

Abstract
GSH is rapidly oxidized by HOCl (hypochlorous acid), which is produced physiologically by the neutrophil enzyme myeloperoxidase. It is converted into, mainly, oxidized glutathione. Glutathione sulfonamide is an additional product that is proposed to be covalently bonded between the cysteinyl thiol and amino group of the γ-glutamyl residue of GSH. We have developed a sensitive liquid chromatography–tandem MS assay for the detection and quantification of glutathione sulfonamide as well as GSH and GSSG. The assay was used to determine whether glutathione sulfonamide is a major product of the reaction between GSH and HOCl, and whether it is formed by other two-electron oxidants. At sub-stoichiometric ratios of HOCl to GSH, glutathione sulfonamide accounted for up to 32% of the GSH that was oxidized. It was also formed when HOCl was generated by myeloperoxidase and its yield increased with the flux of oxidant. Of the other oxidants tested, only hypobromous acid and peroxynitrite produced substantial amounts of glutathione sulfonamide, but much less than with HOCl. Chloramines were able to generate detectable levels only when at a stoichiometric excess over GSH. We conclude that glutathione sulfonamide is sufficiently selective for HOCl to be useful as a biomarker for myeloperoxidase activity in biological systems. We have also identified a novel oxidation product of GSH with a molecular weight two mass units less than GSH, which we have consequently named dehydroglutathione. Dehydroglutathione represented a few percent of the total products and was formed with all of the oxidants except H2O2.
Keywords: biomarker, dehydroglutathione, glutathione, glutathione sulfonamide, myeloperoxidase, hypochlorous acid, peroxynitrite
Abbreviations: DTNB, 5,5-dithiobis-2-nitrobenzoic acid; GSA, glutathione sulfonamide; HOBr, hypobromous acid; HOCl, hypochlorous acid; HOI, hypoiodous acid; HOSCN, hypothiocyanous acid; LC–ESI–MS, liquid chromatography–electrospray ionization–MS; LC–MS/MS, liquid chromatography-tandem MS; MPO, myeloperoxidase; NEM, N-ethylmaleimide, NH2Cl, ammonia monochloramine; TauNCl, taurine chloramine; TNB, 5-thio-2-nitrobenzoic acid
INTRODUCTION
Neutrophils are a major physiological source of reactive oxygen species in mammals. These cells generate substantial amounts of the strong oxidant HOCl (hypochlorous acid) via MPO (myeloperoxidase)-catalysed oxidation of chloride by hydrogen peroxide [1]. Other halides such as bromide, iodide and the pseudo-halide thiocyanate are also substrates for MPO, and are converted into HOBr (hypobromous acid), HOI (hypoiodous acid) and HOSCN (hypothiocyanous acid). At physiological concentrations of these halides, formation of HOCl and HOSCN is favoured [2,3]. These oxidants are implicated in injury in a wide range of inflammatory diseases. A key requirement for clinical investigation is a sensitive marker for detecting oxidant generation. 3-Chlorotyrosine is commonly used as a marker of HOCl generation and although specific, it is a minor product and forms indirectly [4]. It would be advantageous to have other specific biomarkers for HOCl.
HOCl reacts with several different functional groups of amino acids. It reacts most favourably with thiols [5–7]. The reaction with the cysteinyl thiol of GSH has a rate constant of greater than 107 M−1·s−1 [8]. GSH is present intracellularly at concentrations ranging from 0.5 to 10 mM and represents at least 90% of the total non-protein low molecular mass thiols [9]. The majority of the GSH is present in the reduced form, with less than 5% of its total as the disulfide, GSSG. Oxidation to GSSG is reversible due to enzymatic reduction. Thus with oxidants that generate GSSG, GSH is able to function as a recyclable antioxidant defence [9,10]. The effectiveness of GSH as an antioxidant defence against HOCl depends on the products formed and whether the parent thiol can be regenerated from these products. Winterbourn [5] noted that just over half of the GSH oxidized by HOCl could be regenerated, which suggested that irreversible products were also generated. GSA (glutathione sulfonamide) has since been identified as a product identified in the following reaction (reaction 1) [11]: Apart from the original identification of GSA, little literature exists on the molecule. It has been detected in neutrophils and endothelial cells treated directly with HOCl either by following the fate of 35S labelled GSH or by direct injection MS [12,13]. GSA has also been observed in PMA-stimulated neutrophils and has been shown to increase with the length of incubation [14].

GSA is a stable, irreversible oxidation product that to date has been observed only with HOCl. It has potential as a specific biomarker of HOCl. This possibility has not been explored because no sensitive method of detection and quantification has been available. GSA is polar and displays no chromophore, making it difficult to retain and detect by traditional reversed phase HPLC techniques. Furthermore, it is not detected by assays involving derivatization with monobromobimane [15] or dansyl chloride [16] because GSA has no free thiol or amino groups. There is a need for a quantitative assay to determine whether GSA is a major product of GSH oxidation and whether it is specific for HOCl. We have developed a sensitive LC–MS/MS (liquid chromatography-tandem MS) method for quantifying GSA, as well as GSH and other oxidation products. The method has been used to measure GSA production from HOCl and from other MPO-derived oxidants: HOBr, HOI, HOSCN and chloramines. We have compared these oxidants with H2O2 and peroxynitrite (ONOO−), other two-electron oxidants not produced by myeloperoxidase. In the current study we demonstrate that HOCl is the most efficient oxidant at generating GSA, and also describe for the first time the generation of dehydroglutathione by all of the MPO-derived oxidants.
