

Abstract
Virtually nothing is known about the interaction of co-translationally active chaperones with nascent polypeptides and the resulting effects on peptide conformation and folding. We have explored this issue by NMR analysis of apomyoglobin N-terminal fragments of increasing length, taken as models for different stages of protein biosynthesis, in the absence and presence of the substrate binding domain of Escherichia coli Hsp70, DnaK-β. The incomplete polypeptides misfold and self-associate under refolding conditions. In the presence of DnaK-β, however, formation of the original self-associated species is completely or partially prevented. Chaperone interaction with incomplete protein chains promotes a globally unfolded dynamic DnaK-β-bound state, which becomes folding-competent only upon incorporation of the residues corresponding to the C-terminal H helix. The chaperone does not bind the full-length protein at equilibrium. However, its presence strongly disfavors the kinetic accessibility of misfolding side-routes available to the full-length chain. This work supports the role of DnaK as a “holder” for incomplete N-terminal polypeptides. However, as the chain approaches its full-length status, the tendency to intramolecularly bury non-polar surface efficiently out-competes chaperone binding. Under these conditions, DnaK serves as a “folding enhancer” by supporting folding of a population of otherwise folding-incompetent full-length protein chains.
Keywords: apomyoglobin, Hsp70, protein folding, DnaK, chaperone
Introduction
Proteins are vectorially synthesized from N to C terminus in living cells. The role of molecular chaperones in polypeptide conformation and folding during translation is of paramount importance for understanding how proteins achieve their three-dimensional structure as they are biosynthesized.1,2 Yet, our current knowledge on how molecular chaperones participate in co-translational folding is limited.
The best-studied chaperone system is the bacterial Hsp70, DnaK. Despite the extensive available information about its biology, biochemistry and functional steps,3–5 there is still little direct evidence about how the binding of Hsp70 to nascent polypeptides assists co-translational and immediately post-translational protein folding.6,7 The published work on DnaK's potential role in folding is mainly based on low-resolution biochemical and immunological assays in cell-free systems. Additionally, in vitro studies utilizing purified components have shown that DnaK exhibits specific binding to non-polar amino acid stretches often flanked by positively charged residues.8
The studies described above and a few biophysical data on the role of DnaK in folding, performed on either small peptides9,10 or relatively large multi-domain proteins (or protein domains),11,12 led to the currently accepted view that DnaK prevents misfolding by binding to the non-polar residues of a protein chain. In the case of full-length proteins or independent protein domains, these experiments are based on DnaK binding to heat-unfolded substrate, followed by analysis at lower temperature. On the other hand, translation occurs at constant temperature and binding to chaperones, for most soluble single-domain proteins, is primarily needed before a polypeptide has reached its full-length status. Therefore, there is still an urgent need to corroborate the above qualitative views by high-resolution biophysical studies, at constant temperature, directly addressing how the DnaK chaperone binds N-terminal incomplete protein sequences rich in non-polar residues. In addition, it is important to provide a correlation between these binding modes and the ability to generate a fully folded protein, as chain elongation gets completed. The present work makes the first steps in this direction.
Three hypotheses can be formulated on the conformational fate of DnaK-bound elongating chains, according to the general models given in Figure 1. These models omit, for simplicity, other co-translationally active chaperones such as the trigger factor (TF), which is usually present during translation in bacteria13,14 and may impose further structural constraints, but is not essential for protein folding.15 Model I postulates that DnaK binding “holds” elongating polypeptides in a globally unfolded state, possibly comprising some local secondary structure, until near completion of protein biosynthesis. Model II proposes that DnaK binding keeps specific polypeptide regions in a temporarily idle state until complementary non-polar folding subunits are biosynthesized and have a chance to promote collapse into structured modules. Finally, model III indicates that this holding function may be necessary only as the polypeptide emerges from the ribosome, to delay folding until a later stage of biosynthesis. This mechanism might apply to any protein and it is particularly appealing for multi-domain proteins or single-domain proteins with distinct sub-domains. Both models II and III allow tertiary structure to form cotranslationally. The models described above are neither necessarily mutually exclusive nor exhaustive of all the possible options. They are primarily meant to convey the most viable mechanistic hypotheses accounting for either the presence or absence of tertiary structure during co-translational folding.
