

Abstract
COX [cyclo-oxygenase; PG (prostaglandin) G/H synthase] oxygenates AA (arachidonic acid) and 2-AG (2-arachidonylglycerol) to endoperoxides that are converted into PGs and PG-Gs (glycerylprostaglandins) respectively. In vitro, 2-AG is a selective substrate for COX-2, but in zymosan-stimulated peritoneal macrophages, PG-G synthesis is not sensitive to selective COX-2 inhibition. This suggests that COX-1 oxygenates 2-AG, so studies were carried out to identify enzymes involved in zymosan-dependent PG-G and PG synthesis. When macrophages from COX-1−/− or COX-2−/− mice were treated with zymosan, 20–25% and 10–15% of the PG and PG-G synthesis observed in wild-type cells respectively was COX-2 dependent. When exogenous AA and 2-AG were supplied to COX-2−/− macrophages, PG and PG-G synthesis was reduced as compared with wild-type cells. In contrast, when exogenous substrates were provided to COX-1−/− macrophages, PG-G but not PG synthesis was reduced. Product synthesis also was evaluated in macrophages from cPLA2α (cytosolic phospholipase A2α)−/− mice, in which zymosan-induced PG synthesis was markedly reduced, and PG-G synthesis was increased approx. 2-fold. These studies confirm that peritoneal macrophages synthesize PG-Gs in response to zymosan, but that this process is primarily COX-1-dependent, as is the synthesis of PGs. They also indicate that the 2-AG and AA used for PG-G and PG synthesis respectively are derived from independent pathways.
Keywords: arachidonic acid, arachidonylglycerol, cyclo-oxygenase, cytosolic phospholipase A2α, glycerylprostaglandin, macrophage
Abbreviations: AA, arachidonic acid; AG, arachidonylglycerol; α-MEM, minimum essential medium-α; COX, cyclo-oxygenase; cPLA2α, cytosolic phospholipase A2α; FCS, fetal calf serum; LC/MS/MS, liquid chromatography tandem MS; LPS, lipopolysaccharide; CaMgfree-PBS, calcium- and magnesium-free PBS; PG, prostaglandin; PG-G, glycerylprostaglandin; PGE2-G, PG-G E2; RPM, resident peritoneal macrophages; WT, wild-type
INTRODUCTION
COX [cyclo-oxygenase; PG (prostaglandin) G/H synthase] oxygenates AA (arachidonic acid) to the hydroxyendoperoxide, PG H2, which is subsequently converted into biologically active PGs [1–4]. There are two COX isoforms, COX-1 and COX-2. COX-1 is usually expressed constitutively, and is broadly distributed, whereas COX-2 is induced in a limited number of cells in response to a variety of inflammatory and proliferative stimuli [5–11]. The two isoforms have nearly superimposable three-dimensional structures, and exhibit essentially the same kinetics with AA in vitro [12–16]. However, subtle differences in the active site have allowed the development of selective inhibitors of each isoform, and enable COX-2 to utilize neutral ester and amide derivatives of AA as substrates more effectively than COX-1 [17–24].
Among the neutral derivatives of AA that are selectively metabolized by COX-2 in vitro is the endocannabinoid, 2-AG (2-arachidonylglycerol). Oxygenation of 2-AG by COX-2 yields PG-Gs (glycerylprostaglandins) [24,25]. Recent studies have shown that picomolar concentrations of one of these metabolites, PGE2-G (PG-G E2), induces Ca2+ mobilization in RAW264.7 cells by a mechanism distinct from that of its free acid analogue, PGE2 [26]. These results suggest that PG-Gs mediate unique physiological functions through binding to specific receptors.
In order to further delineate a possible biological role for PG-Gs, we have undertaken to identify conditions under which primary cell populations produce these metabolites in response to physiologically relevant stimuli. To that end, we recently reported that murine RPM (resident peritoneal macrophages) produce PGE2-G and PGI2-G in response to zymosan phagocytosis [27]. However, although 2-AG is a very poor substrate for purified COX-1 in vitro [23], we found that RPM that had not been pretreated with LPS (lipopolysaccharide) to induce COX-2 expression produced significant quantities of PG-Gs, and that LPS pretreatment resulted in only a 30% increase in PG-G formation. Furthermore, experiments using the COX-2-selective inhibitor, SC236 {4-[5-(4-chlorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide}, indicated that up to 50% of PG-G formation in LPS-pretreated RPM was COX-1-dependent [27]. These results suggested that PG-G formation in zymosan-stimulated RPM is not a primarily COX-2-dependent process as would be predicted from prior in vitro kinetic data [23].
Determining whether PG-Gs play a significant physiological role in vivo depends on a complete understanding of the pathways by which they are synthesized, including the specific COX isoform(s) involved. The finding that COX-1 is a major source of PG-Gs in zymosan-treated RPM was unexpected in light of the in vitro results showing that 2-AG is a COX-2-selective substrate. Consequently, it was important to further explore the relative contributions of COX-1 and COX-2 to both PG-G and PG formation in zymosan-stimulated RPM. To this end, we have used RPM from COX-1−/− or COX-2−/− mice. We also have investigated PG-G and PG synthesis by RPM from cPLA2α (cytosolic phospholipase A2α)−/− mice in order to ascertain if the 2-AG used for PG-G synthesis is released independently of AA used for PG synthesis.
MATERIALS AND METHODS
RPM cultures
All studies involving animals were done with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University. Male WT (wild-type) CD-1 mice (25–30 g) were obtained from Charles River Laboratories (Wilmington, MA, U.S.A.). Male COX-1−/−, COX-2−/− and cPLA2α−/− mice on a CD-1 background (25–30 g) were bred in the Vanderbilt University animal care facility as described in [28]. The mice were killed with carbon dioxide, and the peritoneal cavities were lavaged with a total of 3 ml of ice-cold CaMgfree-PBS (calcium- and magnesium-free PBS) [29]. Peritoneal cells were collected by centrifugation of lavage fluid and resuspended at a concentration of (2–3)×106 cells/ml in α-MEM/FCS [minimum essential medium-α supplemented with Gluta-Max (Gibco, Grand Island, NY, U.S.A.), containing 10% (v/v) heat-inactivated fetal calf serum (Summit Biotechnologies, Fort Collins, CO, U.S.A.) plus 100 units/ml penicillin and 0.10 mg/ml streptomycin (Sigma, St. Louis, MO, U.S.A.)]. The cell suspension was plated on to 35 mm tissue culture dishes at 2 ml/dish and incubated for 2 h at 37 °C in a humidified 5% CO2 atmosphere. Non-adherent cells were removed by washing the plates four times with CaMgfree-PBS, and the cultures were then incubated overnight in fresh α-MEM/FCS. The mean protein content of RPM cultures was 100±10 μg/dish [(8.2±0.8)×105 cells/dish].
