advanced cystic collecting tubule disease in the bpk mouse, as described previously (13). Clinical parameters assessed included KW, BW, and KW/BW%. BUN was assessed by colorimetric assay using blood obtained by orbital puncture just before sacrifice. Data on clinical and laboratory parameters were presented as mean ± SD and differences between groups were analyzed by two-tailed t test.
To assess histologic changes in the appearance of cystic kidney disease in mice with and without TGF-α, immunohistology was performed on 4-μM sections obtained from paraformaldehyde-fixed plastic-embedded cystic kidneys, as described (with modifications) (7). Severity of cystic kidney disease was qualitatively assessed by staining serial sections with biotinylated lectins specific for collecting tubule (CT) [Dolichos biflorus agglutinin (DBA)] and proximal tubule (PT) [Lotus tetragonolobus (LTA)], and counterstained with hematoxylin, as described (2). The ratio of cystic CT to PT was also assessed, as described (2).
In our previous study involving treatment of cystic bpk mice with the TACE inhibitor, WTACE2, only the kidney disease severity was assessed (2). Therefore, the effect of TACE inhibition (i.e. TGF-α inhibition) on the liver phenotype was not known. However, treatment with a novel inhibitor of EGFR function has been shown to slow progression of the liver disease in this model (7). In the current study, we examined the impact of the absence of TGF-α on the liver disease in addition to the kidney disease. Livers were obtained at sacrifice and embedded as described above. Hematoxylin staining was performed on 4-μM sections and hepatic (biliary) pathology was qualitatively assessed in cystic mice with and without TGF-α. Sections were examined for the presence of the characteristic bile duct ectasia and proliferation previously described in this model (13).
Expression of other EGF-related growth factors and their common receptor
Protein expression of the EGF-related growth factors, AR, EGF, and HB-EGF, and their receptors, EGFR and ErbB4, was examined by Western analysis. Total cellular protein was isolated from kidneys obtained at sacrifice, as described (2). Protein content of all samples was determined using the BCA protein assay kit (Pierce Biotechnology, Rockford, IL). For AR and HB-EGF Western analysis, whole kidney lysates were subjected to heparin column purification, as described (15), before Western analysis. Samples were resolved by SDS-PAGE electrophoresis, transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), and hybridized with blocking buffer (TBS/0.05% Tween 20/5% dry milk) followed by overnight incubation with rabbit polyclonal anti-human AR (Ab-1) (NeoMarkers, Fremont, CA) or rabbit polyclonal anti-rat HB-EGF (a gift from Dr. Raymond Harris, Vanderbilt University). Membranes were washed, hybridized with the appropriate peroxidase conjugated secondary antibodies, developed with ECL (Amersham Pharmacia Biotech, Inc., Piscataway, NJ), and exposed to autoradiography film. For EGF, EGFR, and ErbB4 analysis, 100 μg of total kidney lysate was diluted in SDS reducing buffer, resolved by SDS-PAGE electrophoresis, and immunoblotted, as above. Antibodies used were goat anti-mouse EGF (Santa Cruz Biochemicals, Santa Cruz, CA), sheep anti-EGFR (Upstate Biotechnology, Lake Placid, NY), and rabbit anti-ErbB4 (Santa Cruz Biochemicals).
Bpk genotyping protocol
To determine the genotype at the bpk locus, genomic DNA samples from tail clips were PCR-amplified using primers designed to flank the site of the bpk mutation on Bicc1 exon 22, as outlined below. PCR reactions contained 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTP, 130 μM each of Bicc1 forward primer (5′-GGCATCTCTCCACT-GCTA-3′) and reverse primer (5′-AGGTGCGTGTCCAGAAGGTA-3′), and 1U Taq Polymerase. PCR cycle parameters were 95°C × 3 min, 94°C × 2 min, 55°C × 2 min (30 cycles), 72°C × 1 min, 72°C × 10 min. Samples were cleaned using QIAquick PCR Kit (QIAGEN, Valencia, CA) and digested with BstU1 (New England Biolabs, Beverly, MA) for 2 h at 60°C. Restriction digest products were resolved on a 3% agarose gel stained with 0.5 μg/mL of ethidium bromide.