EXPERIMENTAL
Reagents
GSH and Glu-Val-Phe were from Sigma. Labelled GSH [(glycine 1,2-13C2, 15N) GSH] was from Cambridge Isotope Laboratories. MPO was from Planta Natural Products. Water of 18-MΩ quality was prepared by a Milli-Q system (Millipore). Acetonitrile and methanol were from Malinckrodt. H2O2 and propan-2-ol were from BDH. The H2O2 concentration was determined using ϵ240=43.6 M−1cm−1 [17]. Sodium hypochlorite was from Sara Lee (Auckland, New Zealand) and its concentration was determined by measuring its absorbance maxima at 292 nm (pH 12, ϵ292=350 M−1cm−1) [18].
Preparation of oxidants
HOBr was freshly prepared by oxidation of bromide by HOCl. This was performed by mixing equal volumes of 20 mM HOCl in PBS (pH 7.4) with 22 mM sodium bromide. The HOBr concentration was determined by measuring its absorbance at 330 nm (pH 12, ϵ330=332 M−1 cm−1) [19].
HOI (hypoiodous acid) was freshly prepared by oxidation of iodide by HOCl. This was done by mixing equal volumes of 10 mM HOCl and 15 mM KI (both in water). The concentration was calculated by measuring the I3− absorbance at 353 nm (pH 1, ϵ353=2.64×104 M−1 cm−1) [20]. Dilutions in PBS were used for the reactions with GSH.
HOSCN was prepared using lactoperoxidase to catalyse the oxidation of thiocyanate by H2O2 [21]. Briefly, lactoperoxidase (5 μg/ml) and H2O2 (300 μM) were added to 1 mM potassium thiocyanate in PBS. After 10 min incubation at room temperature (23 °C), catalase (5 μg/ml) was added to quench residual H2O2. The concentration of the HOSCN stock was determined using TNB (5-thio-2-nitrobenzoic acid) as described previously [22]. The maximum HOSCN concentration that could be generated was 200–300 μM. This was due to inactivation of the enzyme at high H2O2 concentrations. This meant that experiments at HOSCN to GSH molar ratios of two and four were not performed.
To prepare both NH2Cl (ammonia monochloramine) and TauNCl (taurine chloramine), HOCl in PBS was added dropwise to solutions of ammonium chloride and taurine. Ammonium chloride and taurine were present at a 10-fold excess over HOCl to ensure only the monochloramine was formed. The chloramine solution was kept on ice and used within a few hours. The concentration was determined using the TNB assay [22].
Peroxynitrite was synthesized by reacting acidified hydrogen peroxide with a nitrite solution followed by rapid quenching with NaOH [23]. Its concentration was determined by measuring the absorbance at 302 nm (pH 12, ϵ302=1670 M−1cm−1) [24].
Oxidation of glutathione
Reactions of GSH (100 μM) with eight oxidants was carried out for 30 min at room temperature (23 °C) over a range of oxidant to GSH molar ratios. The reactions were all performed in PBS at pH 7.4. The actual oxidizing species at pH 7.4 is dictated by the pKa values of the various oxidants [pKa (HOSCN/OSCN−)=5.3, pKa (ONOOH/ONOO−)=6.8, pKa (HOCl/OCl−)=7.6, pKa (HOBr/OBr−)=8.7, pKa (HOI/OI−)=10.4] [25–27]. The reactions were initiated by addition of oxidant. Addition of a 10-fold molar excess of NEM (N-ethylmaleimide) after 30 min blocked the free thiol of any unreacted GSH and afforded a stable adduct. The sample was diluted 20-fold in PBS and an internal standard (0.5 μM) was added before LC–MS/MS analysis and quantification. Thiol loss was also quantified using DTNB (5,5-dithiobis-2-nitrobenzoic acid) to compare with and validate the LC–MS/MS method [28].
Preparation of GSA
GSA was synthesized by treating 100 mM GSH in PBS (pH 7.4) with a bolus of HOCl at an equimolar ratio. The sulfonamide was formed on mixing and was separated on a Phenomenex Jupiter semi-preparative column (250 mm×4.6 mm) using an isocratic solvent (50 mM formic acid) with UV detection at 222 nm. GSA gave a weak absorbance at this wavelength and eluted at 8 min. The collected fraction was freeze dried to give a fluffy white powder that was used to construct a standard curve based on mass. GSA solid dissolved readily in buffer and was stable over a period of several months at −20 °C. Standard curves for reagent GSH and GSSG were also based on mass.