LPS and zymosan treatment of RPM cultures
Cultures of RPM were washed twice with CaMgfree-PBS at 37 °C, and then overlaid with 2 ml of fresh α-MEM/FCS with or without 100 ng/ml LPS (Escherichia coli 011:B4; Calbiochem, San Diego, CA, U.S.A.). Cells were incubated for 6 h at 37 °C, washed twice again with CaMgfree-PBS and overlaid with 2 ml of serum-free α-MEM. Unopsonized zymosan A (Sigma) was prepared as described by Bonney et al. [30] and suspended at 160 mg/ml in CaMgfree-PBS. Aliquots (10 μl) were added directly to macrophage culture medium as required, and cells were incubated as indicated for individual experiments.
Metabolism of exogenous AA and 2-AG by RPM cultures
RPM cultures were prepared and incubated in the presence or absence of LPS as described above. Cells were then washed twice with CaMgfree-PBS and overlaid with 1 ml of serum-free α-MEM containing 1 μM of either AA or 2-AG (Cayman, Ann Arbor, MI, U.S.A.). Cells were incubated for 30 min at 37 °C. For addition to culture medium, AA and 2-AG were prepared as 1 mM stock solutions in DMSO and added to medium so that the total DMSO concentration contributed by the addition of lipids was 0.1% for all samples.
Analysis of PGs and PG-Gs in RPM culture medium
Following incubation with desired stimuli or substrates under conditions specified in the descriptions for individual experiments, the medium was removed from RPM cultures, placed on ice, and combined with 20 μl of methanol containing 5 pmol each of PGE2-G-d5 and 6-keto-PGF1α-G-d5 (synthesized as described in [23]) plus 50 pmol each of PGE2-d4 and 6-keto-PGF1α-d4 (both from Cayman) which served as internal standards. Samples were stored at −20 °C prior to solid phase extraction and analysis by selected reaction monitoring of the ammoniated ions by positive-ion electrospray ionization LC/MS/MS (liquid chromatography tandem MS) as described in [31]. The major PGs produced by RPM are PGE2 and PGI2 (identified as its stable hydrolysis product, 6-keto-PGF1α). Similarly, the major PG-Gs were PGE2-G and PGI2-G (identified as 6-keto-PGF1α-G). The relative proportions of PGE2 and PGE2-G increase with LPS pretreatment [27]. Although minor amounts of thromboxane A2 and PGF2α are produced by RPM, those products were not quantified in these studies and total PGs and PG-Gs refer to the sums of PGE2 plus 6-keto-PGF1α, and PGE2-G plus 6-keto-PGF1α-G respectively.
Analysis of cellular AA and total AG
For the measurement of free AA and total AG [1(3)-AG and 2-AG], the medium was removed from the cell cultures and the cells were scraped twice into a total volume of 1 ml of ice-cold methanol. The resulting cell lysate was combined with 1 ml of ice-cold acetonitrile containing 10 ng of 2-AG-d8 and 100 ng of AA-d8 (Cayman) and stored at −20 °C for analysis.
Samples were subjected to liquid extraction followed by silica solid phase extraction prior to analysis for total AG and AA by silver ion co-ordination LC/MS/MS as described in [27,32]. In typical samples, both 2-AG and 1(3)-AG were detected. In most cases, 2-AG was the predominant isomer, and the relative ratio of 2-AG to 1(3)-AG was similar to that in the internal standard which was added as 100% 2-AG-d8 to the samples. Since non-specific acyl migration of 2-AG to a mixture of 2-AG and 1(3)-AG occurs during sample workup, it is not possible to determine exactly how much of each isomer was present in the cells originally, so the two isomers were quantified together.
Immunoblotting for COX-2 protein expression
For the determination of COX-1, COX-2 and cPLA2α protein expression, RPM cultures were incubated with the desired stimuli under conditions specified in the descriptions for individual experiments, the medium was removed, and the cells were then washed twice with ice-cold CaMgfree-PBS. Cell monolayers were scraped into 200 μl of lysis buffer [50 mM Tris/HCl, pH 7.5, plus 150 mM NaCl, 4 mM EDTA, 50 mM NaF, 0.1 mM Na3VO4, 0.2% Triton X-100, 0.1% Nonidet P40, 0.5% sodium deoxycholate, 1 mM dithiothreitol, 1 mg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, and 5 μg/ml each of antipain, leupeptin, chymostatin and pepstatin (all components from Sigma)]. Cell lysates were kept for 30 min on ice with occasional vortex-mixing, and particulate material was then removed by centrifugation for 10 min at 16000 g. Samples were stored at −80 °C until analyses could be completed.
The protein concentrations of 20 μl aliquots of macrophage cell lysates were determined using a BCA (bicinchoninic acid) Protein Assay kit (Pierce, Rockford, IL, U.S.A.) according to the manufacturer's directions. Macrophage lysate samples containing 15 μg of protein were then subjected to SDS/PAGE and immunoblot analysis as described in [33], with the exception that the rabbit polyclonal antibody directed against murine COX-2 (Cayman) was used at a dilution of 1:10000 instead of 1:30000.
Data analysis
Statistical significance was determined by two-way ANOVA with Bonferroni post-hoc tests or by one-way ANOVA with Newman–Keuls multiple comparison test using GraphPad Prism software, as appropriate.