RESULTS
Absence of TGF-α does not prevent or reduce the severity of cystic kidney disease or liver disease
As shown in Figure 1, no differences were seen in the gross appearance of cystic or noncystic kidneys with TGF-α present compared with their TGF-α null counterparts. Table 1 summarizes the clinical parameters of each of the experimental groups. No significant differences in KW, BW, KW/BW%, or BUN values were seen in cystic mice with one or two copies of TGF-α compared with those without. Similarly, no changes in KW/BW% were seen in the noncystic mice with one or two copies of TGF-α versus those without. Noncystic TGF-null mice, however, did have lower KW (0.11 ± 0.02 versus 0.14 ± 0.02, p = 0.003) and BW (6.87 ± 1.23 g versus 8.84 ± 0.88 gm, p = 0.001) compared with normal mice with TGF-α. Figure 2A demonstrates the histologic appearance of cystic kidneys with TGF-α (left panel) and without TGF-α (right panel). The severity of CT cysts, as shown by DBA staining, was similar in both cystic groups. A minority of cystic tubules in both cystic groups was PT in origin, as illustrated in the figure. The cystic CT to PT ratios were similar in mice with versus without TGF-α and the values were similar to those previously reported in kidneys from untreated cystic mice in our previous TACE inhibitor study (2). Figure 2B shows representative histology of livers from cystic mice with (left panel) and without (right panel) TGF-α, showing similar degrees of bile duct ectasia and proliferation.
Figure 1.
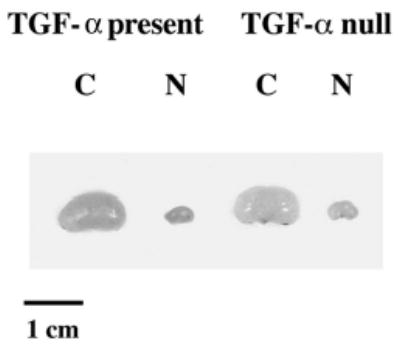
Whole kidney micrographs from d-21 cystic (C) bpk mice and noncystic (N) littermates with TGF-α present or absent. Micrograph demonstrating relative kidney sizes of cystic bpk mice and noncystic mice with TGF-α (TGF-α present) or without (TGF-α null). Cystic kidneys with or without TGF-α are similar in size and markedly enlarged compared their noncystic counterparts.
Table 1.
Clinical parameters of experimental groups
| Phenotype/genotype | No. | KW (g) | BW (g) | KW/BW (%) | BUN (mg/dL) |
|---|---|---|---|---|---|
| (A) Cystic/TGF-α present | 22 | 2.23 ± 0.64 | 8.92 ± 1.18 | 24.8 ± 5.1 | 93 ± 30 |
| bpk−/− TGF +/− | 18 | 2.22 ± 0.67 | 8.90 ± 1.26 | 24.8 ± 5.1 | 89 ± 19 |
| bpk−/− TGF +/+ | 4 | 2.26 ± 0.54 | 9.13 ± 0.87 | 24.8 ± 6.0 | 108 ± 52 |
| (B) Noncystic/TGF-α present | 48 | 0.14 ± 0.02 | 8.84 ± 0.88 | 1.60 ± 0.13 | 22 ± 4 |
| bpk+/− TGF +/− | 25 | 0.14 ± 0.02 | 8.71 ± 0.74 | 1.57 ± 0.13 | 23 ± 5 |
| bpk+/− TGF +/+ | 11 | 0.15 ± 0.02 | 9.29 ± 1.00 | 1.62 ± 0.14 | 22 ± 4 |
| bpk+/+ TGF +/− | 8 | 0.14 ± 0.02 | 8.74 ± 1.08 | 1.61 ± 0.11 | 20 ± 4 |
| bpk+/+ TGF +/+ | 4 | 0.14 ± 0.02 | 8.62 ± 0.86 | 1.64 ± 0.17 | 22 ± 6 |
| (C) Cystic/TGF-α null | 11 | 2.18 ± 0.51 | 8.5 ± 1.0 | 25.6 ± 5.2 | 106 ± 21 |
| bpk−/− TGF −/− | |||||
| (D) Noncystic/TGF-α null | 15 | 0.11 ± 0.02* | 6.87 ± 1.23** | 1.65 ± 0.10 | 26 ± 3 |
| bpk+/− TGF −/− | 11 | 0.11 ± 0.02* | 6.89 ± 1.29** | 1.66 ± 0.09 | 26 ± 3 |
| bpk+/+ TGF −/− | 4 | 0.11 ± 0.02* | 6.81 ± 1.24** | 1.62 ± 0.11 | 24 ± 3 |
p = 0.003, noncystic TGF absent vs present.
p = 0.001, noncystic TGF absent vs present.