LC–ESI–MS (liquid chromatography–electrospray ionization–MS)
The developed method was partially based on that described previously by Erve et al. [14]. LC–ESI–MS analyses were performed in the positive ion mode with a Thermo Finnigan LCQ Deca XP Plus ion trap mass spectrometer coupled to a Thermo Finnigan Surveyor HPLC system. A Thermo Hypercarb column (100 mm×2.1 mm) was used for chromatographic separation using 100% water (0.5% formic acid) as Solvent A and 50% acetonitrile and 50% propan-2-ol (0.1% formic acid) as Solvent B. A flow rate of 0.2 ml/min was used and 20 μl was injected onto the column. Glutathione and its oxidation products were eluted using a linear gradient of 0–20% solvent B over 20 min. At 20 min the column was flushed with 100% solvent B for 5 min before reequilibration at initial conditions. The electrospray needle was held at 4500 V. Nitrogen, the sheath gas, was set at 45 units. The collision gas was helium. The temperature of the heated capillary was 275 °C. Selective reaction monitoring of GSA (338→263), GSSG (613→484), glutathione–NEM conjugate (433→304) and Glu-Val-Phe (394→229) was performed with the collision energies optimized for each compound. The run was divided into four scan events with each scan event dedicated to a single species. Peak areas were normalized by dividing the area of the compound of interest by the area of the internal standard. To observe other products of the GSH oxidation reactions, undiluted reaction mixture was injected into the LC–MS instrument and monitored in the Full Scan mode. The LC conditions remained the same as for the quantification of GSA, GSSG and GSH. Spectra were monitored between m/z of 250–650.
RESULTS
Development of a LC–MS/MS method for monitoring GSH, GSSG and GSA
An LC method was developed for the separation of the NEM-adduct of GSH, GSSG and GSA. For this a Thermo Hypercarb column was used to retain the polar analytes, with detection by positive electrospray ionization. Optimized conditions gave baseline separation of all species with an elution order of GSA, GSSG and GSH. The tripeptide Glu-Val-Phe was added to the mixture as an internal standard and eluted after GSH (Figure 1). Derivatization of GSH with NEM stabilized the thiol against oxidation and improved separation and sensitivity. Each of the four species was fragmented by collision-induced dissociation and the most intense fragment ion was used for quantification (Figure 2). GSA gave an m/z 338 parent ion that was consistent with what had been observed previously [11]. The protonated parent ion (m/z 338) was fragmented and the daughter ion (m/z 263) monitored. The observed neutral loss of 75 Da corresponds to the loss of the glycinyl tail from GSA (see reaction 1). Supporting evidence for this fragmentation pattern comes from GSA generated from GSH containing isotopically labelled glycine (13C2, 15N). Upon fragmentation an identical daughter ion was observed from both the labelled and unlabelled GSA. This indicated that the labelled glycine residue had come off. Both the protonated GSSG (m/z 613) and GSH–NEM conjugate (m/z 433) gave fragment ions (m/z 484 and 304 respectively) that represented a neutral loss of 129 Da. This neutral loss is a well-known fragment of GSH and its conjugates, corresponding to the loss of pyroglutamate from the γ-glutamyl residue [29,30]. Calibration curves were linear (r2>0.99 in each case) over a concentration range of at least 0–240 pmol for GSH, 0–40 pmol for GSSG, and 0–40 pmol for GSA. Limits of detection were 50 fmol, 100 fmol and 100 fmol for GSH, GSSG and GSA respectively based on a signal-to-noise ratio of ten.
Figure 1. Extracted chromatograms of GSH and oxidized species from the optimized LC–MS/MS method.

The species monitored were GSA, GSSG, GSH–NEM and Glu-Val-Phe (internal standard).
Figure 2. Structures of glutathione species and internal standard monitored by LC–MS/MS.

Arrows associated with structures designate bond cleavage sites when undergoing fragmentation by collision-induced dissociation and portion of the molecule monitored during quantification (the ionizing proton is not shown). The masses associated with each species represent the [M+H]+ ion and the protonated mass of the fragment ion monitored.
Quantification of GSA formation from GSH and HOCl
The LC–MS/MS method was used to quantify GSA and GSSG formation when GSH was treated with HOCl. The extent of GSH loss, determined at 30 min when the reaction was complete, was the same when measured by either LC–MS/MS or the DTNB assay (results not shown). At an equimolar ratio of GSH to HOCl, 53% of the GSH was lost. GSA was produced in a similar yield to GSSG and accounted for 32% of the reacted GSH (Figure 3a). At lower HOCl to GSH molar ratios, relatively more GSSG and less GSA were formed. At higher ratios, GSSG formation declined and the amount of GSA formed progressively increased. At a HOCl to GSH molar ratio of four, where all of the GSH was consumed, 62% of the total GSH was converted into GSA.
Figure 3. Product profiles of the reaction between GSH and various oxidants.

GSH (●), GSSG (□) and GSA (▲). Glutathione (100 μM) was reacted with increasing oxidant to GSH molar ratios in PBS (pH 7.4). Reactions were left for 30 min at room temperature (23 °C) before adding an excess of NEM. Analysis of the glutathione species was by LC–MS/MS. GSSG data points have been calculated as GSH equivalents. Results are expressed as the mean±S.D. for a minimum of three experiments.
HOCl generated using the MPO–H2O2–Cl− system oxidized GSH to give GSA (Figure 4). As the myeloperoxidase concentration increased, the amount of GSA observed also increased. The observed increase in GSA with increasing MPO was independent of the amount of oxidant produced, as the amount of GSH that was oxidized remained the same (65.6±4.8%).
Figure 4. GSA formed by the MPO–H2O2–Cl− system.

Glutathione (100 μM) was combined with H2O2 (50 μM), Cl− (140 mM) and various amounts of MPO (0–250 nM) in a final volume of 1 ml in PBS (pH 7.0). Reactions were left for 60 min (when all the H2O2 had been consumed) at room temperature (23 °C) before adding excess NEM. Results are expressed as the mean and range for two experiments.
