

Abstract
The Plasmodium falciparum cysteine peptidases FP-2 (falcipain-2) and FP-3 (falcipain-3), members of the papain-like CAC1 family, are essential haemoglobinases and are therefore potential anti-malarial drug targets. To facilitate a rational drug discovery programme, in the current study we analysed the synthetic substrate and model inhibitor profiles of FP-2 and FP-3 as well as BP-2 (berghepain-2), an orthologue from the rodent parasite Plasmodium berghei. With respect to substrate catalysis, FP-2 exhibited a promiscuous substrate profile based around a consensus non-primeside motif, FP-3 was somewhat more restricted and BP-2 was comparatively specific. Substrate turnover for FP-2 was driven by a basic or acidic P1 residue, whereas for FP-3 turnover occurred predominately through a basic P1 residue only, and for BP-2, turnover was again mainly through a basic P1 residue for some motifs and surprisingly a glycine in the P1 position for other motifs. Within these P1 binding elements, additional recognition motifs were observed with subtle nuances that switched substrate turnover on or off through specific synergistic combinations. The peptidases were also profiled against reversible and irreversible cysteine peptidase inhibitors. The results re-iterated the contrasting kinetic behaviour of each peptidase as observed through the substrate screens. The results showed that the substrate and inhibitor preferences of BP-2 were markedly different from those of FP-2 and FP-3. When FP-2 and FP-3 were compared to each other they also displayed similarities and some significant differences. In conclusion, the in vitro data highlights the current difficulties faced by a peptidase directed anti-malarial medicinal chemistry programme where compounds need to be identified with potent activity against at least three peptidases, each of which displays distinct biochemical traits.
Keywords: cysteine protease, inhibitor, malaria, mapping, substrate
Abbreviations: Abz, 2-amino benzoic acid; AMC, 7-amino-4-methyl coumarin; BP, berghepain; DTT, dithiothreitol; Fmoc/tBu, fluoren-9-ylmethoxycarbonyl/t-butyl; FP, falcipain; FRET, fluorescence resonance energy transfer; HBTU, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOBt, 1-hydroxybenzotriazole; Nle, norleucine; NMM, N-methylmorpholine
INTRODUCTION
Malaria, a disease caused by several species of obligate protozoan parasites, remains one of the most prevalent and persistent diseases to affect the global population, with hundreds of millions of people infected and approx. 1–2 million deaths each year [1]. This situation is compounded by the increasing resistance of the malaria parasites, particularly Plasmodium falciparum, to conventional drugs. There is therefore a great need to identify new therapeutically amenable targets and/or mechanisms with a view to developing small molecule inhibitors and/or vaccines to treat malaria [1].
Malaria parasites utilize a wealth of processes to complete the various stages of their lifecycle [1], for which a range of peptidases are essential for the completion of one or more of these stages [2]. In the human host, processes such as erythrocyte invasion, erythrocyte rupture upon escape, as well as nutrient acquisition by degradation of host proteins, particularly haemoglobin, rely on the action of peptidases [3]. Previous studies have demonstrated the importance of these peptidases, since in the presence of peptidase inhibitors parasite development could be arrested whereas removal of inhibitor resulted in resumption of development. Biochemical studies, complemented by genomic [4] and proteomic studies, have enabled temporal [2] and spatial identification of the various peptidases to be made, allowing their precise biological role(s) to be addressed. To date members of the aspartic, metallo, serine and cysteine peptidase classes have been described from at least one or more species, a subject that has been reviewed previously [3].
The papain-like cysteine peptidases derived from P. falciparum, the most prolific human malaria parasite, are termed FP (falcipains) and comprise four peptidases, namely FP-1 [5], FP-2 [6], FP-2′ [7] and FP-3 [8]. FP-1, located on chromosome 14, shares approx. 38–40% sequence identity with FP-2 and FP-3, and has been implicated in erythrocyte invasion by merozoites [9]. Gene disruption studies suggest that FP-1 is not essential to the erythrocytic stage of P. falciparum [5,10], although gene silencing studies have alluded to a functional biological role for FP-1 [9,11]. FP-2 and FP-2′ are approx. 96% identical, share approx. 68% identity with FP-3 and are encoded on chromosome 11. FP-2, FP-2′ and FP-3 are food vacuole haemoglobinases [6,8,12] and their joint expression, in concert with aspartic peptidases, seems to be key to efficient hydrolysis [8]. All three enzymes have been cloned, over-expressed and biochemically characterized [6,8,12–14]. Gene disruption [15], coupled with inhibitor studies [3], have established a critical role for FP-2 in haemoglobin hydrolysis by P. falciparum and as such it has been pursued as an anti-malarial target. However, it seems that functional redundancy exists between the various falcipain peptidases and therefore, attention has to be paid to more than one enzyme during drug discovery and development [12,16]. Coupled to this, the advancement of compounds inevitably requires demonstration of efficacy, first in cell-based assays and secondly in a relevant disease-related animal model [17]. It is here that cysteine peptidase anti-malarial drug discovery faces a conundrum. The initial animal models of choice are rodent-based, however non-engineered rodents do not support infection by human P. falciparum [18]. Instead, rodents are infected by various species-specific malaria parasites that are employed to model the human disease [17]. The in vitro and in vivo screening cascades are further complicated since the analogous rodent parasite-infected cell-based assays are not currently viable. Thus a rational drug discovery design programme that utilizes optimization of compounds against falcipains both in vitro then through cell-based assays of infection, is subsequently required to show efficacy in rodent-based models where the target peptidase is functionally analogous, but may be biochemically distinct [14]. This potential dilemma has been highlighted previously, prompting the engineering of an immunocompromised mouse capable of hosting P. falciparum infection [18]. Although a major step forward in peptidase anti-malarial discovery, this model is yet to receive widespread application. Reported in the current study is an extensive examination of the synthetic substrate and CAC1 peptidase inhibitor (commercially available and literature-based) profiles for FP-2, FP-3 and BP-2 (berghepain-2). The results clearly indicate that, although FP-2 and FP-3 share some common features, there are both subtle and significant differences in their biochemical behaviour. Moreover, the data also highlights that BP-2 was markedly different from FP-2 and FP-3. Thus, the difficulties faced by a medicinal chemistry and drug discovery programme are clearly evident even at the initial in vitro screening stages. The results of the current study are important if the intent is to develop approaches to simultaneously target both FP-2 and FP-3, and yet demonstrate efficacy by targeting BP-2, a related but distinct enzyme. The data suggests that it would be inappropriate to use the rodent in vivo Plasmodium berghei model for testing falcipain inhibitors unless transgenic parasites, in which the BP-2 gene had been replaced by the falcipain genes, can be used.