Figure 2.
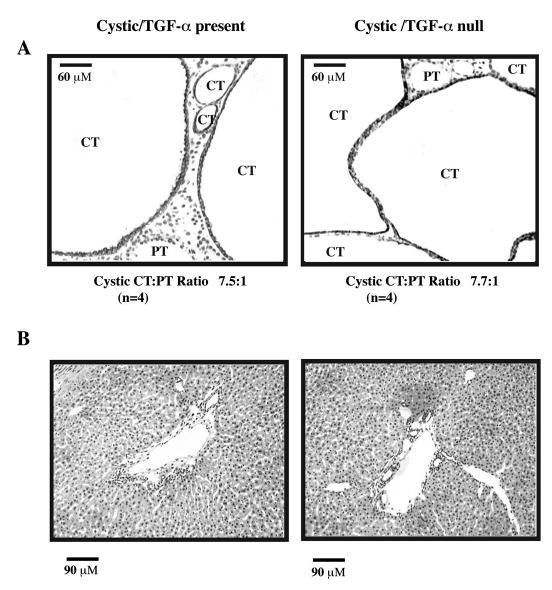
Histologic appearance of d-21 kidneys and livers from cystic bpk mice with TGF-α present or absent. (A) Serial kidney sections from cystic mice with TGF-α (Cystic/TGF-α present) or without TGF-α (Cystic/TGF-α null) were stained with biotinylated Dolichos biflorus agglutinin (DBA), a CT marker or biotinylated Lotus tetragonolobus (LTA), a PT marker and counter-stained with hematoxylin as described in the text. Representative kidney histologies are shown. CT cysts (DBA+)/LTA− are indicated by dark staining and predominate in both groups studied. A minority of cystic PT (DBA−/LTA+) is present. The cystic CT:PT ratio is indicated below each panel. Neither the overall severity of cystic CT disease nor the appearance or proportion of cystic PT appears to be modulated by the presence or absence of TGF-α. CT, collecting tubule cyst; PT, proximal tubule cyst. Original magnification 20×. (B) Serial liver sections from cystic mice with TGF-α (Cystic/TGF-α present) or without TGF-α (Cystic/TGF-α null) were stained with hematoxylin as described in the text. Representative liver histologies are shown and demonstrate that the severity of bile duct ectasia and proliferation was qualitatively similar in mice with versus without TGF-α. Original magnification 20×.
Absence of TGF-α does not modulate the abnormal expression of other EGF-related growth factors and their receptors in cystic kidney disease
As noted previously, abnormal TGF-α and EGFR expression have been reported in both ARPKD and ADPKD. EGF expression abnormalities in PKD have also been described (16–20). In addition, we recently demonstrated abnormal AR and HB-EGF kidney expression in bpk mice (15). To determine whether the absence of TGF-α resulted in changes in the expression levels of other EGF-related growth factors or their receptors, Western analysis for AR, EGF, HB-EGF, EGFR, and ErbB4 was performed on whole kidney lysates obtained from cystic and noncystic mice with and without TGF-α. As shown in Figure 3, cystic mice (C) with TGF-α (TGF-α present) or without (TGF-α null) consistently showed similar abnormalities in the expression of all three growth factors as well as EGFR and ErbB4 when compared with their noncystic (N) counterparts. The abnormal expression of AR, HB-EGF, and EGFR is consistent with published data in ARPKD models (15,21). The EGF immunoreactive bands at approximately 18 and 28 kD differ in size from the more commonly reported protein species of 6, 47, and 170 kD (22,23); however, multiple different EGF species have been reported in PKD (16,17). Furthermore, the 18 and 28 kD species are overexpressed to an equal extent in cystic mice with versus without TGF-α and may represent an alternatively processed form of the protein. The ErbB4 abnormalities are consistent with our previous observations for ErbB4 (manuscript in preparation). Prominent expression of the full-length (170 kD) receptor, as well as the constitutively cleaved (80 kD) protein species is present in cystic mice with and without TGF-α and only minimally visible in their noncystic counterparts.
Figure 3.