Reaction of GSH with other MPO-derived oxidants
The reaction products of GSH with other hypohalous acids, NH2Cl and TauNCl were also examined. NH2Cl and TauNCl are mild chlorinated oxidants that are produced from the reaction of HOCl with ammonia and taurine respectively. Reactions were performed at pH 7.4 and assayed after 30 min when all the oxidant had been consumed. This was confirmed by observing the same GSH loss and oxidation product formation at a later time point.
HOBr caused greater losses of GSH than with comparable HOCl to GSH molar ratios (Figure 3b). GSA was produced in the reaction. The amount increased with increasing HOBr to GSH molar ratios but was much less than with HOCl. At an equimolar ratio, GSA accounted for 8% of the GSH oxidized. At an oxidant to GSH molar ratio of two, where all of the GSH was consumed, 25% of GSH was converted into GSA. With the milder halogenated oxidants NH2Cl (Figure 3c) and TauNCl (Figure 3d), measurable amounts of GSA were observed, but only when the oxidants were at a stoichiometric excess over GSH. At an oxidant to GSH molar ratio of four, GSA accounted for <2% of the GSH lost with both of these oxidants. Neither HOI (Figure 3e) nor HOSCN (Figure 3f) produced detectable GSA at any of the oxidant to GSH molar ratios tested.
Oxidation of GSH by peroxynitrite and H2O2
The specificity of GSA as a product from the reaction between GSH and two-electron oxidants was further tested using peroxynitrite and H2O2. All of the GSH was consumed at a peroxynitrite to GSH molar ratio of two (Figure 3g). GSA was detected at all oxidant concentrations tested and accounted for a maximum of 5% of the original GSH at a peroxynitrite to GSH molar ratio of four. Peroxynitrite reacts with GSH directly, but in the presence of CO2 it reacts rapidly to form carbonate and nitrogen dioxide radicals that account for much of the GSH loss [31]. When the reaction with peroxynitrite was run in the presence of bicarbonate, substantially less GSH was lost and there was half as much GSA formed. The decrease in GSH oxidation is consistent with the work of Bonini and Augusto [32]. The lower yield of GSA implies that it was formed directly from peroxynitrite rather than by radical-mediated oxidation. H2O2 reacts slowly with GSH and was not all consumed during the 30 min reaction [33]. Therefore, the stoichiometry could not be related to the amount added, as with the other oxidants. With H2O2, GSSG accounted for all of the GSH lost. No GSA was detected even when a 10-fold excess of oxidant was used (Figure 3h).
Qualitative analysis of the reaction mixtures by full-scan LC–MS
Apart from with H2O2, the amounts of GSSG and GSA formed from the oxidation of GSH did not always account for all of the GSH lost (Table 1). Therefore, we scanned the full mass range to obtain a more complete representation of the total product profiles. Reaction mixtures chosen for analysis represented the oxidant to GSH molar ratio where approximately half of the GSH was consumed.
Table 1. Percentage of GSH loss accounted for as GSSG or GSA.
Products monitored at the oxidant to GSH mole ratio where approximately half of the GSH was consumed. The molar ratios were 1 (HOCl), 0.5 (HOBr), 0.25 (NH2Cl), 0.5 (TauNCl), 0.25 (HOI), 0.25 (HOSCN), 0.5 (ONOO−) and 4 (H2O2). nd indicates that no GSA was detectable at the specified molar ratio.
| Oxidant | GSSG | GSA | Total |
|---|---|---|---|
| HOCl | 40 | 32 | 72 |
| HOBr | 44 | 4 | 48 |
| NH2Cl | 80 | nd | 80 |
| TauNCl | 66 | nd | 66 |
| HOI | 97 | nd | 97 |
| HOSCN | 98 | nd | 98 |
| ONOO− | 73 | 2 | 75 |
| H2O2 | 100 | nd | 100 |
GSH alone gave a single peak eluting at 11.1 min with an m/z 433 ion, that represented the NEM adduct. For GSH treated with HOCl, the spectra of six different species could be extracted from the total ion chromatogram (Figure 5). The earliest eluting peak at 7.2 min (a) gave an ion with m/z 340, which corresponds to glutathione sulfinic acid (GSO2H). At HOCl to GSH molar ratios greater than one, glutathione sulfonic acid (GSO3H, m/z 356) was also observed co-eluting with this peak. The peak eluting at 8.0 min (b) gave an m/z 306 ion. This ion represents a singly protonated species two mass units less than that of protonated GSH. As this must represent the loss of two protons, we have named it dehydroglutathione. GSA eluted at 8.9 min with an m/z 338 ion (c). GSSG eluted at 9.9 min (d) and was identified by the presence of both singly (m/z 613) and doubly (m/z 307) charged ions in the extracted spectra. Reduced GSH eluted at 11.1 min (e). A broad peak eluted between 12.5 and 13.0 min (f) and gave an m/z 568 ion. This mass is consistent with an aldehyde that would be formed by the decarboxylation and deamination of one of the γ-glutamyl groups of GSSG. A possible route to this ion would be via breakdown of GSSG monochloramine, which could be formed by the reaction of GSSG with HOCl. No chloramine peak was evident from the total ion chromatogram, and to see the aldehyde, the chloramine would have to break down either in solution or in the mass spectrometer. To test this, GSSG was reacted with HOCl and the oxidation products were monitored using the LC–MS method. A broad peak with m/z 568 was observed, indicating it had formed from the oxidation of GSSG by HOCl.