EXPERIMENTAL
Materials
Unless otherwise stated, all general chemicals and biochemicals were purchased from Sigma or Fisher Scientific U.K. Detergents (e.g. CHAPS, zwittergents) were purchased from Merck Biosciences U.K. Stock solutions of substrate or inhibitor were made up to 10 mM in 100% DMSO (Rathburns) and diluted in buffer as required. In all cases the DMSO concentration in the assays was maintained at less than 1% (v/v).
Solid-phase peptide synthesis
All solid-phase synthesis was performed using an ‘Fmoc/tBu’ (fluoren-9-ylmethoxycarbonyl/t-butyl) procedure [19] using standard solid-phase synthesis resin washing protocols. All solvents were purchased from ROMIL at SpS (Super Purity Solvent) or ‘Hi-Dry’ grade unless otherwise stated. General peptide synthesis reagents, amino acids and/or derivatives were purchased from Chem-Impex International, Bachem U.K., Merck Biosciences or Neosystems and were of the L-configuration unless otherwise stated. Peptide synthesis was carried out in repetitive cycles consisting of a coupling step, a reagent wash step, a piperidine/DMF (dimethylformamide; 20:80) Fmoc de-protection step, then a further extensive wash step followed by the next coupling round. Between each step excess reagent and solvent were removed by application of a vacuum. Each coupling step was initiated by activating a five molar excess of Fmoc-amino acid (with respect to total solid phase loading capacity) in the presence of a five molar excess HBTU (2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; Merck Biosciences), a five molar excess HOBt (1-hydroxybenzotriazole; Merck Biosciences) and a ten molar excess of NMM (N-methylmorpholine; Sigma).
Synthesis of the spatially addressed FRET (fluorescence resonance energy transfer) substrate library synthesis
The FRET substrate library was based on the utilization of the Abz (2-amino benzoic acid)/Tyr(3-NO2) FRET pair [20]. Solid-phase library syntheses were performed using gears (Mimotope SynPhase GAP 1.3 μmol; Perbio Sciences U.K.) pre-derivatised with RINK-amide linker, in an eight by twelve 96-well microtitre plate format following standard coupling, washing, deprotection and cleavage protocols [21]. Peptide synthesis was based on the manufacturer's protocols (Protocol STN 002-2; http://www.synphase.com/). Using the cyclic repetitive protocol described above, gears were elaborated with coupling cycles of Fmoc–Glu–OH followed by Fmoc–Tyr(3-NO2)–OH (Chem-Impex) to yield NH2–Tyr(3-NO2)–Glu–solid phase. Library construction was carried out using a split-and-mix strategy comprised of four rounds of synthesis (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/399/bj3990047add.htm).
Large-scale peptide synthesis
Large-scale peptide syntheses were performed manually using lanterns (Mimotope SynPhase GAP 8 μmol; Perbio Sciences U.K.) pre-derivatized with RINK-amide linker, following standard coupling, washing, deprotection and cleavage protocols [21]. Upon completion of the sequence, crude product was cleaved with a cocktail of 92.5% TFA (trifluoroacetic acid)/2.5% triisopropylsilane/2.5% water/2.5% ethanedithiol for 90 min after which the resin was removed by filtration and the filtrate concentrated by sparging with nitrogen gas. The crude products were precipitated by addition of 50 ml cold MTBE (methyl t-butyl ether) and precipitates were collected by centrifugation (4960 g for 5 min). The supernatant was discarded and the process repeated. The final precipitate was solubilized using HPLC buffer and purified as required.
Inhibitor compound synthesis
The inhibitors shown in Tables 1 and 2 N-[1-(cyanomet-hylcarbamoyl)cyclohexyl]-4-[1-(2-methoxyethyl)piperidin-4-yl] benzamide (1) [22], (S)-N-[1-(cyanomethylamino)-4-methyl-1-oxopentan-2-yl] biphenyl-4-carboxamide (2) [23], N-(S)-4-methyl-1-oxo-1-{[R]-3-oxo-1-(pyridin-2-ylsulfonyl)azepan-4-ylaminopentan-2-yl}benzofuran-2-carboxamide (3) [23], N-(S)-4-methyl-1-oxo-1-{[S]-3-oxo-1-(pyridin-2-ylsulfonyl)azepan-4-ylaminopentan-2-yl}benzofuran-2-carboxamide (4) [23], benzyl (2S,2′S)-1,1′-[2,2′-carbonylbis(hydrazine-2,1-diyl)]bis(4-methyl-1-oxopentane-2,1-diyl)dicarbamate (5) [24], Cbz-Leu-NH-NH-C(O)-O-Bz (6) [25], Cbz-Leu-NH-NH-C(O)-O-Ph (7) [25] and N-(S)-4-methyl-1-oxo-1-{[S,E]-5-phenyl-1-(phenylsulfonyl)pent-1-en-3-ylaminopentan-2-yl} morpholine-4-carboxamide (LHVS) (12) [26] were synthesized using previously published methods.
Table 1. The inhibition constants of selected reversible compounds against FP-2, FP-3, BP-2 and selected human cathepsin peptidases.
 |
Table 2. Second-order inactivation rate constants for selected irreversible compounds against FP-2, FP-3 and BP-2.
 |
HPLC and HPLC–MS analysis
In all cases, solvent A consisted of 0.1% (v/v) TFA in water and solvent B consisted of 90% acetonitrile mixed with 10% solvent A. Analytical HPLC was conducted using a Jupiter C4 column [5μ, 300 Å (1 Å=0.1 nm), 250 mm×4.6 mm; Phenomenex] on a manual HP1100 system (Agilent Technologies) with data collection at 215 nm. Unless otherwise stated, a linear increasing gradient of 10–90% solvent B over 25 min at 1.5 ml/min was used for column elution. Semi-preparative HPLC purification of crude samples was performed on a Jupiter C4 column [5μ, 300 Å, 250 mm×10 mm; Phenomenex] using a linear increasing gradient of solvent B in solvent A using empirically derived gradients at 4 ml/min. The eluant absorbance was monitored at 230 nm and desired fractions analysed and then freeze-dried (Heto Holten; Thermo Electron).