Figure 1.
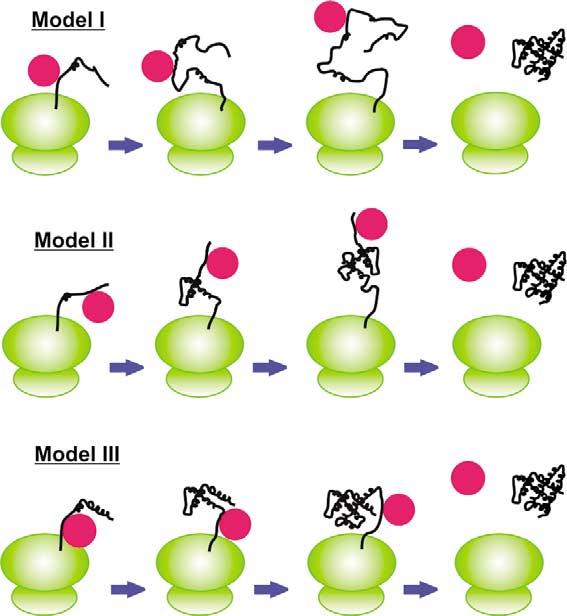
Cartoon representation of co-translational protein folding models in the presence of Hsp70 chaperones (red circles). Ribosomes and nascent polypeptides are drawn in green and black, respectively. The stoichiometry of substrate–chaperone binding illustrates a representative example in this scheme and is not meant to be mandatory.
In order to study which of the above models best describes the role of Hsp70, we have examined the effect of DnaK-β on the conformation of apomyoglobin (apoMb), a representative soluble single-domain all-a-helical protein, and its N-terminal fragments of increasing length, which are known to misfold and self-associate in the absence of chaperone to a degree inversely proportional to their fractional chain length.2 The structure of apoMb and its N-terminal fragments used in this work are illustrated in Figure 2. apoMb is characterized by two significant sets of long-range interactions. The first is a series of contacts among its N-terminal A helix and its C-terminal H helix, and the second comprises interactions among residues belonging to the B and G helices. These topological constraints argue in favor of model II, which allows the chaperone to “hold” the residues in helix A until biosynthesis of the residues corresponding to helix H is complete. Similar arguments apply to the collapse of helices B and G, which might occur once the residues belonging to the G helix have been synthesized. Shorter-range sub-domain-type interactions are present among helices B and E, and G and H, supporting folding according to model III.
Figure 2.
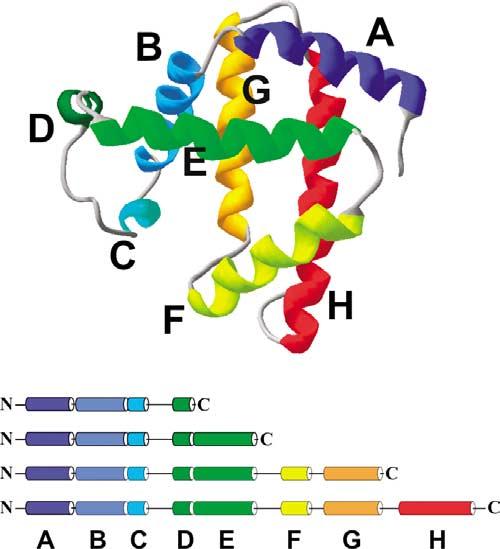
Ribbon diagram illustrating the three-dimensional structure of sperm whale myoglobin. The helices are color-coded and labeled A to H (from N to C terminus). Three-dimensional coordinates were derived from the PDB file of carbonmonoxymyoglobin.48,49 The image was created with the SwissPdbViewer software.50 The bottom panel represents the N-terminal fragments investigated in this work. The chain length and color coding match that of the corresponding helices in the native state.