Figure 5. Total ion chromatogram and extracted spectra of the products formed from the reaction between GSH and HOCl.

An equimolar reaction mixture of GSH and HOCl (both 100 μM) was treated with NEM after 30 min and monitored by positive electrospray ionization full scan LC–MS. Masses were monitored between m/z 250 and 650. All spectra show the [M+H]+ ion, with GSSG also displaying the [M+2H]2+ ion. The extracted spectra represent (a) glutathione sulfinic acid, (b) dehydroglutathione, (c) glutathione sulfonamide, (d) GSSG, (e) GSH–NEM and (f) putative aldehydic breakdown product of GSSG–chloramine.
The total ion chromatogram generated with HOBr had an identical product profile to that with HOCl but differed in absolute amounts (Figure 6). There was a significantly larger signal for glutathione sulfinic acid (a), a similar signal for dehydroglutathione (b) and a smaller signal for GSA (c). The broad m/z 568 peak between 12 and 13 min (f) remained and could have arisen from the breakdown of the GSSG bromamine, which would also give an aldehyde of the same mass. This mass was also observed, albeit at a lower level, with the two chloramines tested. With these two milder oxidants, dehydroglutathione (b) was a significant oxidation product, in contrast to glutathione sulfinic acid which was a detectable but minor product. An additional product with m/z 629 was observed in the total ion chromatograms generated with the chloramines and HOI. This mass eluted just before GSSG (g) and represents GSSG with the addition of a single oxygen. For HOSCN, the only oxidation product observed was GSSG. Dehydroglutathione, a previously unrecognized GSH oxidation product, was formed with all of the MPO-derived oxidants and peroxynitrite (Figure 7). It was also observed when using the MPO–H2O2–Cl− system as in Figure 4. The amount of dehydroglutathione formed with an equivalent amount of HOCl added as a bolus was approximately half that observed in the MPO system. There was only a trace of dehydroglutathione with HOSCN and none was detected with H2O2. The amount formed increased as the oxidant to GSH molar ratio increased. Most oxidants had a threshold, where additional oxidant resulted in loss of the dehydroglutathione signal. This suggests that dehydroglutathione is an intermediate species capable of being further oxidized. In contrast to this, the amount of dehydroglutathione formed with HOI kept increasing as the amount of oxidant increased. HOI was capable of forming this species but was unable to oxidize it further. At a HOI to GSH molar ratio of 0.5, where approximately half of the GSH had been consumed, GSSG was the major oxidation product, accounting for 97% of the GSH lost (Table 1). The majority of the remaining 3% was dehydroglutathione. This indicates that although a strong signal was observed, dehydroglutathione is not a major oxidation product of GSH, even under the most favourable circumstances. Fragmentation of dehydroglutathione (m/z 306) by collision-induced dissociation gave a daughter ion (m/z 177) representing a neutral loss of 129 Da. This is the same loss as was observed with the GSH–NEM conjugate.
Figure 6. Total ion chromatograms from the reaction of GSH with oxidants.

LC–MS chromatograms represent the oxidant to GSH molar ratio where approximately half of the GSH was consumed and are normalized to GSSG. Masses were monitored between m/z 250 and 650. Species observed were (a) glutathione sulfinic acid, (b) dehydroglutathione, (c) GSA, (d) GSSG, (e) GSH-NEM, (f) putative aldehydic breakdown product of GSSG–chloramine and (g) GSSG with the addition of one oxygen.
Figure 7. Formation of dehydroglutathione from GSH and MPO-derived oxidants.
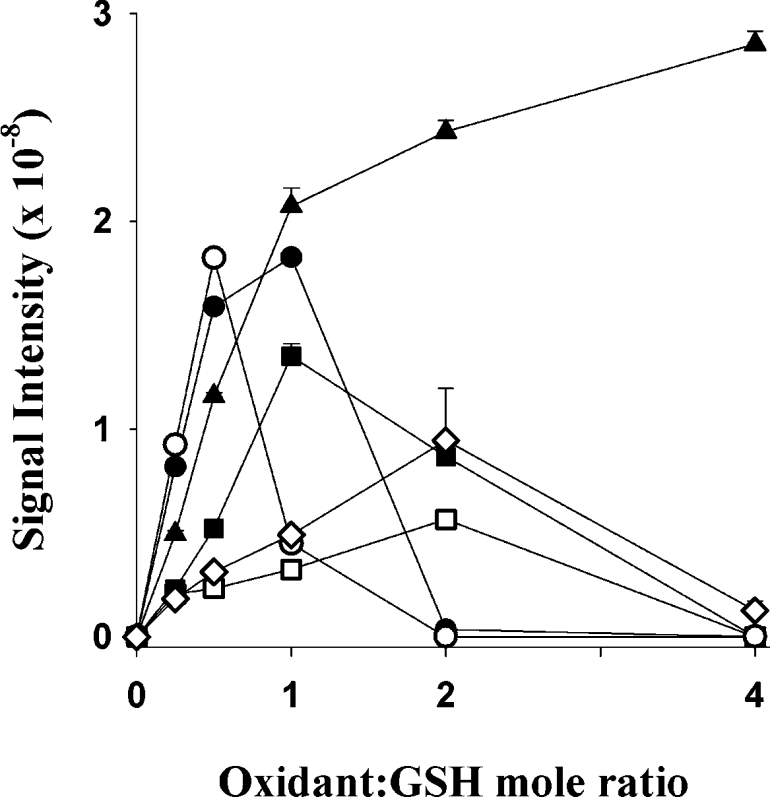
Each plot represents the signal intensity of dehydroglutathione formed when GSH (100 μM) was reacted with increasing concentrations of oxidant. The oxidants were HOCl (□), HOBr (■), HOI (▲), NH2Cl (○), TauNCl (●) and ONOO− (◇). HOSCN formed only very small amounts of dehydroglutathione and is not shown. H2O2 formed no detectable dehydroglutathione. Dehydroglutathione (m/z 306) was monitored by selective reaction monitoring, with the most intense fragment ion (m/z 177) representing a neutral loss of 129 Da. Reactions were treated with NEM after 30 min to alkylate any unreacted GSH.