HPLC–MS analysis was performed on an Agilent HP1100 series LC/MSD machine with a linear increasing gradient of 10–90% solvent B in solvent A over 10 min on a Columbus C8 column (5μ, 300 Å, 50 mm×2.0 mm; Phenomenex) at 0.6 ml/min. Absorbance data were collected using a diode-array detector, and spectra for all peaks were stored as part of the chromatogram. The mass spectrometer was set to API-ES (atmospheric pressure ionization with electrospray) mode, positive polarity; scanning in the 200–2000 Da mass range with the gas temperature set to 350 °C, the nebulizer pressure of 414 kPa and the drying gas flow set at 12 l/min.
The cleavage sites for the FRET peptides were identified by incubating substrate (100 μM) with enzyme at room temperature for 3–6 h prior to analysis by HPLC–MS.
Peptidase activity assays
Unless otherwise stated, all kinetic experiments were performed at 25±0.5 °C. Fluorescence high-throughput assays were carried out in either 384-well microtitre plates (Corning Costar 3705 plates, Fisher Scientific) or 96-well ‘U’ bottomed Microfluor W1 microtitre plates (Thermo Labsystems). Fluorescence assays were monitored using a SpectraMax Gemini fluorescence plate reader (Molecular Devices). Absorbance assays were carried out in flat-bottomed 96-well plates (Spectra; Greiner Bio-One) using a SpectraMax PLUS384 plate reader (Molecular Devices). In all cases, SOFTmax Pro software version 3.1.2 was used for data collection and rate analysis. For substrates employing the coumarin fluorophores, assays were monitored at an excitation wavelength of 365 nm and an emission wavelength of 450 nm and the fluorescence plate reader calibrated with AMC (7-amino-4-methyl coumarin) in assay buffer. For substrates employing an Abz fluorophore, assays were monitored at an excitation wavelength of 310 nm and an emission wavelength of 445 nm; the fluorescence plate reader was calibrated with 2-amino-benzamide (Fluka) in assay buffer. Unless otherwise indicated, all the peptide-coumarin substrates were purchased from Bachem U.K. Z–LR–AMC and Z–VLR–AMC were synthesized by Amura Therapeutics Limited (see Supplementary Information at http://www.BiochemJ.org/bj/399/bj3990047add.htm). The FRET peptide concentrations were calibrated at 445 nm in 20 mM sodium phosphate (pH 9.0) using the nitro-tyrosine chromophore [20].
Recombinant FP-2, FP-3 and BP-2 [6,8,27] were obtained from Professor P. J. Rosenthal, Department of Medicine, University of California, San Francisco, U.S.A. Briefly, proteins were produced in Escherichia coli, solubilized from inclusion bodies, purified by Ni-NTA (Ni2+-nitriloacetate) chromatography, refolded and further purified by anion-exchange (Q-Sepharose) chromatography. Purity was demonstrated by the detection of a single protein band after SDS/PAGE and staining with Coomassie Blue [28]. The active-site titrations of FP-2 and FP-3 were carried out using Mu-Phe-hPhe-FMK (N-methoxysuccinyl-phenylalanyl-homohenylalanyl-fluoromethyl ketone; Sigma), whereas the active titration of BP-2 was carried out using E-64 (Bachem U.K.). Routine peptidase assays were carried out in 100 mM sodium acetate (pH 5.75) containing 1 mM EDTA and 10 mM L-cysteine. Peptidase activity assays were routinely monitored for 30 min and the rates determined by the linear increase in fluorescence signal over time. Initial velocities were used for calculations of the kinetic constants and steady-state velocities for calculations of the inhibition constants. Data were routinely analysed using Prism version 4.03 (GraphPad).
Cathepsin K, B, S (Merck Biosciences), V (R&D Systems) and L (Athens Research and Technology) were assayed according to methods described previously [29].
FRET-based substrate library screening
For high-throughput screening of the FRET substrate library, assays were routinely carried out by dispensing 10 μl of assay buffer followed by 1 μl of a 250 μM solution of the substrate library diluted in 1 mM sodium acetate (pH 5.75). Assays were initiated by the addition of 10 μl of enzyme in assay buffer. Peptidase activity was monitored for 30 min and the rate determined by the increase in fluorescence signal over time. SigmaPlot version 8.0 (Systat Software U.K.) was used to plot the rate data as a colour density array utilizing the colour transition toolbox. Each peptidase dataset was treated independently of the other with zero rates set as black and the maximum observed rate set as yellow.
Measurement of the inhibition constants
The inhibition constant (Ki) for test compounds was determined on the assumption that inhibition was reversible and occurred by a pure-competitive mechanism (see Supplementary Figure 2 at http://www.BiochemJ.org/bj/399/bj3990047add.htm). The Ki values were calculated from the dependence of enzyme activity as a function of inhibitor concentration, by direct non-linear regression analysis (Prism version 4.03) using the equation
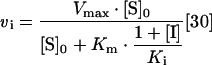 |
where vi is the observed residual activity, Vmax is the observed maximum activity (i.e. in the absence of inhibitor), Km is the apparent macroscopic binding constant for the substrate, [S]0 is the initial substrate concentration, Ki is the dissociation constant and [I] is the inhibitor concentration.
In situations where the dissociation constant (Ki) approached the enzyme concentration, the Ki values were calculated using a quadratic solution in the form described by
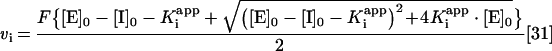 |
In this case vi is the observed residual activity, F is the difference between the maximum activity (i.e. in the absence of inhibitor) and minimum enzyme activity, [E]0 is the total active enzyme concentration, Kiapp is the apparent dissociation constant and Io is the inhibitor concentration. Curves were fitted by non-linear regression analysis (Prism version 4.03) using a fixed value for the enzyme concentration. The relationship
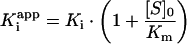 |
was used to account for the substrate kinetics, where Ki is the inhibition constant.