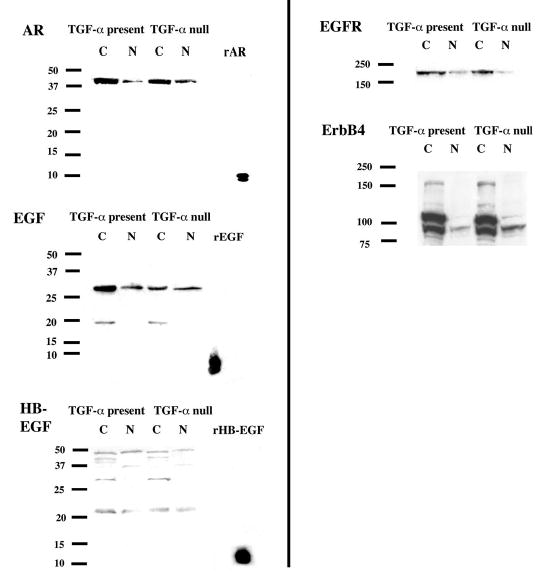
AR, EGF, HB-EGF, EGFR, and ErbB4 expression in cystic bpk mice and noncystic littermates, TGF-α present or absent. Whole kidney lysates from d-21 cystic (C) and noncystic (N) mice with TGF-α (TGF-α present) or without (TGF-α null) were prepared as described in the text. Western analysis for AR, EGF, HB-EGF, EGFR, and ErbB4 protein expression was performed, also as described. AR and HB-EGF whole kidney lysates were subjected to heparin column purification before loading. Recombinant purified growth factor was loaded as a positive control for EGF-related growth factor immunoblots. Expression of AR, EGF, HB-EGF, EGFR, and ErbB4 is increased to similar degrees in cystic mice with or without TGF-α when compared with their noncystic counterparts.
Rapid genotyping of bpk litters
The bpk mutation has been previously identified in exon 22 of Bicc1 (10), however, no published protocol exists for the rapid identification of this mutation from mouse genomic DNA. Using publicly available data from the Ensembl Mouse Genome Server (http://www.ensembl.org/Mus_musculus/) and from Genbank, genomic DNA sequences from the bpk locus and the Bicc1 cDNA sequence (accession number NM_031397) were obtained and PCR was performed on DNA samples from known wild-type and homozygous mutant animals using primers flanking exon 22. Forward and reverse sequencing of the PCR products confirmed a CG insertion in bpk mutants, resulting in a frameshift mutation and loss of a stop codon in exon 22 as well as creation of a unique BstU1 restriction site (Fig. 4A). Primers were developed to flank the insertion/restriction site and genomic DNA samples from known wild-type, heterozygous, and homozygous mutant mice (n = 3 for each group) were amplified and cut with BstU1. As shown in Figure 4B, PCR amplification of the wild-type allele yields a 213 bp product, which is uncut by BstU1 digestion; whereas amplification of the mutant allele yields a 215 bp product, which is cut by BstU1 into 124 bp and 91 bp fragments.
Figure 4.
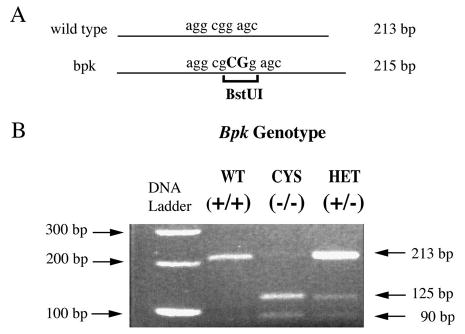
Rapid genotype analysis of bpk. (A) Schematic representation of Bicc1 exon 22 showing the location of the bpk mutation (CG insertion) and creation of a unique BstU1 restriction site. (B) PCR amplification with primers flanking the mutation site, followed by BstU1 restriction digest of PCR products clearly distinguishes between wild-type, homozygous, and heterozygous DNA samples. The wild-type allele yields a 213 bp product, which is not cut by BstU1. The bpk mutant allele yields a 215 bp product, which is cut into 125 bp and 90 bp fragments. WT, wild type; CYS, cystic; HET, heterozygote.