DISCUSSION
We have developed an LC–MS/MS assay for the detection and quantification of GSH and its oxidation products. It is a sensitive method enabling quantification of GSA for the first time, as well as GSH and GSSG, with 100 fmol detection limits. It can be readily adapted to monitor other oxidation products of GSH, such as the sulfinic and sulfonic acids, by using the full-scan mode of the mass spectrometer. This adaptation led to identification of the novel product, dehydroglutathione. GSA was quantified by relating to an external standard curve constructed from the purified compound. The method also has the potential to detect GSA and other oxidation products of GSH in more complex biological samples. However, for this purpose it would be preferable to use isotopically labelled internal standards for accurate quantification.
GSA has been previously identified as a product of the reaction between HOCl and GSH [11–13]. By using the LC–MS/MS method we have been able to show that it is a significant product of the reaction. GSA was formed at all HOCl to GSH molar ratios tested and was the major product at the higher ratios. At low HOCl to GSH molar ratios, which are more likely to represent the physiological situation, GSA was still formed and accounted for a substantial percentage of the GSH lost. GSA was also generated when GSH was treated with the MPO–H2O2–Cl− system. The amount of GSA formed from the peroxidase system was lower than with a bolus of oxidant but did increase as the MPO concentration increased. This lower yield probably reflects a lower steady-state concentration of HOCl reached with the enzymatic system compared with the bolus addition. As the increase in GSA with increasing MPO concentrations was independent of the total amount of HOCl produced, it suggests that it is the HOCl flux that dictates the amount of GSA generated.
GSA was produced with other oxidants but in lower yields than with HOCl. Most was seen with HOBr, but at concentrations that oxidized less than half the GSH, HOBr gave approximately 10-fold less GSA than was observed with HOCl. The chloramines gave no detectable GSA at these concentrations and only at higher concentrations did it become a minor product. Peroxynitrite also produced small amounts of GSA. Therefore, although not absolutely specific, GSA is a potentially good biomarker for HOCl. As MPO is the physiological source of HOCl, GSA measurements may provide a useful index of MPO activity. Eosinophil peroxidase-derived HOBr and peroxynitrite will need to be considered, but inefficient GSA production by these oxidants makes them less likely to contribute to GSA formation. In vivo, the reaction of peroxynitrite with carbon dioxide would further limit its contribution. Currently the favoured biomarkers for HOCl, HOBr and peroxynitrite are chloro-, bromo- and nitro-tyrosine respectively. Potential advantages of GSA over chlorotyrosine lie in GSH being a more favoured target for HOCl than tyrosine [6]. This should result in much more GSH being converted into GSA in contrast to the 1:1000 or less chlorotyrosine/tyrosine ratios that have been measured in clinical samples [4,34,35]. Also, the GSA assay is simpler and less prone to artifact formation. We now have the required sensitivity with the LC–MS/MS assay to see if GSA is detectable. Nanomolar levels of GSA have been observed in endotracheal aspirate samples from pre-term infants with respiratory distress (results not shown), indicating that GSA can be quantified in clinical samples from an inflammatory condition. Studies relating GSA to MPO and tyrosine markers will need to be performed to assess its sensitivity and selectivity for detecting MPO activity, and to assess its value as a biomarker.
Mechanistically, the formation of GSA from GSH oxidation by HOCl proceeds through three oxidation steps (see reaction 1). We have previously proposed a ring structure linked through the sulfur and amine groups [11,12]. The first step of the reaction involves the oxidation of the cysteinyl thiol to a sulfenyl-chloride intermediate [8,36]. This either becomes further oxidized with subsequent ring closure or ring closure precedes further thiol oxidation. Irrespective of which comes first, ring closure proceeds through an intramolecular reaction involving nucleophilic attack of the terminal amine of the γ-glutamyl moiety to the sulfur to form an intramolecular sulfonamide and release a proton and chloride ion. Oxidation of GSH by other halogenated oxidants is thought to generate an analogous sulfenyl–halide intermediate. The lower yield of GSA observed with these oxidants is likely to be due to their reduction potentials being too low to oxidize the sulfenyl–halide intermediate through to the sulfonamide. Hydrolysis of intermediates to give a sulfenic or sulfinic acid could also be a factor. Fragmentation data obtained for GSA by collision-induced dissociation MS supports the proposed ring structure. A neutral loss of 75 Da was observed, indicating cleavage of the glycinylcysteine peptide bond and removal of the tail.