RESULTS
Effect of selective deletion of COX-1 and COX-2 on zymosan-induced PG-G and PG formation by RPM
RPM were isolated from WT, COX-1−/− and COX-2−/− mice as described in the Materials and methods section. Following a 6 h incubation in the presence or absence of LPS (to achieve maximal induction of COX-2 protein [33]), cells were washed and challenged with zymosan (160 μg). Cultures were incubated for 2 h and the medium was analysed for PG-G and PG formation. WT RPM produced PG-Gs in quantities comparable with those reported previously [27], showing a significant 1.4-fold increase in PG-G formation as a result of LPS pretreatment (Figure 1A). In the absence of LPS pretreatment, COX-2−/− RPM produced essentially the same quantities of PG-Gs as did WT RPM, but no increase resulted from LPS treatment in these cells. In contrast, PG-G formation was undetectable in COX-1−/− RPM in the absence of LPS pretreatment, and even though LPS treatment resulted in an increase, levels achieved were only 9.6% as great as those observed in LPS-pretreated WT cells.
Figure 1. Formation of PGs and PG-Gs by RPM in response to zymosan.

Cultures of RPM from WT, COX-1−/− or COX-2−/− mice in 35 mm dishes were pre-incubated for 6 h in the absence (−LPS) or presence (+LPS) of 100 ng/ml bacterial LPS. Cells were washed, transferred to serum-free medium, and then incubated for 2 h with 160 μg of zymosan. The medium was then removed and analysed for PG-Gs (A, B) and PGs (C, D) by LC/MS/MS. Values are given for total product (A, C) or for PGE2-G or PGE2 (B, D), the remainder being PGI2-G or PGI2. In the absence of zymosan, WT cells pre-incubated in the absence of LPS produced 56±11 pmol of PGs/107 cells and those pre-incubated in the presence of LPS produced 360±60 pmol/107 cells. Formation of PG-Gs was undetectable in the absence of a zymosan stimulus. Asterisk indicates that results are significantly different from WT RPM of the same treatment. The star symbol indicates that results from LPS-pretreated RPM are significantly different from RPM of the same genotype that were not treated with LPS (P<0.05). The results, including values given for PG formation in the absence of zymosan, are the means±S.D. for three separate experiments in which triplicate determinations were made.
WT RPM produced quantities of PGs that were similar to our previously reported values [27], and LPS pretreatment had no effect on the total amounts synthesized (Figure 1C). In COX-2−/− RPM, a reduction in total PG synthesis was observed, although this decrease did not reach statistical significance. LPS pretreatment had no effect on total PG synthesis in these cells. As in the case of PG-G formation, PG synthesis in COX-1−/− RPM was nearly undetectable without LPS pretreatment. Although LPS treatment resulted in a significant increase in PG formation, levels produced were only 14% of those achieved in LPS-pretreated WT cells.
As reported previously [27], the primary PGs produced by RPM were PGE2 and PGI2 (identified as its stable hydrolysis product, 6-keto-PGF1α). Similarly, the primary PG-Gs were PGE2-G and PGI2-G. Because LPS induces the expression of microsomal PGE synthase in RPM [33], the relative proportion of PGE2 and PGE2-G was higher in LPS-pretreated RPM than in those cells not treated with LPS [27]. As shown in Figure 1(D), the selected deletion of COX-1 and COX-2 had no significant effect on the relative proportion of PGE2, indicating that the effects observed on total PG synthesis could not be explained by an alteration of the expression of the specific synthase enzymes. The relative proportion of PGE2-G was significantly higher (P<0.02) in LPS-pretreated COX-1−/− RPM than in LPS-pretreated WT RPM (86%±10% versus 74%±8% respectively) (Figure 1B). However, the quantities of PG-Gs synthesized by these cells were near the limit of detection of the assay, so it is not clear whether this relatively small increase is truly significant.
The levels of product formation in COX-1−/− RPM (Figures 1A and 1C) suggest that the contribution of COX-2 to zymosan-stimulated PG-G and PG synthesis was lower than previously estimated using the selective COX-2 inhibitor, SC236, in WT RPM [27]. However, before such conclusions could be drawn, it was important to verify that the reduced product formation in COX-1−/− RPM (as compared with WT RPM) was not due to low availability of the immediate precursors of PG and PG-G synthesis. We have previously shown that zymosan stimulation leads to an increase in the levels of AA (the precursor of PGs) and AG (the precursor of PG-Gs) [27]. Both lipids reach maximal levels at 60 min, and remain elevated for as long as 120 min after zymosan addition [27]. Consequently, the levels of cellular AA and AG were measured in RPM 2 h after the addition of zymosan in order to determine whether selected deletion of either COX isoform causes a major change in the availability of these critical lipids. The 2 h time point was chosen, since it allowed the determination of maximal product synthesis and cellular lipids from the same cultures, thus minimizing the number of animals required for the experiments. As seen in Figures 2(A) and 2(B), LPS pretreatment resulted in reduced levels of both AA and AG in zymosan-treated WT cells. These differences were shown to be statistically significant in prior studies using large numbers of independent experiments [27]. A similar effect of LPS pretreatment on AA levels was seen in COX-1−/− RPM. In addition, COX-1−/− RPM showed a marked increase in AA levels, and no significant difference in total AG levels when compared with WT cells. In COX-2−/− RPM, LPS pretreatment had no effect on zymosan-induced AA or AG levels. However, AA levels in these cells were not significantly different from those in WT cells regardless of prior treatment with LPS. AG levels were significantly higher in LPS-pretreated COX-2−/− RPM than in LPS-pretreated WT cells (Figure 2A), although this clearly did not lead to augmented PG-G synthesis in the COX-2−/− RPM (Figure 1A). In conclusion, although some significant differences were noted in precursor levels between the RPM populations, under no circumstances were substrate levels lower in COX-1−/− or COX-2−/− cells when compared with WT cells. Therefore reduced PG and PG-G synthesis in RPM bearing targeted COX deletions cannot be attributed to reduced precursor availability.
Figure 2. AG and AA levels in zymosan-treated RPM.

Experimental conditions are exactly as described for Figure 1. Following the 2 h incubation with zymosan, the cells were scraped into methanol and analysed for AG (A) and AA (B) content by LC/MS/MS. Asterisk indicates that results are significantly different (P<0.05) from WT RPM of the same treatment. The star symbol indicates that results from LPS-pretreated RPM are significantly different (P<0.01) from RPM of the same genotype that were not treated with LPS. The results are the means±S.D. for three separate experiments in which duplicate determinations were made.