DISCUSSION
To definitively test the hypothesis that TGF-α was an absolute requirement for renal cystogenesis and disease progression, we examined the impact of the loss of TGF-α expression on the development and severity of cystic kidney disease as well as biliary disease in the bpk model. Somewhat unexpectedly, the histologic and biochemical data of this study demonstrate that TGF-α is not required for renal cystogenesis in murine ARPKD and that the absence of TGF-α does not affect the severity of cystic kidney disease or biliary disease. One possible explanation for the lack of an effect on renal cyst formation in this model is that other EGF-related growth factors were up-regulated in response to the “loss” of TGF-α. We assessed kidney expression of EGF, AR, and HB-EGF as well as their receptors, EGFR and ErbB4, and did not find any significant differences in the expression of these growth factors and receptors in cystic mice with TGF-α versus those without. However, it should be noted that all of the growth factors and receptors studied were consistently abnormally expressed in cystic kidneys compared with noncystic kidneys, regardless of the presence or absence of TGF-α. This finding suggests that abnormal expression of one or more EGF-related growth factors is sufficient to induce cystogenesis and disease progression even in the absence of TGF-α.
These data further highlight the potential redundancy of the EGF-related growth factors/receptors in this disease. As noted previously, mice that overexpress TGF-α develop renal cysts (5). Our data suggest that up-regulation of one or more of the other EGF-related growth factors could have a similar effect. Similar findings were reported in studies examining the impact of single or combinatorial loss of AR, EGF, and TGF-α on mammary gland development (24). Despite the lack of all three growth factors studied, triple null mice were healthy and fertile. Furthermore, those lacking all three growth factors did not show significant changes in cellular proliferation within terminal buds. To our knowledge, transgenic mice that over-express AR and HB-EGF in the kidney have not been reported, however, transgenic mice that overexpress AR or HB-EGF in the pancreas demonstrate abnormal proliferation of ductal epithelium (25,26). Similarly, mice that overexpress EGFR in the mammary glands develop mammary epithelial hyperplasia (27). In contrast, mice that widely overexpress EGF in multiple organs did not show any abnormalities (28), suggesting that not all of the EGF-related growth factors are able to initiate renal cystogenesis.
Modulation of disease by loss of TGF-α has been examined in several studies involving other organ systems. In a study of bleomycin-induced lung injury and fibrosis, TGF-α null mice showed a significant reduction in pulmonary fibrosis compared with mice with intact TGF-α (29). In that study, increased expression of HB-EGF was noted in both wild-type and TGF-α null mice treated with bleomycin compared with untreated controls but the protective effect of TGF-α loss was still seen. In a study of gastric ulceration in normal and TGF-α null mice, those without TGF-α failed to show the expected temporal increase trefoil peptide expression, suggesting that the latter stages of injury repair in this model had a TGF-α-dependent component (30). In contrast, in a study of peripheral nerve injury, TGF-α null mice had the same degree of response as those with intact TGF-α (31). These studies highlight the complexity of this growth factor/receptor axis and suggest that individual EGF-related growth factors may have specific and unique functions in certain tissues and cells, while showing redundancy of function in other organs.
In addition to causing renal cysts, TGF-α overexpression has also been implicated in worsening progression of disease in the pcy mouse, a purported model of human ADPKD (6). The fact that our data did not show any change in cystic phenotype would seem to be at odds with this previous report. However, it should be noted that although TGF-α overexpression accelerated disease progression in that model, neither the timing of initial cyst development nor the final degree of kidney enlargement were impacted by the presence of the TGF-α transgene (6). Of note, recent genetic studies have shown that the pcy mouse is actually a model of juvenile nephronophthisis (32), a cystic kidney disease of childhood that is genetically and phenotypically distinct from ARPKD. Furthermore, the fact that the overexpression of TGF-α accelerated disease in that model, does not mean that its absence would retard disease, given the redundancy of this growth factor system as discussed above. The timing of overexpression during the course of development may also impact the effect of growth factor on disease progression. In the bpk mouse, administration of exogenous EGF before 9 d of age improved renal function but not cystic kidney appearance, whereas, when treatment was continued beyond 9 d, mortality actually increased in treated versus untreated cystic mice (33).