In the reaction of GSH with oxidants such as the chloramines, GSSG is usually considered as the only product. However, it is clear from the total ion chromatograms of the reaction mixtures that additional species are formed. We observed these even at low oxidant concentrations when some GSH had not reacted. Small amounts of higher oxidation states, particularly the sulfinic acid, were seen with HOCl and HOBr, and to a lesser extent with the chloramines. We also saw formation of an aldehyde of GSSG, that appears to arise from breakdown of its chloramine. The most novel finding was that all of the MPO-derived oxidants and peroxynitrite gave rise to a previously unreported product, dehydroglutathione. Formation of dehydroglutathione, and GSSG, both require one oxidizing equivalent. To form GSSG, the initial sulfenyl–halide reacts with a second GSH molecule. Formation of dehydroglutathione must involve internal elimination of HX (where X is the halide) with the net loss of two protons. The most likely outcome would be a cyclic product. One possibility would be condensation with the amine group as for GSA. However fragmentation of this molecule gave the common neutral loss of 129 Da, consistent with loss of the γ-glutamyl portion of the molecule rather than loss of glycine as seen with GSA. This suggests that oxidation occurred in the cysteinylglycine portion of the molecule and that, unlike GSA, the amine group was unaffected. Oxidation of the sulfur was confirmed by the inability of NEM to alkylate the product. A possible explanation for the observed results is that the sulfenyl–halide intermediate could undergo an intramolecular reaction with the adjacent amide of the cysteinylglycine peptide bond to form a five membered cyclic sulfenamide. Although not previously documented for GSH the generation of cyclic dehydromethionine from the iodine oxidation of methionine has been described [37,38]. We are currently isolating dehydroglutathione to establish its structure and properties.
GSSG is commonly referred to as ‘oxidized glutathione’, and is often considered as the only product of GSH oxidation. Indeed this is the case with H2O2. However, it is apparent from this work that physiological oxidants can give a variety of other stable products, some of which cannot be reduced back to GSH. This finding has important implications for cells that experience high levels of oxidative stress. With continued recycling of GSSG, irreversible oxidation products would accumulate, leading to a depletion of GSH and a decline in its antioxidant defences.
Acknowledgments
This work was supported by the New Zealand Health Research Council, the Lottery Health Grants Board and the National Research Centre for Growth and Development. We would also like to thank Dr Liz Ledgerwood (The Department of Biochemistry, University of Otago, Dunedin, New Zealand) for kindly supplying peroxynitrite for experiments with glutathione.
References
- 1.Klebanoff S. J. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J. Bacteriol. 1968;95:2131–2138. doi: 10.1128/jb.95.6.2131-2138.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Dalen C. J., Whitehouse M. W., Winterbourn C. C., Kettle A. J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senthilmohan R., Kettle A. J. Bromination and chlorination reactions of myeloperoxidase at physiological concentrations of bromide and chloride. Arch. Biochem. Biophys. 2006;445:235–244. doi: 10.1016/j.abb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Hazen S. L., Crowley J. R., Mueller D. M., Heinecke J. W. Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: A sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radical Biol. Med. 1997;23:909–916. doi: 10.1016/s0891-5849(97)00084-1. [DOI] [PubMed] [Google Scholar]
- 5.Winterbourn C. C. Comparative reactivities of various biological compounds with myeloperoxidase-hydrogen peroxide-chloride, and similarity of the oxidant to hypochlorite. Biochim. Biophys. Acta. 1985;840:204–210. doi: 10.1016/0304-4165(85)90120-5. [DOI] [PubMed] [Google Scholar]
- 6.Pattison D. I., Davies M. J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins C. L., Pattison D. I., Davies M. J. Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids. 2003;25:259–274. doi: 10.1007/s00726-003-0016-x. [DOI] [PubMed] [Google Scholar]
- 8.Folkes L. K., Candeias L. P., Wardman P. Kinetics and mechanisms of hypochlorous acid reactions. Arch. Biochem. Biophys. 1995;323:120–126. doi: 10.1006/abbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 9.Meister A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 10.Sies H. Glutathione and its role in cellular functions. Free Radical Biol. Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 11.Winterbourn C. C., Brennan S. O. Characterization of the oxidation products of the reaction between reduced glutathione and hypochlorous acid. Biochem. J. 1997;326:87–92. doi: 10.1042/bj3260087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pullar J. M., Vissers M. C., Winterbourn C. C. Glutathione oxidation by hypochlorous acid in endothelial cells produces glutathione sulfonamide as a major product but not glutathione disulfide. J. Biol. Chem. 2001;276:22120–22125. doi: 10.1074/jbc.M102088200. [DOI] [PubMed] [Google Scholar]