Another factor that could affect the quantities of PGs and PG-Gs produced by COX-1−/− and COX-2−/− RPM is the level of expression of the remaining COX isoform. As shown in Figure 3(A), COX-1 expression in COX-2−/− RPM was not significantly different from WT RPM, although it did show a small reduction. In contrast, COX-2 expression in COX-1−/− RPM was significantly reduced (41%) from that in WT cells, a factor that may have contributed to the relatively low quantities of PGs produced by these cells. Also of note is the finding that cPLA2α expression was decreased (although not significantly) in COX-1−/− RPM (Figure 3C) despite the much higher levels of free AA found in those cells after the zymosan stimulus (Figure 2B).
Figure 3. Expression of COX-1, COX-2 and cPLA2α in zymosan-treated RPM.
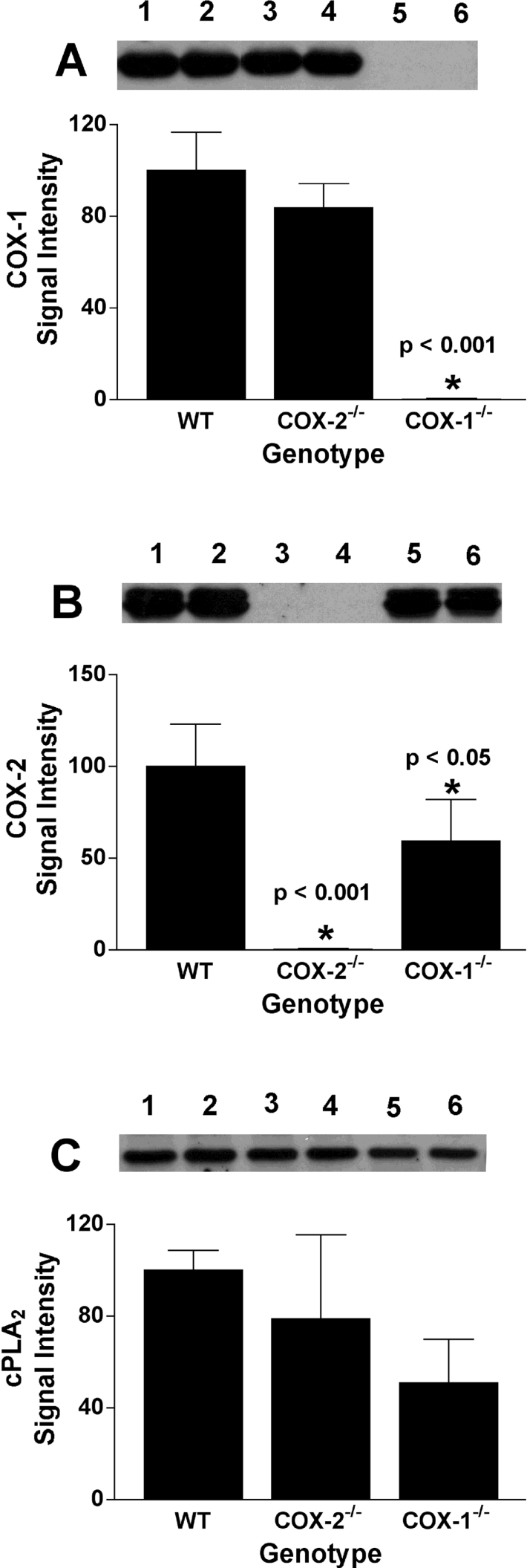
RPM cultures were prepared and incubated with LPS and zymosan as described in the legend to Figure 1. Following the zymosan incubation, the medium was removed, and the cells were washed and scraped into lysis buffer for immunoblot analysis of COX-1 (A), COX-2 (B) or cPLA2α (C) expression. Chemiluminescence was measured using a Fluor-S Max imager (Bio-Rad, Hercules, CA, U.S.A.), and results were normalized to the expression of WT enzyme in each experiment. Results are the means±S.D. for three separate experiments in which single or duplicate determinations were made. Asterisk indicates that results are significantly different from WT RPM at the indicated P value. Example blots are shown for each enzyme. Lane identification numbers are as follows: 1 and 2, WT cells; 3 and 4, COX-2−/− cells; 5 and 6, COX-1−/− cells.
PG and PG-G synthesis by RPM from exogenous substrates
In zymosan-challenged RPM, the formation of PGs and PG-Gs is dependent on the availability of released endogenous AA and AG respectively. Therefore total product formation does not necessarily reflect the total PG synthetic capacity (total COX activity) of the cells, or the changes in synthetic capacity resulting from LPS pretreatment (through induction of COX-2 expression). Therefore we explored the effects of LPS pretreatment on product formation from exogenously supplied substrates. These experiments utilized 1 μM AA or 2-AG to avoid cellular toxicity [34]. At these substrate concentrations, total PG synthesis is significantly lower than that observed with zymosan. However, we have previously shown that high substrate concentrations (20 μM) produce identical changes in PG synthesis patterns in response to LPS while yielding the same amount of PGs as are observed with a zymosan stimulus [27]. Similar to prior results, the production of PGs from exogenous AA by WT RPM increased 1.9-fold as a result of LPS pretreatment (Figure 4). No increase was observed with LPS pretreatment in COX-2−/− RPM. Furthermore, total PG synthesis from exogenous AA was reduced by 68 and 79% in the absence and presence of LPS pretreatment respectively in COX-2−/− RPM when compared with WT RPM. These reductions were greater than had been observed for PG synthesis from endogenous substrates by zymosan-treated cells. For COX-1−/− RPM, LPS pretreatment evoked an 11-fold increase in PG synthesis from exogenous AA. However, the pretreated cells produced only 28% as much PGs as LPS-pretreated WT RPM.
Figure 4. Synthesis of PGs by RPM from exogenous AA.