The fact that TGF-α is not an absolute requirement for renal cystogenesis and that multiple EGF-related growth factors are abnormally expressed in murine ARPKD raises the question of how the previously studied TACE inhibitor, WTACE2, exerted its therapeutic effects in this model (2). As mentioned above, TACE is a metalloproteinase that mediates ectodomain shedding of TGF-α (34). More recent data have shown that other members of the EGF-related growth factor “family” also undergo metalloproteinase-mediated proteolytic cleavage to release the mature “secreted” growth factor [reviewed in (35) and (36)]. One possible reason for the observed therapeutic efficacy of WTACE2 was its actions on the shedding of other growth factors. Published data demonstrate that AR is a target of TACE (37). More recent studies demonstrate that HB-EGF and ErbB4 (which is unique among the ErbB receptors in that it undergoes proteolytic cleavage) are also targets of TACE (38,39).
Although WTACE2 is a potent inhibitor of TACE, it is not completely specific for TACE. Other metalloproteinases that are targets of the WTACE2 include MMP-3 and MMP-9 (2), both of which are overexpressed in PKD (40). Thus, the therapeutic effects of WTACE2 could have been due, in part, to inhibition of several other metalloproteinases in addition to TACE.
The bpk mouse is a well-characterized model of murine ARPKD and has consistent phenotype. These investigations highlight its usefulness in studying the pathogenesis of ARPKD. The PCR/restriction digest protocol developed in this study allows for rapid genotyping of whole animals, tissues, and cells. This will facilitate breeding experiments, future studies of therapeutic interventions, and studies involving cell lines derived from this model (41).
Acknowledgments
The authors thank Dr. Ellis Avner and William E. Sweeney, Jr., for their thoughtful comments and suggestions.
Footnotes
This study was presented at the 2003 meeting of the American Society of Nephrology (San Diego, CA) and published in abstract form.
Supported by National Institutes of Health grants K08DK-59488 (K.M.D.) and P50DK-57306 (Rainbow Center for Childhood PKD, Ellis D. Avner, PI).
References
- 1.Dell KM, Avner ED. Polycystic kidney disease. In: Avner ED, Harmon WE, Niaudet P, editors. Pediatric Nephrology. Lippincott, Williams & Wilkins; Philadelphia: 2003. pp. 675–699. [Google Scholar]
- 2.Dell KM, Nemo R, Sweeney WE, Jr, Levin JI, Frost P, Avner ED. A novel inhibitor of tumor necrosis factor-alpha converting enzyme ameliorates polycystic kidney disease. Kidney Int. 2001;60:1240–1248. doi: 10.1046/j.1523-1755.2001.00963.x. [DOI] [PubMed] [Google Scholar]
- 3.Lee DC, Chan KW, Chan SY. Expression of transforming growth factor alpha and epidermal growth factor receptor in adult polycystic kidney disease. J Urol. 1998;159:291–296. doi: 10.1016/s0022-5347(01)64084-9. [DOI] [PubMed] [Google Scholar]
- 4.Klingel R, Dippold W, Storkel S, Meyer zum Buschenfelde KH, Kohler H. Expression of differentiation antigens and growth-related genes in normal kidney, autosomal dominant polycystic kidney disease, and renal cell carcinoma. Am J Kidney Dis. 1992;19:22–30. doi: 10.1016/s0272-6386(12)70198-1. [DOI] [PubMed] [Google Scholar]
- 5.Lowden DA, Lindemann GW, Merlino G, Barash BD, Calvet JP, Gattone VH., II Renal cysts in transgenic mice expressing transforming growth factor-alpha. J Lab Clin Med. 1994;124:386–394. [PubMed] [Google Scholar]
- 6.Gattone VH, II, Kuenstler KA, Lindemann GW, Lu X, Cowley BD, Jr, Rankin CA, Calvet JP. Renal expression of a transforming growth factor-alpha transgene accelerates the progression of inherited, slowly progressive polycystic kidney disease in the mouse. J Lab Clin Med. 1996;127:214–222. doi: 10.1016/s0022-2143(96)90081-5. [DOI] [PubMed] [Google Scholar]