Determination of the rate of reaction for irreversible inhibitors
Where applicable, the concentration dependence of the observed rate of reaction (kobs) of ligand with enzyme was analysed by determining the rate of enzyme inactivation under pseudo-first-order conditions in the presence of substrate. Assays were carried out by the addition of various concentrations of inhibitor to assay buffer containing substrate. Assays were initiated by the addition of enzyme to the reaction mixture and the change in fluorescence monitored over time. During the course of the assay (60 min) less than 10% of the substrate was consumed. The activity fluorescence progress curves were fitted by non-linear regression analysis (Prism version 4.03) using the equation
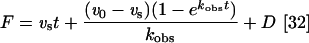 |
where F is the fluorescence response, t is time, v0 is the initial velocity, vs is the equilibrium steady-state velocity, kobs is the observed pseudo first-order rate constant and D is the intercept at time zero (i.e. the ordinate displacement of the curve). The second-order rate constant was obtained from the slope of the line of a plot of kobs versus the inhibitor concentration (i.e. kobs/[I]). The equation
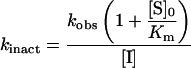 |
RESULTS
Optimization of the peptidase activity assay conditions
In order to facilitate the screening effort, robust assays for each peptidase, amenable to automation and high-throughput format, were required. Using methods previously described [6,8,12,14], we screened a set of approx. 1100 assay conditions encompassing different buffers (e.g. phosphate, Bistris propane), various buffer compositions (e.g. mixed buffers like acetic acid/Mes/Tris), pH ranges from pH 4.5 to 10, as well as the presence of a range of additives (e.g. detergents, polyethylene glycol, glycerol and alkali salts). In all three cases, the highest peptidase activity was observed in the range pH 5–6, with activity decreasing sharply above pH 7.5. Although a number of diverse buffer systems could be accommodated, as determined by enzyme activity, there was no significantly better assay buffer system than that which had been previously reported [6,8]. In contrast to some cathepsins, the presence of detergent(s) did not significantly enhance peptidase activity. Although the inclusion of DTT (dithiothreitol) in the assay buffer produced higher protease activity at relatively lower concentrations, the use of higher concentrations of L-cysteine or 2-mercaptoethanol could compensate for this. We decided to employ L-cysteine as a reductant since, under prolonged incubation, the dithiane oxidation product of DTT [33] quenched the absorbance of the Abz chromophore, especially considering that DTT was employed at millimolar concentrations and the FRET substrates were at micromolar concentrations. Based on these results, the standard assay buffer employed for monitoring all three peptidase activities was 100 mM sodium acetate (pH 5.75) containing 1 mM EDTA and 10 mM L-cysteine.
Substrate screen: AMC-based substrate screen
In order to quickly and efficiently establish an activity assay, a set of 57 peptide–AMC substrates were screened against all three proteases. Coumarin-based substrates provide a sensitive method by which peptidase activity can be monitored. The synthesis of coumarin-based substrates in combinatorial library format has been reported previously [34], and positional scanning AMC library screens have been reported previously for vivapain-2 and vivapain-3 [35] as well as vinckepain-2 [27] along with a wealth of data for individual AMC substrates against FP-2 and FP-3, and to a lesser extent BP-2 [6,8,12,14]. The AMC screening data (Figure 1) are in general agreement with those previously reported but also highlight a number of novel peptide–AMC substrates. The data demonstrated common and unique substrate preferences between the three enzymes. On the basis of the peptide–AMC screening data, with substrates screened at a fixed concentration (10 μM) against all three peptidases, the specific activity of FP-2 was approx. 100-fold greater than that of FP-3 and approx. 10-fold greater than that of BP-2. Substrates utilized by all peptidases included Z–LR–AMC, Z–VLR–AMC and boc–VLK–AMC. Z–LR–AMC was most preferred by FP-2, Z–VLR–AMC was most preferred by FP-3 and BP-2, and boc–VLK–AMC was relatively well catalysed by all three peptidases. Substrates common to two of the three enzymes included Z–LQ–AMC, utilized by FP-2 and FP-3 only; whereas Bz–FVR–AMC, and surprisingly, Ac–DEVD–AMC were utilized by FP-3 and BP-2. In line with its less stringent substrate preference, FP-2 utilized substrates that were not hydrolysed by FP-3 or BP-2, such as Z–FR–AMC, D–VLK–AMC and to a lesser extent Z–LE–AMC, PFR–AMC and Ac–FR–AMC. Z–GPR–AMC was the only substrate unique to FP-3 and Ac–NlePNleD–AMC was the only substrate unique to BP-2, although the rate of catalysis in this latter case was consistently but reproducibly very low (approx. 0.8% of the rate observed with Z–VLR–AMC). As reported previously [14], we found that BP-2 cleaved the tri-peptide substrate Z–VLR–AMC significantly more efficiently than the di-peptide substrates (e.g. Z–LR–AMC). Since the residue positions were fixed relative to the cleavage site for the coumarin-based substrates, this precluded an understanding of prime-side binding information. In this respect, we sought an additional screening method providing synergistic residue interplay and which also spanned the active site. The use of FRET-based peptide substrates [20] was ideal for this purpose.
Figure 1. The utilization of AMC-based peptide substrates by FP-2, FP-3 and BP-2.

The rates of substrate catalysis were determined using fixed concentrations of peptide–AMC substrates (10 μM final concentration) in the presence of either FP-2 (1 nM), FP-3 (10 nM) or BP-2 (5 nM).