- 13.Carr A. C., Winterbourn C. C. Oxidation of neutrophil glutathione and protein thiols by myeloperoxidase-derived hypochlorous acid. Biochem. J. 1997;327:275–281. doi: 10.1042/bj3270275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erve J. C., Svensson M. A., von Euler-Chelpin H., Klasson-Wehler E. Characterization of glutathione conjugates of the remoxipride hydroquinone metabolite NCQ-344 formed in vitro and detection following oxidation by human neutrophils. Chem. Res. Toxicol. 2004;17:564–571. doi: 10.1021/tx034238n. [DOI] [PubMed] [Google Scholar]
- 15.Cotgreave I. A., Moldeus P. Methodologies for the application of monobromobimane to the simultaneous analysis of soluble and protein thiol components of biological systems. J. Biochem. Biophys. Methods. 1986;13:231–249. doi: 10.1016/0165-022x(86)90102-8. [DOI] [PubMed] [Google Scholar]
- 16.Martin J., White I. N. Fluorimetric determination of oxidised and reduced glutathione in cells and tissues by high-performance liquid chromatography following derivatization with dansyl chloride. J. Chromatogr. 1991;568:219–225. doi: 10.1016/0378-4347(91)80356-h. [DOI] [PubMed] [Google Scholar]
- 17.Beers R. F., Jr, Sizer I. W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 18.Gazda M., Margerum D. W. Reactions of monochloramine with Br2, Br3−, HOBr, and OBr−: formation of bromochloramines. Inorg. Chem. 1994;33:118–123. [Google Scholar]
- 19.Kumar K., Margerum D. W. Kinetics and mechanism of general-acid-assisted oxidation of bromide by hypochlorite and hypochlorous acid. Inorg. Chem. 1987;26:2706–2711. [Google Scholar]
- 20.Kumar K., Day R. A., Margerum D. W. Atom-transfer redox kinetics: General-acid-assisted oxidation of iodide by chloramines and hypochlorite. Inorg. Chem. 1986;25:4344–4350. [Google Scholar]
- 21.Aune T. M., Thomas E. L. Accumulation of hypothiocyanite ion during peroxidase-catalyzed oxidation of thiocyanate ion. Eur. J. Biochem. 1977;80:209–214. doi: 10.1111/j.1432-1033.1977.tb11873.x. [DOI] [PubMed] [Google Scholar]
- 22.Kettle A. J., Winterbourn C. C. Assays for the chlorination activity of myeloperoxidase. Methods Enzymol. 1994;233:502–512. doi: 10.1016/s0076-6879(94)33056-5. [DOI] [PubMed] [Google Scholar]
- 23.Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes M. N., Nicklin H. G. The chemistry of pernitrites. Part 1. Kinetics of decomposition of pernitrous acid. J. Am. Chem. Soc. 1968;2:450–452. [Google Scholar]
- 25.Prutz W. A., Kissner R., Nauser T., Koppenol W. H. On the oxidation of cytochrome c by hypohalous acids. Arch. Biochem. Biophys. 2001;389:110–122. doi: 10.1006/abbi.2001.2321. [DOI] [PubMed] [Google Scholar]
- 26.Pruitt K. M., Tenovuo J., Mansson-Rahemtulla B., Harrington P., Baldone D. C. Is thiocyanate peroxidation at equilibrium in vivo? Biochim. Biophys. Acta. 1986;870:385–391. doi: 10.1016/0167-4838(86)90245-1. [DOI] [PubMed] [Google Scholar]
- 27.Pryor W. A., Squadrito G. L. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995;268:699–722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 28.Mendelson D. S., Metz E. N., Sagone A. L., Jr Effect of phagocytosis on the reduced soluble sulfhydryl content of human granulocytes. Blood. 1977;50:1023–1030. [PubMed] [Google Scholar]
- 29.Stockigt D. Identification of collision-induced dissociation fragments from a protonated glutathione conjugate by isotope-specific MS3 experiment in an ion trap. Rapid Commun. Mass Spectrom. 1998;12:273–275. [Google Scholar]
- 30.Pearson P. G., Howald W. N., Nelson S. D. Screening strategy for the detection of derivatized glutathione conjugates by tandem mass spectrometry. Anal. Chem. 1990;62:1827–1836. [Google Scholar]
- 31.Bonini M. G., Radi R., Ferrer-Sueta G., Ferreira A. M. D. C., Augusto O. Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J. Biol. Chem. 1999;274:10802–10806. doi: 10.1074/jbc.274.16.10802. [DOI] [PubMed] [Google Scholar]
- 32.Bonini M. G., Augusto O. Carbon dioxide stimulates the production of thiyl, sulfinyl, and disulfide radical anion from thiol oxidation by peroxynitrite. J. Biol. Chem. 2001;276:9749–9754. doi: 10.1074/jbc.M008456200. [DOI] [PubMed] [Google Scholar]
- 33.Winterbourn C. C., Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radical Biol. Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 34.Hazen S. L., Heinecke J. W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buss I. H., Senthilmohan R., Darlow B. A., Mogridge N., Kettle A. J., Winterbourn C. C. 3-Chlorotyrosine as a marker of protein damage by myeloperoxidase in tracheal aspirates from preterm infants: association with adverse respiratory outcome. Pediatr. Res. 2003;53:455–462. doi: 10.1203/01.PDR.0000050655.25689.CE. [DOI] [PubMed] [Google Scholar]
- 36.Armesto X. L., Canle L., Fernandez M. I., Garcia M. V., Santabella J. A. First steps in the oxidation of sulfur-containing amino acids by hypohalogenation: Very fast generation of intermediate sulfenyl halides and halosulfonium cations. Tetrahedron. 2000;56:1103–1109. [Google Scholar]
- 37.Young P. R., Hsieh L. General base catalysis and evidence for a sulfurane intermediate in the iodine oxidation of methionine. J. Am. Chem. Soc. 1978;100:7121–7122. [Google Scholar]
- 38.Lambeth D. O., Swank D. W. Oxidative cyclization of 3-(amino)thioethers to form S-substituted isothiazolidinium salts. J. Org. Chem. 1979;44:2632–2636. [Google Scholar]