- 7.Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57:33–40. doi: 10.1046/j.1523-1755.2000.00829.x. [DOI] [PubMed] [Google Scholar]
- 8.Torres VE, Sweeney WE, Jr, Wang X, Qian Q, Harris PC, Frost P, Avner ED. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int. 2003;64:1573–1579. doi: 10.1046/j.1523-1755.2003.00256.x. [DOI] [PubMed] [Google Scholar]
- 9.Sweeney WE, Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int. 2003;64:1310–1319. doi: 10.1046/j.1523-1755.2003.00232.x. [DOI] [PubMed] [Google Scholar]
- 10.Cogswell C, Price SJ, Hou X, Guay-Woodford LM, Flaherty L, Bryda EC. Positional cloning of jcpk/bpk locus of the mouse. Mamm Genome. 2003;14:242–249. doi: 10.1007/s00335-002-2241-0. [DOI] [PubMed] [Google Scholar]
- 11.Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell. 1993;73:249–261. doi: 10.1016/0092-8674(93)90227-h. [DOI] [PubMed] [Google Scholar]
- 12.Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF-α deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–278. doi: 10.1016/0092-8674(93)90228-i. [DOI] [PubMed] [Google Scholar]
- 13.Nauta J, Ozawa Y, Sweeney WE, Jr, Rutledge JC, Avner ED. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 1993;7:163–172. doi: 10.1007/BF00864387. [DOI] [PubMed] [Google Scholar]
- 14.Nakanishi K, Sweeney WE, Jr, Zerres K, Guay-Woodford LM, Avner ED. Proximal tubular cysts in fetal human autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2000;11:760–763. doi: 10.1681/ASN.V114760. [DOI] [PubMed] [Google Scholar]
- 15.MacRae Dell K, Nemo R, Sweeney WE, Jr, Avner ED. EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int. 2004;65:2018–2029. doi: 10.1111/j.1523-1755.2004.00623.x. [DOI] [PubMed] [Google Scholar]
- 16.Lakshmanan J, Eysselein V. Hereditary error in epidermal growth factor prohormone metabolism in a rat model of autosomal dominant polycystic kidney disease. Biochem Biophys Res Commun. 1993;197:1083–1093. doi: 10.1006/bbrc.1993.2589. [DOI] [PubMed] [Google Scholar]
- 17.Lakshmanan J, Fisher DA. An inborn error in epidermal growth factor prohormone metabolism in a mouse model of autosomal recessive polycystic kidney disease. Biochem Biophys Res Commun. 1993;196:892–901. doi: 10.1006/bbrc.1993.2333. [DOI] [PubMed] [Google Scholar]
- 18.Moskowitz DW, Bonar SL, Liu W, Sirgi CF, Marcus MD, Clayman RV. Epidermal growth factor precursor is present in a variety of human renal cyst fluids. J Urol. 1995;153:578–583. doi: 10.1097/00005392-199503000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Munemura C, Uemasu J, Kawasaki H. Epidermal growth factor and endothelin in cyst fluid from autosomal dominant polycystic kidney disease cases: possible evidence of heterogeneity in cystogenesis. Am J Kidney Dis. 1994;24:561–568. doi: 10.1016/s0272-6386(12)80212-5. [DOI] [PubMed] [Google Scholar]
- 20.Gattone VH, Jr, Andrews GK, Niu FW, Chadwick LJ, Klein RM, Calvet JP. Defective epidermal growth factor gene expression in mice with polycystic kidney disease. Dev Biol. 1990;138:225–230. doi: 10.1016/0012-1606(90)90192-l. [DOI] [PubMed] [Google Scholar]
- 21.Orellana SA, Sweeney WE, Neff CD, Avner ED. Epidermal growth factor receptor expression is abnormal in murine polycystic kidney. Kidney Int. 1995;47:490–499. doi: 10.1038/ki.1995.62. [DOI] [PubMed] [Google Scholar]
- 22.Sheflin LG, Brooks EM, Keegan BP, Spaulding SW. The cytosolic 47-kilodalton protein that binds to the 3′ untranslated region of epidermal growth factor transcripts responds to orchiectomy in a tissue-specific fashion. Endocrinology. 1996;137:5616–5623. doi: 10.1210/endo.137.12.8940392. [DOI] [PubMed] [Google Scholar]
- 23.Mroczkowski B, Reich M, Chen K, Bell GI, Cohen S. Recombinant human epidermal growth factor precursor is a glycosylated membrane protein with biological activity. Mol Cell Biol. 1989;9:2771–2778. doi: 10.1128/mcb.9.7.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luetteke NC, Qui TH, Fenton SE, Troyer KL, Riedel RF, Chang A, Lee DC. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development. 1999;126:2739–2750. doi: 10.1242/dev.126.12.2739. [DOI] [PubMed] [Google Scholar]