Substrate screen: FRET-peptide based substrate screen
We synthesized a 1536 member FRET peptide substrate library (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/399/bj3990047add.htm) that was screened against all three peptidases. In order to better present the wealth of data generated, the rates of fluorescence generation were converted into a colour transition intensity plot from a maximum rate, shown as yellow, to a zero rate, shown as black (Figure 2). The data, presented as the library was synthesized (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/399/bj3990047add.htm), comprised sixteen 96-well plates. Each peptidase data set was processed independently of the others, with the maximum rate observed for each peptidase used as the maximum intensity. A simple visual assessment clearly shows that FP-2 displayed much greater diversity in its substrate preference compared with FP-3, whereas BP-2 displayed more restricted substrate preferences as indicated by significantly fewer peptides displaying substrate activity. In order to avoid an in depth and lengthy description of the data, a summary of the salient features has been presented with the obvious caveat that synergistic exceptions may be present in the data (see Supplementary Table 1 at http://www.BiochemJ.org/bj/399/bj3990047add.htm As a general principle, the presence of particular amino acids (e.g. leucine) at a given position, allowed greater residue tolerance at other positions, whereas the presence of other amino acids (e.g. proline or valine) obliterated substrate activity. Based on the screening data, some of the highest ranking FRET peptide substrates were then selected for kinetic and structural analysis (Table 3). Also, the FRET substrate library did not contain a Leu–Arg containing sequence, and mindful of the peptide–AMC data (Figure 1), a representative set of Leu–Arg containing FRET peptides was additionally synthesized. The results clearly indicated that the Leu–Arg-containing peptides were much better substrates, with second-order rate constants generally 10-fold higher as a result of a reduction in Km as well as an increase in kcat (Table 3). The absence of Leu–Arg-containing peptides in the substrate library may have serendipitously had a benefit, since the cleavage rates for these peptides would have dominated the data so as to mask the more subtle information. FP-2 exhibited a clear preference for arginine and glutamic acid as the P1 residue in combination with leucine and, to a lesser extent, tyrosine as the P2 residue. The β-branched isoleucine was moderately well accepted as the P2 residue whereas the acidic aspartic acid clearly was not. Within this Leu/(Tyr)–Arg/Glu–P1′–P2′ motif many combinations of P1′–P2′ were accommodated with the exception of proline at the P1′ position. FP-3 exhibited a clear preference for arginine or lysine as the P1 residue and, to a lesser extent, 2-thienylalanine, threonine and glutamine in combination with leucine and, to an equal extent, tyrosine as the P2 residue. Clearly not accepted as the P2 residue were the β-branched residue valine and the acidic residue glutamic acid. Within this P3–Leu/Tyr–Arg/Lys/(2-thienylalanine)/(Thr)/(Gln)–P1′ motif, combinations containing proline, aspartic acid and isoleucine at the P1′ position were not accommodated. BP-2 exhibited a clear preference for arginine or lysine and glycine as the P1 residue and occasionally the aromatic 2-thienylalanine residue. The arginine/lysine P1 residue occurs in combination with, predominantly, leucine as the P2 residue but, in stark contrast to FP-3, the β-branched valine was also accommodated. The P1 glycine occurred in combination with, predominantly, the β-branched threonine or aromatic 2-thienylalanine as the P2 residue, and acidic or larger aromatics were clearly not accepted in this particular motif. This preference for P1 glycine is very unusual for papain family peptidases. However a similar result was observed for vinckepain-2 through a positional scanning tetra-peptide AMC library [27].
Figure 2. Graphical representation of the rates of FRET peptide substrate catalysis as intensity plots.

The rates of fluorescence generation (relative fluorescence units/s) from the FRET peptide library screen (10 μM final concentration), in the presence of either FP-2 (1 nM), FP-3 (10 nM) or BP-2 (5 nM), were independently converted into colour-transition plots for each data set. The library format, construction and key have been described in detail elsewhere (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/399/bj3990047add.htm).
Table 3. Kinetic and structural characterization of selected FRET peptide substrates.
The P2 (bold), P1 (white on black background) and P1′ (italics) positions have been highlighted relative to the cleavage site (▼).
 |
Inhibitor profile
To extend the peptide substrate analysis, we tested a number of literature-based inhibitors against FP-2, FP-3, BP-2 and a range of human cathepsin peptidases. The inhibitors were split into two class types based on whether they were reversible or irreversible inhibitors (Tables 1 and 2). As a generalization, the results reflected aspects of the substrate screening data, where certain binding elements (e.g. leucine at the P2 position) were required for recognition and other elements added to selectivity. As with the substrate results, the inhibitor data clearly highlighted differences between the inhibition of FP-2, FP-3 and BP-2 by the various compounds analysed. For example nitrile (1) [22] inhibited FP-2 and BP-2 in approx. the 500 nM range, whereas FP-3 was not significantly inhibited at 2000 nM. Azepanone (4) [23] was a relatively potent inhibitor of FP-2 compared to FP-3 and BP-2, although the diacylcarbohydrazide (5) [36] was even more selective for FP-2 compared to FP-3 and BP-2. The azapeptides (6) and (7) [25] served to highlight the dramatic changes in potency and selectivity due to relatively minor structural changes. The aldehyde leupeptin (8) was a potent inhibitor of all three peptidases. The vinyl sulfone (12) (LHVS) [26], which has been utilized as a probe compound to correlate falcipain inhibition with antimalarial effect [37,38], clearly demonstrated good potency against FP-2, FP-3 and BP-2, but when tested against a number of mammalian cathepsin peptidases it was non-selective (e.g. Ki values versus cathepsin B, S, K, L and V were 314.8±19.9 nM, 0.21±0.05 nM, 1.1±0.1 nM, 10±1 nM, 0.08±0.03 nM respectively). Although in general the augmentation of compounds with potent electrophilic centres resulted in increased potency, these changes tended to reduce specificity, as judged by the inhibition profile against the common human cathepsin peptidases. As an additional analysis of inhibition profile we tested a range of irreversible inhibitors against all three peptidases. The results of the rates of inhibition and relative potencies of the irreversible inhibitors against FP-2, FP-3 and BP-2 were even more marked than those observed with the reversible inhibitors. For example E-64 (9) and E-64c (10) were relatively fast, potent inhibitors of BP-2 and relatively slow, poor inhibitors of FP-3 and FP-2 (Table 2). Conversely, the halomethylketones [11,13–16] (examples of which have been used to correlate the inhibition of falcipains with antimalarial activity [39,40]) were fast, potent inhibitors of FP-2 and FP-3 and relatively slow, poor inhibitors of BP-2.