The fate of the apoMb N-terminal polypeptides has been followed until evolution to full-length natively folded chain, in the absence and presence of the substrate-binding domain of bacterial Hsp70, DnaK-β. This is the smallest DnaK subunit retaining full substrate-binding ability. The structure of a DnaK-β construct similar to the one employed here was previously investigated by NMR9 and found to be entirely similar to the complex of DnaK-β with a small model peptide substrate,10 and to a related free chaperone construct containing an additional helical lid sub-domain.16 Although the affinity of DnaK-β for small peptide substrates is 15-fold weaker than that of full-length DnaK in the presence of ATP,9 DnaK-β is able to convert from an open to a closed conformation upon substrate binding, similarly to the full-length chaperone.17,18 DnaK-β is used here as a model for the Hsp70 chaperones, consistently with several prior biophysical investigations on the substrate-binding role of DnaK.9,10,16
Results and Discussion
Gel analysis of chaperone binding to N-terminal fragments
All the N-terminal fragments investigated here have specific potential binding sites for DnaK, as we predicted using the algorithm reported by Bukau and co-workers.8 This prediction has been confirmed experimentally by cellulose-bound apoMb peptide library scanning (to be published elsewhere).
In order to investigate binding of DnaK-β to apoMb N-terminal polypeptides and gain specific experimental evidence for complex formation, native gel analysis was carried out. The apoMb polypeptides were analyzed in the absence and presence of chaperone (100 μM each, 1:1 molar ratio), under conditions similar to those employed in the NMR experiments. Figure 3 shows the resulting native gel bands at pH 5.8 and 4 °C. The presence of DnaK-β in the refolding buffer significantly alters the properties of the N-terminal incomplete chains. As shown by the gel band shifts, these are predominantly recruited to different species, in the presence of DnaK-β (Figure 3). In contrast, the sample containing full-length apoMb and DnaK-β displays two distinct bands corresponding to free protein and chaperone, indicating that there is no complex formation at equilibrium.
Figure 3.
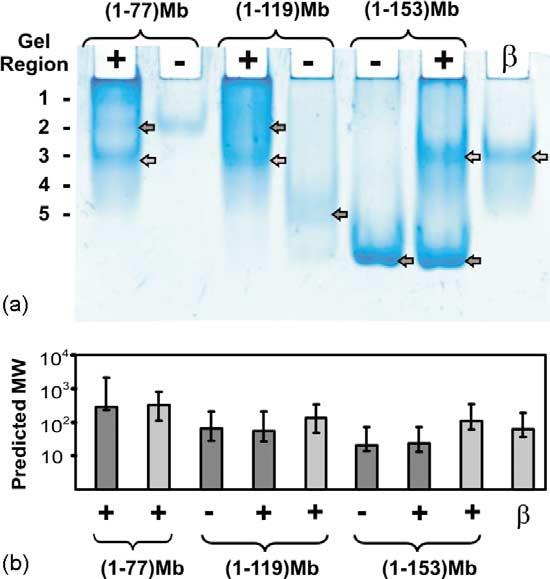
Native gel analysis (non-denaturing 7.5% gel) of N-terminal apoMb fragments. (a) Peptides of increasing length in the absence and presence of DnaK-β. The signs -, + and β denote pure polypeptide fragment, polypeptide–DnaK-β mixture, and pure DnaK-β, respectively. The numbers on the left denote the gel regions analyzed by SDS-PAGE in Figure 4. The bands used for Ferguson analysis are labeled by arrows. Light gray denotes bands whose running endpoints match those of free DnaK-β. (b) Results of Ferguson analysis for gel bands with arrows in (a). The plot displays the predicted apparent molecular mass for each species.