Cultures of RPM from WT, COX-1−/− and COX-2−/− mice in 35 mm dishes were pre-incubated for 6 h in the absence (−LPS) or presence (+LPS) of 100 ng/ml LPS. Cells were washed and transferred to serum-free medium, containing 1 μM AA. The cells were incubated for 30 min, and the medium was then removed and analysed for PGs by LC/MS/MS. The results are the means±range for two separate experiments in which duplicate determinations were made. Asterisk indicates that results are significantly different from WT RPM of the same treatment. The star symbol indicates that results from LPS-pretreated RPM are significantly different from RPM of the same genotype that were not treated with LPS (P<0.05).
RPM produce both PGs and PG-Gs from exogenous 2-AG. Prior results show that the PGs are most likely formed by hydrolysis of 2-AG followed by COX-dependent oxygenation of the resulting free AA [27]. PG synthesis from 2-AG in WT RPM was similar to that reported previously [27], including a 3.2-fold increase resulting from LPS pretreatment (Figure 5A). No such increase was observed with LPS pretreatment in COX-2−/− RPM, and as was seen with exogenous AA, PG synthesis from 2-AG was reduced to a greater extent (as compared with WT) than was observed for PG synthesis from endogenous substrate in response to zymosan. Notably, LPS-pretreated COX-1−/− RPM produced 28% as much PGs from 2-AG as WT RPM, exactly the same as was observed when AA was used as the substrate. Cellular levels of AA and total AG at the end of the incubation period were not significantly different among the three different types of cell populations (results not shown).
Figure 5. Synthesis of PGs and PG-Gs by RPM from exogenous 2-AG.

Experimental conditions are exactly as described in the legend to Figure 4, except that cells were transferred to serum-free medium, containing 1 μM 2-AG. The medium was analysed for PGs (A) or PG-Gs (B) by LC/MS/MS. The results are the means±range for two separate experiments in which duplicate determinations were made. Asterisk indicates that results are significantly different from WT RPM of the same treatment. The star symbol indicates that results from LPS-pretreated RPM are significantly different from RPM of the same genotype that were not treated with LPS (P<0.05).
WT RPM produced PG-Gs from 2-AG in quantities similar to those previously reported [27], and LPS pretreatment evoked a 4.8-fold increase (Figure 5B). When compared with WT cells, PG-G formation from 2-AG in COX-2−/− RPM showed a similar decrease as was seen for PG formation from 2-AG. However, LPS-pretreated COX-1−/− RPM produced only 11% as much PG-Gs as LPS-pretreated WT cells, a much smaller percentage than was observed for PG formation from either AA or 2-AG.
The cells used for these experiments were evaluated for COX-1 and COX-2 expression. As was observed in the case of zymosan-treated RPM (Figure 2), COX-1 expression was slightly reduced (21%) in COX-2−/− RPM, but COX-2 expression was significantly reduced (51%) in COX-1−/− RPM. These lower levels of protein expression must be considered when interpreting the quantities of PGs and PG-Gs produced by each population of cells.
Role of cPLA2α in PG and PG-G synthesis in response to zymosan
AA for zymosan-induced PG synthesis is provided primarily through the action of cPLA2α [35]. Since cPLA2α hydrolyses the ester bond between AA and the glycerol backbone of the phospholipid, it is unlikely that 2-AG formation would be a direct result of this pathway. However, it is conceivable that 2-AG could be formed indirectly through the action of cPLA2α. In order to explore this possibility, we measured zymosan-dependent PG and PG-G formation in LPS-pretreated RPM from cPLA2α−/− mice. Following zymosan addition to WT RPM, AA levels increased markedly, reached a peak at approx. 60 min, and then gradually decreased, reaching baseline after 6 h (Figure 6A). No such increase was observed in RPM from cPLA2α−/− mice. Total AG also increased in WT RPM, following a similar time course as was observed for AA. In cPLA2α−/− RPM, AG levels rose higher, reached a peak later, and remained elevated for a longer time (Figure 6B) than in WT cells, although these changes were not statistically significant. WT RPM produced large quantities of PGs in response to zymosan, and PG synthesis was essentially complete after 2 h (Figure 7A). In contrast, PG formation by cPLA2α−/− RPM was markedly reduced, although not totally eliminated, and a gradual accumulation of PGs continued for the entire 10 h incubation, reaching levels equivalent to 6.7% of those generated by WT cells. Both WT and cPLA2α−/− RPM produced PG-Gs in response to zymosan. PG-G formation in cPLA2α−/− RPM was somewhat higher than in WT RPM, although considerable variability was observed between experiments, and the differences were not statistically significant. Immunoblot analysis revealed that COX-1 and COX-2 levels in cPLA2α−/− RPM were similar to those in WT cells (results not shown). These results demonstrate that cPLA2α is not required for mobilization of 2-AG or PG-G formation in zymosan-stimulated RPM.
Figure 6. Time course of substrate release by WT and cPLA2α−/− RPM during zymosan phagocytosis.

RPM from WT and cPLA2α−/− mice in 35 mm dishes were incubated for 6 h in the presence of 100 nM LPS. Cells were then washed and overlaid with serum-free medium. Zymosan (160 μg) was added to each dish, and the cells were incubated for the indicated times. The cells were scraped into methanol for the LC/MS/MS analysis of AA (A) and total AG (B). Results are the means±range for two experiments in which duplicate determinations were made. Asterisk indicates that results are significantly different between WT and cPLA2α−/− RPM at the same time point.
Figure 7. Time course of PG and PG-G formation by WT and cPLA2α−/− RPM during zymosan phagocytosis.

Experimental conditions are exactly as described in the legend to Figure 6. At the indicated times after zymosan addition, the medium was removed for analysis by LC/MS/MS of PG (A) and PG-G (B) content. Results are the means±range for two experiments in which duplicate determinations were made. Asterisk indicates that results are significantly different between WT and cPLA2α−/− RPM at the same time point.