DISCUSSION
The papain-like cysteine peptidases FP-2 and FP-3 have been shown to play critical roles in haemoglobin hydrolysis as an essential requirement for parasite growth and development. As such they have been investigated as possible therapeutic targets for the treatment of malaria. In order to facilitate a medicinal chemistry programme, we analysed the substrate and inhibitor profiles of the two essential P. falciparum cysteine peptidases and, since the progression of compounds requires demonstration of efficacy in a disease-related animal model, a rodent orthologue of FP-2, termed BP-2, was also investigated.
The screen of AMC-based peptide substrates confirmed previously observed results and we were able to significantly extend the range of substrates analysed. We confirmed that Z–LR–AMC was most efficiently cleaved by FP-2 whereas Z–VLR–AMC was most efficiently cleaved by FP-3 [6,8]. The data obtained for BP-2 also agreed with results reported previously [14], even though this study had only analysed two substrates. New peptide–AMC substrates for all three peptidases were also identified. In this case the data obtained with the peptide–AMC substrate complemented the FRET data and provided valuable information relating to sequences absent from the FRET library. The peptide–AMC substrates also proved to be excellent probes with which to study the effects of the N-terminal capping group on substrate catalysis and specificity. The FRET peptide substrate data clearly demonstrated that FP-2 utilized a greater diversity of substrates than FP-3, and that BP-2 was relatively specific in its substrate preference. In both cases, FP-2 was shown to have significantly higher specific activity against the substrates tested than FP-3. Although a similar observation has been previously reported [8], it has been noted that on a molar basis, FP-3 was a more efficient haemoglobinase than FP-2 [8]. It is therefore possible that the in vitro assay conditions artificially favoured FP-2 peptidase activity, whereas in the natural context these peptidases exhibit equipotent activity against protein substrates, where activity may be affected by the cellular environment [8]. Another consideration is that although FP-2 has been shown to be an efficient haemoglobinase, it is also capable of effectively cleaving erythrocyte membrane ankyrin and protein 4.1 [41]. There is, therefore, biochemical evidence to suggest that FP-2 can accommodate a wider selection of biological substrates, perhaps augmenting its primary role by assisting in other cellular functions. Recent evidence does however strongly suggest the primary role of FP-2 is the digestion of haemoglobin [42]. The homology models of FP-2 and FP-3 [43–45], as well as the structure of FP-2 [42] have been reported previously, however the absence of publicly available crystallographic data prevented a structure-based interpretation of the results. Although it was clear from amino acid sequence alignments that FP-2 and FP-3 were significantly different from BP-2 (results not shown), this data did not greatly facilitate our interpretations without gross assumptions. Irrespective of such interpretations, the data clearly indicated that the FP-2 substrate profile was dominated by leucine in the P2 position and the presence of glutamic acid or especially arginine in the P1 position greatly enhanced the cleavage rates. FP-2 therefore had a specific binding preference for arginine in the P1 position as opposed to just being able to accommodate arginine in this position and also showed a selective preference for glutamic acid when compared with FP-3 and BP-2.
In contrast, the FP-3 substrate preference indicated a more extended substrate binding pocket with a preference for β-branched or hydrophobic residues in the P3 position. In this case the substitution of the P1 residue with arginine had a relatively modest effect on the rate of catalysis, suggesting that other factors augmented substrate binding. Perhaps in this case backbone interactions provided a greater contribution to substrate binding. The FRET peptide substrates most efficiently cleaved by BP-2 were of two distinct classes: one comprised capped tri-peptides and the other comprised capped tetra-peptides with, in the latter cases, an absolute preference for glycine in the P1 position. Once again this suggested an extended binding mode for BP-2 substrates with at least two types of mode being observed. Again perhaps backbone interactions contributed significantly to substrate binding and recognition.
The characterization of the substrate and inhibitor profiles of FP-2, FP-3 and BP-2 will significantly aid the biochemical understanding of the enzymes and also facilitate aspects of the structurebased drug discovery process. Our results emphasize the complexity of parasite biology, highlighting not only subtle differences between highly similar enzymes from the same species but also significant differences between similar enzymes from different species. The malaria cysteine peptidase drug discovery process has therefore to take into account the inhibition of more than one target. Clearly for P. falciparum, FP-2 and FP-3 will need to be inhibited whereas for P. berghei, BP-2 will need to be inhibited. Since we and others have obtained data demonstrating clear and marked differences between these enzymes, consideration has to be given to the idea that a single compound with potent inhibition of FP-2 and FP-3, other than those that contain a potent electrophilic centre, may be difficult to achieve, whilst inhibition of all three peptidases is likely to prove even more challenging. Another level of complication are the variations observed for an enzyme from different parasite strains. Coupled to this, the data would suggest that an inhibitor of FP-2 and/or FP-3 may demonstrate efficacy in a P. falciparum cell-based assay and yet possibly show no efficacy in a P. berghei disease-related animal model. Clearly the current results, as well as other previously published reports, indicate that a degree of caution must be exercised when interpreting data generated from the P. berghei disease-related animal model when testing FP-2 and FP-3 inhibitors. It has been proposed that BP-2 does not fulfil the battery of roles as does FP-2 [14], and it has been suggested that the P. berghei disease-related animal model may not represent an adequately predictive model for the in vivo analysis of P. falciparum inhibitors [14,46]. Although the current animal mouse models provide a framework for the testing and development of anti-malarial therapeutics [17], continued effort should be directed towards the development of more pertinent models (e.g. transgenic or immunocompromised models [18]). Given the current restrictions on the types of cell-based assays and disease related animal models readily available for malaria research, care should be exercised when evaluating the efficacy of compounds, whether they are cysteine protease inhibitors or even other therapeutics targets. In the worst case scenario, it is possible that a compound aimed at treating P. falciparum infection in the human host is rejected on the basis that it shows no efficacy in a P. berghei disease-related mouse animal model.
Online data
Acknowledgments
We thank the staff at Amura Therapeutics Limited for advice and pre-submission manuscript review. We are indebted to Professor P. J. Rosenthal for provision of malarial peptidases, pre-submission manuscript review, advice and support.