In order to further investigate the presence of complex formation and the nature of populated species, native gels bands were analyzed by denaturing polyacrylamide gels (SDS-PAGE). The denaturing conditions cause disruption of intermolecular complexes and separation of the species that co-migrate in the native gel, allowing their unambiguous identification. Gel bands from different native gel regions (labeled in Figure 3; horizontal regions numbered from top to bottom) were excised, cut into small pieces and incubated in 10% (w/v) SDS. The gel pieces were then analyzed by SDS-PAGE. As shown in Figure 4, the denaturing gel distinctly separates apoMb fragments and DnaK-β chaperone into individual bands, showing whether the two species co-migrate under non-denaturing conditions. Specifically, the denaturing gel shows that pure (1–77)apoMb, detected only in one native gel is able to spread across regions 1 to 3 in the presence of DnaK-β. Moreover, the relative amounts of both N-terminal polypeptide and chaperone in region 2 increase relative to their pure forms, indicating formation of a complex. Similarly for (1–119)apoMb, most of the polypeptide population in regions 4 and 5, and the chaperone population in region 3, are recruited to regions 1 and 2, in the sample containing both apoMb fragment and chaperone. We also carried out a similar analysis on full-length apomyoglobin in the absence and presence of chaperone, as a control (data not shown). No significant changes in the amounts of species present in different region of the native gel were observed, for the free and mixed samples, confirming the absence of a complex at equilibrium. In summary, the native gel data illustrate the presence of complex formation between DnaK-β and incomplete N-terminal fragments. This conclusion was further supported by analysis of gel bands by in-gel tryptic digestion followed by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. MALDI peaks corresponding to both polypeptide and chaperone were identified in the gel bands corresponding to the intermolecular complex. The polypeptide–chaperone complexes are peculiar, in that they do not appear as a unique band in native gels, but they are spread over more than one gel region. This supports the presence of either multiple complexes with different stoichiometry or a unique complex with intrinsic conformational heterogeneity. A combination of the above is also possible. The broad appearance of the gel bands supports the presence of conformational/site exchange on a timescale comparable to the protein migration timescale (millisecond to second). Therefore, we denote the chaperone–polypeptide association as a “dynamic complex”, in light of the above-mentioned characteristics.
Figure 4.

SDS-PAGE analysis of bands excised from different regions (listed in Figure 3) of a native gel similar to that in Figure 3. The excised gel pieces were incubated in 10% SDS, and then loaded on a 16.5% Tris–tricine SDS-PAGE gel. Detection was done by silver staining.
Protein migration in native gels depends on molecular mass, charge and shape. In order to factor out the electrostatic effect and study the size of different species, Ferguson analysis was carried out.19,20 Samples were run on native gels of different polyacrylamide concentration and compared to several protein standards. Analysis of the shortest (1–60)apoMb fragment is precluded by its low solubility. Figure 3(b) shows that the remaining fragments do not largely differ in terms of overall size in the absence and presence of DnaK-β. This is consistent with the partial (or total) compensation of the decrease in mass associated with preventing aggregation, by the increase in mass acquired upon complex formation with DnaK-β. Results for full-length apoMb are consistent with the presence of a predominantly monomeric protein chain both in the presence and absence of chaperone, at equilibrium.
NMR characterization of N-terminal fragments
The conformation of 15N-labeled N-terminal apoMb fragments of increasing length was investigated at 4 °C under three different conditions (unfolded state at pH 2.5, and refolding conditions into pH 6.0 buffer, either in the presence or absence of DnaK-β) by 1H,15N sensitivity-enhanced Carr–Purcell–Meiboom–Gill heteronuclear single quantum coherence (SE-CPMG-HSQC).21 The data collected for different polypeptide chain lengths, starting from identical stock solutions, are displayed in matrix format in Figure 5. SE-CPMG-HSQC is a powerful probe of global polypeptide backbone conformation, due to the high sensitivity of 1H and 15N resonance frequencies to electronic environment. The CPMG pulse train factors out line-broadening contributions due to chemical exchange occurring in the upper-microsecond to millisecond timescale regime. Therefore, chemical exchange is ruled out as a primary source of spectral line broadening. Initial binding data for (1–77)apoMb in the presence of either DnaK-β or full-length DnaK showed that resonances are broader and several peaks are missing in the case of full-length DnaK (data not shown). Therefore, we focused on DnaK-β for high-resolution spectroscopic analysis.
Figure 5.

1H,15N-SE-CPMG-HSQC spectra for N-terminal apoMb polypeptides and full-length protein under different conditions, at low pH and at pH 6 in the absence and presence of DnaK-β chaperone. Row labels denote polypeptide lengths, and column labels denote experimental conditions. Labels to columns 2 and 3 denote refolding conditions starting from a polypeptide (pH 2.5) stock solution.