DISCUSSION
Effect of COX-2 deletion on zymosan-induced PG and PG-G synthesis
Theoretically, in the absence of LPS pretreatment, product synthesis by COX-2−/− RPM should resemble that of WT cells, since the process would be COX-1-dependent in both cases. Furthermore, because no additional COX activity can be induced, one would expect that LPS pretreatment would have little effect on product synthesis in COX-2−/− cells. Technically this was true, since the quantities of PGs and PG-Gs produced by COX-2−/− RPM were unaffected by LPS pretreatment, and were not significantly different from those of WT cells in the absence of prior LPS treatment. In every experiment, however, the quantities of PGs (but not PG-Gs) produced by COX-2−/− RPM were lower than those produced by WT cells, so it is likely that in a larger study, these differences would reach statistical significance. After corrections are made for the small decrease in COX-1 expression in the COX-2−/− cells, PG formation was 77 and 80% of that of WT cells in the absence and presence of LPS pretreatment respectively. Thus it appears that COX-2 deletion leads to additional changes in the cells that cause a mild suppression of COX-1 activity.
COX-2−/− RPM produced essentially the same amount of PG-Gs as did WT RPM. Specifically, when corrected for differences in COX-1 expression, COX-2−/− RPM produced 136 and 104% as much PG-Gs as did WT RPM in the absence and presence of LPS pretreatment respectively. These results suggest that COX-2 is unnecessary for zymosan-dependent PG-G formation by these cells, a remarkable finding in light of the fact that 2-AG is preferentially oxidized by COX-2 (∼20-fold greater activity than COX-1) in vitro [23].
Effect of COX-1 deletion on zymosan-induced PG and PG-G synthesis
COX-1−/− RPM produced nearly undetectable levels of PGs or PG-Gs in the absence of LPS pretreatment, consistent with the absence of COX-1 and extremely low levels of COX-2 expression in the cells. Although COX-2 is induced in RPM in response to zymosan, significant protein levels do not appear until after the period of maximal PG and PG-G synthesis is over [27]. LPS pretreatment resulted in a significant increase in zymosan-dependent product formation in COX-1−/− RPM; however, these cells produced only a small fraction of the PG-Gs and PGs synthesized by LPS-pretreated WT cells. Since COX-2 expression was reduced by approx. 40% in the COX-1−/− cells, it is likely that the quantities of products synthesized underestimate the COX-2 contribution by approximately this much. If this correction is applied, LPS-pretreated COX-1−/− RPM produced 23% as much PGs and 14% as much PG-Gs as did LPS-pretreated WT RPM.
Effect of COX isoform deletion on the metabolism of exogenous substrates
In our prior studies [27], we investigated the metabolism of exogenous AA and 2-AG by control and LPS-pretreated RPM in order to assess the ability of LPS to increase PG-G and PG synthetic capacity distinct from its effect on substrate availability. Using WT RPM, and the selective COX-2 inhibitor, SC236, those experiments suggested that COX-2 played a greater role in the metabolism of exogenous substrates than in the metabolism of endogenous substrates. Thus, for the addition of either AA or 2-AG, LPS pretreatment caused a significant increase in product formation that was eliminated by SC236, suggesting that the augmented synthesis was due to the de novo expression of active COX-2 [27]. In the present studies, WT macrophages behaved similarly. Furthermore, results from COX-2−/− and COX-1−/− RPM met expectations in that LPS pretreatment had no effect on PG and PG-G synthesis in COX-2−/− RPM, and COX-1−/− RPM produced almost no products in the absence of LPS pretreatment. However, the quantities of products formed were lower than expected in COX-2−/− and COX-1−/− RPM. Again, one would predict that, in the absence of LPS pretreatment, COX-2−/− RPM would display PG and PG-G synthetic capacities similar to those of WT cells. However, even after correction for the 21% reduction in COX-1 expression, values for PG synthesis from AA and 2-AG by COX-2−/− RPM were 41 and 61% of those from WT cells respectively, and the corrected quantity of PG-Gs formed from 2-AG was 69% of that of WT cells. Similarly, one would expect that LPS-pretreated COX-1−/− RPM would synthesize products in quantities comparable with the increase observed in WT cells as a result of the LPS treatment. This prediction was reasonably accurate for PG synthesis from AA and 2-AG (113 and 79% respectively of the LPS-dependent increment in WT RPM) after correcting for the 48% reduction in COX-2 expression in COX-1−/− RPM. However, the quantities of PG-Gs produced by LPS-pretreated COX-1−/− RPM, after correction for reduced COX-2 expression, corresponded to only 27% of the LPS-dependent increase in PG-G formation observed in WT cells. Together, these results suggest that deletion of COX-2 results in a reduction in COX-1 activity that appears more profound when exogenous rather than endogenous substrate is used. This reduction in activity affects the utilization of AA more significantly than the utilization of 2-AG. In contrast, deletion of COX-1 appears to have little effect on the ability of COX-2 to convert exogenous AA or 2-AG into PGs, but it markedly reduces PG-G formation from 2-AG. There is no obvious explanation for these results. Possibly, the two COX isoforms synergize in some fashion. More likely, deletion of either isoform leads to compensatory changes in the mice that alter the overall metabolism of the exogenous substrates, resulting in reduced levels of oxygenation by the remaining isoform.
Effect of cPLA2α deletion on PG and PG-G formation
Consistent with prior studies showing that cPLA2α is the major enzyme responsible for the release of AA for PG synthesis [35–40], cPLA2α−/− RPM showed no increases in AA levels following addition of zymosan. AG levels were not diminished in cPLA2α−/− RPM when compared with WT cells, verifying that cPLA2α is not required for 2-AG formation. Furthermore, the finding that PG-G formation is not reduced in cPLA2α−/− RPM strongly suggests that PG-G synthesis occurs via a distinct pathway from that of PG synthesis.
Despite the absence of an elevation of AA, cPLA2α−/− RPM produced significant, although markedly reduced, quantities of PGs. Since RPM possess minimal PG-G hydrolytic activity [27], it is unlikely that the PGs are formed through the degradation of PG-Gs.
We have previously shown that 2-AG is readily hydrolysed in WT RPM, and that the resulting AA is subject to COX-dependent oxygenation [27]. Therefore it is possible that the PGs produced by cPLA2α−/− RPM result from this pathway, and that oxygenation of AA derived from 2-AG may represent a second, although likely minor, pathway for PG formation.