References
- 1.Various authors. Nature insight-Malaria. Nature (London) 2002;415:669–715. [Google Scholar]
- 2.Bozdech Z., Llinas M., Pulliam B. L., Wong E. D., Zhu J., DeRisi J. L. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:E5. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenthal P. J. Protease inhibitors. Antimalarial Chemotherapy. In: Rosenthal P. J., editor. Mechanisms of action, resistance, and new directions in drug discovery. Totowa, NJ, U.S.A.: Humana Press; 2001. [DOI] [PubMed] [Google Scholar]
- 4.Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., Carlton J. M., Pain A., Nelson K. E., Bowman S., et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature (London) 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sijwali P. S., Kato K., Seydel K. B., Gut J., Lehman J., Klemba M., Goldberg D. E., Miller L. H., Rosenthal P. J. Plasmodium falciparum cysteine protease falcipain-1 is not essential in erythrocytic stage malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8721–8726. doi: 10.1073/pnas.0402738101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shenai B. R., Sijwali P. S., Singh A., Rosenthal P. J. Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. J. Biol. Chem. 2000;275:29000–29010. doi: 10.1074/jbc.M004459200. [DOI] [PubMed] [Google Scholar]
- 7.Singh N., Sijwali P. S., Pandey K. C., Rosenthal P. J. Plasmodium falciparum: biochemical characterization of the cysteine protease falcipain-2′. Exp. Parasitol. 2006;112:187–192. doi: 10.1016/j.exppara.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Sijwali P. S., Shenai B. R., Gut J., Singh A., Rosenthal P. J. Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochem. J. 2001;360:481–489. doi: 10.1042/0264-6021:3600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenbaum D. C., Baruch A., Grainger M., Bozdech Z., Medzihradszky K. F., Engel J., DeRisi J., Holder A. A., Bogyo M. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science. 2002;298:2002–2006. doi: 10.1126/science.1077426. [DOI] [PubMed] [Google Scholar]
- 10.Eksi S., Czesny B., Greenbaum D. C., Bogyo M., Williamson K. C. Targeted disruption of Plasmodium falciparum cysteine protease, falcipain 1, reduces oocyst production, not erythrocytic stage growth. Mol. Microbiol. 2004;53:243–250. doi: 10.1111/j.1365-2958.2004.04108.x. [DOI] [PubMed] [Google Scholar]
- 11.Malhotra P., Dasaradhi P. V., Kumar A., Mohmmed A., Agrawal N., Bhatnagar R. K., Chauhan V. S. Double-stranded RNA-mediated gene silencing of cysteine proteases (falcipain-1 and -2) of Plasmodium falciparum. Mol. Microbiol. 2002;45:1245–1254. doi: 10.1046/j.1365-2958.2002.03105.x. [DOI] [PubMed] [Google Scholar]
- 12.Singh N., Sijwali P. S., Pandey K. C., Rosenthal P. J. Plasmodium falciparum: biochemical characterization of the cysteine protease falcipain-2′. Exp. Parasitol. 2005;112:187–192. doi: 10.1016/j.exppara.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Sim T. S., Loke P., Lee M. A., Singh M., Flotow H. Cloning and sequence characterization of falcipain-2 from Plasmodium falciparum Gombak A strain (Malaysia) Parasitol. Res. 2001;87:683–686. doi: 10.1007/s004360100444. [DOI] [PubMed] [Google Scholar]
- 14.Chan C., Goh L. L., Sim T. S. Differences in biochemical properties of the Plasmodial falcipain-2 and berghepain-2 orthologues: implications for in vivo screens of inhibitors. FEMS Microbiol. Lett. 2005;249:315–321. doi: 10.1016/j.femsle.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 15.Sijwali P. S., Rosenthal P. J. Gene disruption confirms a critical role for the cysteine protease falcipain-2 in hemoglobin hydrolysis by Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4384–4389. doi: 10.1073/pnas.0307720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahl E. L., Rosenthal P. J. Biosynthesis, localization, and processing of falcipain cysteine proteases of Plasmodium falciparum. Mol. Biochem. Parasitol. 2005;139:205–212. doi: 10.1016/j.molbiopara.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Fidock D. A., Rosenthal P. J., Croft S. L., Brun R., Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug Discov. 2004;3:509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 18.Moreno A., Badell E., Van Rooijen N., Druilhe P. Human malaria in immunocompromised mice: new in vivo model for chemotherapy studies. Antimicrob. Agents Chemother. 2001;45:1847–1853. doi: 10.1128/AAC.45.6.1847-1853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atherton E., Sheppard R. C. Oxford, U.K.: IRL Press; 1989. Solid phase peptide synthesis: a practical approach. [Google Scholar]
- 20.Meldal M., Breddam K. Anthranilamide and nitrotyrosine as a donor-acceptor pair in internally quenched fluorescent substrates for endopeptidases: multicolumn peptide synthesis of enzyme substrates for subtilisin Carlsberg and pepsin. Anal. Biochem. 1991;195:141–147. doi: 10.1016/0003-2697(91)90309-h. [DOI] [PubMed] [Google Scholar]
- 21.Grabowska U., Rizzo A., Farnell K., Quibell M. 5-(hydroxymethyl)oxazoles: versatile scaffolds for combinatorial solid-phase synthesis of 5-substituted oxazoles. J. Comb. Chem. 2000;2:475–490. doi: 10.1021/cc0000186. [DOI] [PubMed] [Google Scholar]
- 22.Missbach M. Dipeptide nitrile cathepsin K inhibitors. In: Organisation W. I. P., editor. World Intellectual Property Organisation. Switzerland: Novartis AG; 2001. [Google Scholar]
- 23.Marquis R. W., Ru Y., LoCastro S. M., Zeng J., Yamashita D. S., Oh H. J., Erhard K. F., Davis L. D., Tomaszek T. A., Tew D., et al. Azepanone-based inhibitors of human and rat cathepsin K. J. Med. Chem. 2001;44:1380–1395. doi: 10.1021/jm000481x. [DOI] [PubMed] [Google Scholar]