- 25.Means AL, Ray KC, Singh AB, Washington MK, Whitehead RH, Harris RC, Jr, Wright CV, Coffey RJ, Jr, Leach SD. Overexpression of heparin-binding EGF-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology. 2003;124:1020–1036. doi: 10.1053/gast.2003.50150. [DOI] [PubMed] [Google Scholar]
- 26.Wagner M, Weber CK, Bressau F, Greten FR, Stagge V, Ebert M, Leach SD, Adler G, Schmid RM. Transgenic overexpression of amphiregulin induces a mitogenic response selectively in pancreatic duct cells. Gastroenterology. 2002;122:1898–1912. doi: 10.1053/gast.2002.33594. [DOI] [PubMed] [Google Scholar]
- 27.Brandt R, Eisenbrandt R, Leenders F, Zschiesche W, Binas B, Juergensen C, Theuring F. Mammary gland specific hEGF receptor transgene expression induces neoplasia and inhibits differentiation. Oncogene. 2000;19:2129–2137. doi: 10.1038/sj.onc.1203520. [DOI] [PubMed] [Google Scholar]
- 28.Wong RW, Kwan RW, Mak PH, Mak KK, Sham MH, Chan SY. Overexpression of epidermal growth factor induced hypospermatogenesis in transgenic mice. J Biol Chem. 2000;275:18297–18301. doi: 10.1074/jbc.M001965200. [DOI] [PubMed] [Google Scholar]
- 29.Madtes DK, Elston AL, Hackman RC, Dunn AR, Clark JG. Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am J Respir Cell Mol Biol. 1999;20:924–934. doi: 10.1165/ajrcmb.20.5.3526. [DOI] [PubMed] [Google Scholar]
- 30.Cook GA, Yeomans ND, Giraud AS. Temporal expression of trefoil peptides in the TGF-alpha knockout mouse after gastric ulceration. Am J Physiol. 1997;272:G1540–G1549. doi: 10.1152/ajpgi.1997.272.6.G1540. [DOI] [PubMed] [Google Scholar]
- 31.Xian CJ, Li L, Deng YS, Zhao SP, Zhou XF. Lack of effects of transforming growth factor-alpha gene knockout on peripheral nerve regeneration may result from compensatory mechanisms. Exp Neurol. 2001;172:182–188. doi: 10.1006/exnr.2001.7771. [DOI] [PubMed] [Google Scholar]
- 32.Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N, Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapato-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 33.Nakanishi K, Gattone VH, II, Sweeney WE, Avner ED. Renal dysfunction but not cystic change is ameliorated by neonatal epidermal growth factor in bpk mice. Pediatr Nephrol. 2001;16:45–50. doi: 10.1007/s004670000495. [DOI] [PubMed] [Google Scholar]
- 34.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 35.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 36.Derynck R. The physiology of transforming growth factor-α. Adv Cancer Res. 1992;58:27–52. doi: 10.1016/s0065-230x(08)60289-4. [DOI] [PubMed] [Google Scholar]
- 37.Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003;22:2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinkle CL, Sunnarborg SW, Loiselle D, Parker CE, Stevenson M, Russell WE, Lee DC. Selective roles for tumor necrosis factor alpha-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J Biol Chem. 2004;279:24179–24188. doi: 10.1074/jbc.M312141200. [DOI] [PubMed] [Google Scholar]
- 39.Rio C, Buxbaum JD, Peschon JJ, Corfas G. Tumor necrosis factor-alpha-converting enzyme is required for cleavage of erbB4/HER4. J Biol Chem. 2000;275:10379–10387. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- 40.Rankin CA, Suzuki K, Itoh Y, Ziemer DM, Grantham JJ, Calvet JP, Nagase H. Matrix metalloproteinases and TIMPS in cultured C57BL/6J-cpk kidney tubules. Kidney Int. 1996;50:835–844. doi: 10.1038/ki.1996.383. [DOI] [PubMed] [Google Scholar]
- 41.Sweeney WE, Jr, Kusner L, Carlin CR, Chang S, Futey L, Cotton CU, Dell KM, Avner ED. Phenotypic analysis of conditionally immortalized cells isolated from the BPK model of ARPKD. Am J Physiol Cell Physiol. 2001;281:C1695–C1705. doi: 10.1152/ajpcell.2001.281.5.C1695. [DOI] [PubMed] [Google Scholar]