The results presented here demonstrate that the effects of COX isoform deletion are complex, leading to changes in both the expression and activity of the remaining isoform. Changes also likely occur in other aspects of cellular metabolism, including the overall response to LPS pretreatment, which can lead to more global effects on lipid metabolism. For example, we have shown that COX-1−/− RPM produce lower quantities of PGs during the LPS response, leading to a failure of PG-dependent suppression of tumour necrosis factor-α secretion [33]. Consequently, interpretation of the effects of COX isoform deletion on PG and PG-G formation must be made with caution. Despite these concerns, however, there can be little doubt that the present findings strongly support our earlier conclusion that the formation of both PGs and PG-Gs in zymosan-stimulated RPM is primarily COX-1-dependent, even in cells that have been pretreated with LPS to induce COX-2.
Previous in vitro studies showed that COX-1 metabolizes 2-AG less efficiently than AA, whereas COX-2 metabolizes these substrates with similar efficiency [23]. Thus the present results lead to the question of why COX-2 contributes so little to zymosan-dependent PG-G formation. Previous kinetic studies suggest the possibility of rapid inactivation of COX-2, contributing to low levels of PG-G and PG formation in zymosan-stimulated RPM [27]. Since zymosan generates reactive oxygen and nitrogen species, and since macrophage activation up-regulates these processes, it is possible that rapid enzyme inactivation occurs during zymosan phagocytosis, especially in LPS-pretreated cells. Although both COX isoforms are subject to peroxide-dependent inactivation, it remains to be determined if COX-2 is more susceptible than COX-1 to this inactivation in intact cells. The fact that PG-Gs are formed at a much lower ratio than PGs in zymosan-stimulated RPM is consistent with the expectation for a primarily COX-1 dependent process. If this is true, then stimuli that induce 2-AG release with preservation of COX-2 activity may result in much higher levels of PG-G biosynthesis.
The results reported here suggest that COX-2 contributes very little to the response of RPM to zymosan, even in the case of PG-G formation. Earlier, we demonstrated that COX-1 is a major contributor to PG synthesis by RPM responding to LPS, a process generally thought to be totally COX-2-dependent [33]. Notably, McAdam et al. [41] demonstrated a significant role for COX-1 in the response to LPS in humans in vivo. Together, these findings call into question the general assumption that COX-2 is the major pro-inflammatory COX isoform and demonstrate the need for further studies to define conditions leading to the selective activation and/or inactivation of each isoform under inflammatory conditions.
Acknowledgments
We thank Samir Saleh (Vanderbilt University School of Medicine, Nashville, TN, U.S.A.) for the synthesis of deuterated PG-G internal standards for LC/MS/MS analyses. This work was supported by NIH (National Institutes of Health; Bethesda, MD, U.S.A.) grants GM15431, DA06668, HD12304, HD33994 and PO1-CA-77839.
References
- 1.Hamberg M., Samuelsson B. Detection and isolation of an endoperoxide intermediate in prostaglandin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1973;70:899–903. doi: 10.1073/pnas.70.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nugteren D. H., Hazelhof E. Isolation and properties of intermediates in prostaglandin biosynthesis. Biochim. Biophys. Acta. 1973;326:448–461. doi: 10.1016/0005-2760(73)90145-8. [DOI] [PubMed] [Google Scholar]
- 3.Hamberg M., Svensson J., Wakabayashi T., Samuelsson B. Isolation and structure of two prostaglandin endoperoxides that cause platelet aggregation. Proc. Natl. Acad. Sci. U.S.A. 1974;71:345–349. doi: 10.1073/pnas.71.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouzer C. A., Marnett L. J. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 2003;103:2239–2304. doi: 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- 5.Smith W. L., DeWitt D. L., Garavito R. M. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 6.Xie W. L., Chipman J. G., Robertson D. L., Erikson R. L., Simmons D. L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kujubu D. A., Fletcher B. S., Varnum B. C., Lim R. W., Herschman H. R. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 8.O'Banion M. K., Winn V. D., Young D. A. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4888–4892. doi: 10.1073/pnas.89.11.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hla T., Neilson K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herschman H. R. Prostaglandin synthase 2. Biochim. Biophys. Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 11.Vane J. R., Bakhle Y. S., Botting R. M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 12.Smith W. L., Garavito R. M., DeWitt D. L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 13.Picot D., Loll P. J., Garavito R. M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature (London) 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 14.Luong C., Miller A., Barnett J., Chow J., Ramesha C., Browner M. F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 15.Kurumbail R. G., Stevens A. M., Gierse J. K., McDonald J. J., Stegeman R. A., Pak J. Y., Gildehaus D., Miyashiro J. M., Penning T. D., Seibert K., et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature (London) 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 16.Laneuville O., Breuer D. K., Xu N., Huang Z. H., Gage D. A., Watson J. T., Lagarde M., DeWitt D. L., Smith W. L. Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z, 13E/Z, 15Z)-octadecatrienoic acids from alpha-linolenic acid. J. Biol. Chem. 1995;270:19330–19336. doi: 10.1074/jbc.270.33.19330. [DOI] [PubMed] [Google Scholar]
- 17.Chen W., Pawelek T. R., Kulmacz R. J. Hydroperoxide dependence and cooperative cyclooxygenase kinetics in prostaglandin H synthase-1 and -2. J. Biol. Chem. 1999;274:20301–20306. doi: 10.1074/jbc.274.29.20301. [DOI] [PubMed] [Google Scholar]