- 24.Thompson S. K., Halbert S. M., Bossard M. J., Tomaszek T. A., Levy M. A., Zhao B., Smith W. W., Abdel-Meguid S. S., Janson C. A., D'Alessio K. J., et al. Design of potent and selective human cathepsin K inhibitors that span the active site. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14249–14254. doi: 10.1073/pnas.94.26.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xing R., Hanzlik R. P. Azapeptides as inhibitors and active site titrants for cysteine proteinases. J. Med. Chem. 1998;41:1344–1351. doi: 10.1021/jm970802d. [DOI] [PubMed] [Google Scholar]
- 26.Palmer J. T., Rasnick D., Klaus J. L., Bromme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995;38:3193–3196. doi: 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- 27.Singh A., Shenai B. R., Choe Y., Gut J., Sijwali P. S., Craik C. S., Rosenthal P. J. Critical role of amino acid 23 in mediating activity and specificity of vinckepain-2, a papain-family cysteine protease of rodent malaria parasites. Biochem. J. 2002;368:273–281. doi: 10.1042/BJ20020753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sijwali P. S., Brinen L. S., Rosenthal P. J. Systematic optimization of expression and refolding of the Plasmodium falciparum cysteine protease falcipain-2. Protein Expr. Purif. 2001;22:128–134. doi: 10.1006/prep.2001.1416. [DOI] [PubMed] [Google Scholar]
- 29.Barrett A. J., Rawlings N. D., Woessner J. F. London: Academic Press; 2000. Handbook of proteolytic enzymes. [Google Scholar]
- 30.Cornish-Bowden A. London, U.K.: Portland Press; 2001. Fundamentals of enzyme kinetics. [Google Scholar]
- 31.Stone S. R., Hofsteenge J. Kinetics of the inhibition of thrombin by hirudin. Biochemistry. 1986;25:4622–4628. doi: 10.1021/bi00364a025. [DOI] [PubMed] [Google Scholar]
- 32.Morrison J. F., Walsh C. T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 33.Cleland W. W. Dithiothreitol, a new protective reagent for Sh groups. Biochemistry. 1964;3:480–482. doi: 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- 34.Maly D. J., Leonetti F., Backes B. J., Dauber D. S., Harris J. L., Craik C. S., Ellman J. A. Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J. Org. Chem. 2002;67:910–915. doi: 10.1021/jo016140o. [DOI] [PubMed] [Google Scholar]
- 35.Na B. K., Shenai B. R., Sijwali P. S., Choe Y., Pandey K. C., Singh A., Craik C. S., Rosenthal P. J. Identification and biochemical characterization of vivapains, cysteine proteases of the malaria parasite Plasmodium vivax. Biochem. J. 2004;378:529–538. doi: 10.1042/BJ20031487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bossard M. J., Tomaszek T. A., Levy M. A., Ijames C. F., Huddleston M. J., Briand J., Thompson S., Halpert S., Veber D. F., Carr S. A., et al. Mechanism of inhibition of cathepsin K by potent, selective 1, 5-diacylcarbohydrazides: a new class of mechanism-based inhibitors of thiol proteases. Biochemistry. 1999;38:15893–15902. doi: 10.1021/bi991193+. [DOI] [PubMed] [Google Scholar]
- 37.Rosenthal P. J., Olson J. E., Lee G. K., Palmer J. T., Klaus J. L., Rasnick D. Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrob. Agents Chemother. 1996;40:1600–1603. doi: 10.1128/aac.40.7.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shenai B. R., Lee B. J., Alvarez-Hernandez A., Chong P. Y., Emal C. D., Neitz R. J., Roush W. R., Rosenthal P. J. Structure-activity relationships for inhibition of cysteine protease activity and development of Plasmodium falciparum by peptidyl vinyl sulfones. Antimicrob. Agents Chemother. 2003;47:154–160. doi: 10.1128/AAC.47.1.154-160.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenthal P. J., Lee G. K., Smith R. E. Inhibition of a Plasmodium vinckei cysteine proteinase cures murine malaria. J. Clin. Invest. 1993;91:1052–1056. doi: 10.1172/JCI116262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenthal P. J., Wollish W. S., Palmer J. T., Rasnick D. Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J. Clin. Invest. 1991;88:1467–1472. doi: 10.1172/JCI115456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dua M., Raphael P., Sijwali P. S., Rosenthal P. J., Hanspal M. Recombinant falcipain-2 cleaves erythrocyte membrane ankyrin and protein 4.1. Mol. Biochem. Parasitol. 2001;116:95–99. doi: 10.1016/s0166-6851(01)00306-1. [DOI] [PubMed] [Google Scholar]
- 42.Pandey K. C., Wang S. X., Sijwali P. S., Lau A. L., McKerrow J. H., Rosenthal P. J. The Plasmodium falciparum cysteine protease falcipain-2 captures its substrate, hemoglobin, via a unique motif. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9138–9143. doi: 10.1073/pnas.0502368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sabnis Y., Rosenthal P. J., Desai P., Avery M. A. Homology modeling of falcipain-2: validation, de novo ligand design and synthesis of novel inhibitors. J. Biomol. Struct. Dyn. 2002;19:765–774. doi: 10.1080/07391102.2002.10506783. [DOI] [PubMed] [Google Scholar]
- 44.Sabnis Y. A., Desai P. V., Rosenthal P. J., Avery M. A. Probing the structure of falcipain-3, a cysteine protease from Plasmodium falciparum: comparative protein modeling and docking studies. Protein Sci. 2003;12:501–509. doi: 10.1110/ps.0228103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goh L. L., Sim T. S. Homology modeling and mutagenesis analyses of Plasmodium falciparum falcipain 2A: implications for rational drug design. Biochem. Biophys. Res. Commun. 2004;323:565–572. doi: 10.1016/j.bbrc.2004.08.130. [DOI] [PubMed] [Google Scholar]
- 46.Humphreys M. J., Moon R. P., Klinder A., Fowler S. D., Rupp K., Bur D., Ridley R. G., Berry C. The aspartic proteinase from the rodent parasite Plasmodium berghei as a potential model for plasmepsins from the human malaria parasite, Plasmodium falciparum. FEBS Lett. 1999;463:43–48. doi: 10.1016/s0014-5793(99)01597-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.