- 18.Guo Q., Wang L. H., Ruan K. H., Kulmacz R. J. Role of Val509 in time-dependent inhibition of human prostaglandin H synthase-2 cyclooxygenase activity by isoform-selective agents. J. Biol. Chem. 1996;271:19134–19139. doi: 10.1074/jbc.271.32.19134. [DOI] [PubMed] [Google Scholar]
- 19.Greig G. M., Francis D. A., Falgueyret J. P., Ouellet M., Percival M. D., Roy P., Bayly C., Mancini J. A., O'Neill G. P. The interaction of arginine 106 of human prostaglandin G/H synthase-2 with inhibitors is not a universal component of inhibition mediated by nonsteroidal anti-inflammatory drugs. Mol. Pharmacol. 1997;52:829–838. doi: 10.1124/mol.52.5.829. [DOI] [PubMed] [Google Scholar]
- 20.Rieke C. J., Mulichak A. M., Garavito R. M., Smith W. L. The role of arginine 120 of human prostaglandin endoperoxide H synthase-2 in the interaction with fatty acid substrates and inhibitors. J. Biol. Chem. 1999;274:17109–17114. doi: 10.1074/jbc.274.24.17109. [DOI] [PubMed] [Google Scholar]
- 21.Kalgutkar A. S., Crews B. C., Rowlinson S. W., Marnett A. B., Kozak K. R., Remmel R. P., Marnett L. J. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2000;97:925–930. doi: 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu M., Ives D., Ramesha C. S. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 1997;272:21181–21186. doi: 10.1074/jbc.272.34.21181. [DOI] [PubMed] [Google Scholar]
- 23.Kozak K. R., Rowlinson S. W., Marnett L. J. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J. Biol. Chem. 2000;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- 24.Kozak K. R., Prusakiewicz J. J., Rowlinson S. W., Prudhomme D. R., Marnett L. J. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid anandamide. Biochemistry. 2003;42:9041–9049. doi: 10.1021/bi034471k. [DOI] [PubMed] [Google Scholar]
- 25.Kozak K. R., Crews B. C., Morrow J. D., Wang L. H., Ma Y. H., Weinander R., Jakobsson P. J., Marnett L. J. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 2002;277:44877–44885. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- 26.Nirodi C. S., Crews B. C., Kozak K. R., Morrow J. D., Marnett L. J. The glyceryl ester of prostaglandin E2 mobilizes calcium and activates signal transduction in RAW264.7 cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1840–1845. doi: 10.1073/pnas.0303950101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rouzer C. A., Marnett L. J. Glycerylprostaglandin synthesis by resident peritoneal macrophages in response to a zymosan stimulus. J. Biol. Chem. 2005;280:26690–26700. doi: 10.1074/jbc.M501021200. [DOI] [PubMed] [Google Scholar]
- 28.Wang H., Ma W. G., Tejada L., Zhang H., Morrow J. D., Das S. K., Dey S. K. Rescue of female infertility from the loss of cyclooxygenase-2 by compensatory up-regulation of cyclooxygenase-1 is a function of genetic makeup. J. Biol. Chem. 2004;279:10649–10658. doi: 10.1074/jbc.M312203200. [DOI] [PubMed] [Google Scholar]
- 29.Cohn Z. A., Benson B. The in vitro differentiation of mononuclear phagocytes. 3. The reversibility of granule and hydrolytic enzyme formation and the turnover of granule constituents. J. Exp. Med. 1965;122:455–466. doi: 10.1084/jem.122.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonney R. J., Wightman P. D., Davies P., Sadowski S. J., Kuehl F. A., Jr, Humes J. L. Regulation of prostaglandin synthesis and of the selective release of lysosomal hydrolases by mouse peritoneal macrophages. Biochem. J. 1978;176:433–442. doi: 10.1042/bj1760433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kingsley P. J., Rouzer C. A., Saleh S., Marnett L. J. Simultaneous analysis of prostaglandin glyceryl esters and prostaglandins by electrospray tandem mass spectrometry. Anal. Biochem. 2005;343:203–211. doi: 10.1016/j.ab.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Kingsley P. J., Marnett L. J. Analysis of endocannabinoids by Ag+ coordination tandem mass spectrometry. Anal. Biochem. 2003;314:8–15. doi: 10.1016/s0003-2697(02)00643-7. [DOI] [PubMed] [Google Scholar]
- 33.Rouzer C. A., Kingsley P. J., Wang H., Zhang H., Morrow J. D., Dey S. K., Marnett L. J. Cyclooxygenase-1-dependent prostaglandin synthesis modulates tumor necrosis factor-α secretion in lipopolysaccharide-challenged murine resident peritoneal macrophages. J. Biol. Chem. 2004;279:34256–34268. doi: 10.1074/jbc.M402594200. [DOI] [PubMed] [Google Scholar]
- 34.Scott W. A., Pawlowski N. A., Andreach M., Cohn Z. A. Resting macrophages produce distinct metabolites from exogenous arachidonic acid. J. Exp. Med. 1982;155:535–547. doi: 10.1084/jem.155.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gijon M. A., Spencer D. M., Siddiqi A. R., Bonventre J. V., Leslie C. C. Cytosolic phospholipase A2 is required for macrophage arachidonic acid release by agonists that do and do not mobilize calcium. Novel role of mitogen-activated protein kinase pathways in cytosolic phospholipase A2 regulation. J. Biol. Chem. 2000;275:20146–20156. doi: 10.1074/jbc.M908941199. [DOI] [PubMed] [Google Scholar]
- 36.Gijon M. A., Leslie C. C. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J. Leukoc. Biol. 1999;65:330–336. doi: 10.1002/jlb.65.3.330. [DOI] [PubMed] [Google Scholar]
- 37.Bonventre J. V., Huang Z., Taheri M. R., O'Leary E., Li E., Moskowitz M. A., Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature (London) 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 38.Uozumi N., Kume K., Nagase T., Nakatani N., Ishii S., Tashiro F., Komagata Y., Maki K., Ikuta K., Ouchi Y., et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature (London) 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 39.Hirabayashi T., Shimizu T. Localization and regulation of cytosolic phospholipase A2. Biochim. Biophys. Acta. 2000;1488:124–138. doi: 10.1016/s1388-1981(00)00115-3. [DOI] [PubMed] [Google Scholar]
- 40.Dennis E. A. Phospholipase A2 in eicosanoid generation. Am. J. Respir. Crit. Care Med. 2000;161:S32–S35. doi: 10.1164/ajrccm.161.supplement_1.ltta-7. [DOI] [PubMed] [Google Scholar]
- 41.McAdam B. F., Mardini I. A., Habib A., Burke A., Lawson J. A., Kapoor S., FitzGerald G. A. Effect of regulated expression of human cyclooxygenase isoforms on eicosanoid and isoeicosanoid production in inflammation. J. Clin. Invest. 2000;105:1473–1482. doi: 10.1172/JCI9523. [DOI] [PMC free article] [PubMed] [Google Scholar]