The HSQC data at pH 2.5 (Figure 5) display small chemical shift dispersion in the 1H dimension, illustrating the predominantly unfolded nature of all the fragments. All peptides are well behaved and soluble under these conditions, and give rise to intense NMR signals with the expected number of resonances, which increase as a function of chain elongation, as expected. Although all species are globally unfolded at low pH, some residual secondary structure may be present, as shown for full-length22 and (1–77)apoMb (S.C. et al., unpublished results).
The N-terminal apoMb fragments self-associate and form non-native b-sheet structure at physiologically relevant pH.2 The degree of misfolding and self-association inversely correlates with the extent of chain elongation. The shortest fragment, (1–60)apoMb aggregates heavily at NMR concentrations, with visible precipitation and no detectable NMR signal. All other free peptides are completely water-soluble. Yet, only a fraction of the expected peaks, i.e. ∼20% and ∼50%, is present in the spectra of (1–77)- and (1–119)apoMb at pH 6.0, respectively. Notably, the (1–119) fragment only lacks the residues belonging to the C-terminal H helix. All detectable resonances display poor 1H chemical shift dispersion. A striking variation in spectral features occurs when the polypeptide is further elongated to include the amino acid residues corresponding to the C-terminal H helix. A spectrum with large 1H chemical shift dispersion is obtained, consistent with the presence of a folded protein. The last C-terminal residues make important long-range tertiary contacts with the rest of the chain to stabilize the folded native structure.23 Hence, incorporation of the residues corresponding to the H helix triggers a compelling driving force for intramolecular folding, leading to tertiary structure formation.
The effect of DnaK-β chaperone on the conformational characteristics of the N-terminal fragments was probed by refolding the 15N-labeled fragments into pH 6.0 buffer containing a stoichiometric amount of unlabeled DnaK-β. The spectral features of the polypeptides improve significantly at all chain lengths (Figure 5, columns 2 and 3), relative to the samples lacking DnaK-β. About ten new resonances become detectable upon refolding (1–60)apoMb in the presence of DnaK-β and the sample retains partial solubility over time. The effect of DnaK-β on the SE-CPMG-HSQC spectrum of (1–77)apoMb is remarkable, resulting in the appearance of new resonances, leading to the recovery of nearly all the expected cross-peaks. The number of detectable resonances for (1–119)apoMb does not vary; upon addition of DnaK-β, however, peak intensities significantly increase.
The small chemical shift dispersion in the 1H dimension for the observable resonances indicates a lack of global tertiary structure for the incomplete chains in the presence of chaperone. Therefore, DnaK prevents misfolding but it does not cause any detectable tertiary folding within incomplete polypeptide chains. This effect on polypeptide conformation may be of general significance, and it may be shared by other chaperone families.24
Additional insights are provided by a comparison of the intensities of well-resolved resonances, for data acquired in the absence and presence of DnaK-β (Figure 6). An average increase in peak intensity by 141(±42)% and 117(±86)% is observed for (1–77)apoMb and (1–119)apoMb, respectively, when DnaK-β is present in the refolding buffer. These results are consistent with the presence of DnaK-β leading to an increased population of chaperone-bound protein. Key supporting evidence for this mechanism is provided by the native and denaturing gel data discussed in the previous section, which indicate the presence of significant populations of chaperone-bound incomplete protein chains. The net effect produced by the above action of DnaK-β is a minimization of the extent of intermolecular misfolding, for these apoMb fragments.
Figure 6.

Comparison of 1H,15N SE-CPMG-HSQC resonance intensities for (1–119) and full-length apoMb at pH 6.0 in the absence and presence of DnaK-β. The bar diagrams display the percentage intensity variations in the presence of chaperone.
In the case of full-length apoMb, there are virtually no variations in either the number of CPMG-HSQC resonances or their chemical shift values, in both the absence and presence of DnaK-β (Figure 5). The large 1H chemical shift dispersion is consistent with the presence of a folded protein. In addition, the HSQC spectrum overlaps with the reported spectrum for native apoMb.25 Therefore, the NMR data show that all the spectroscopically detectable population of full-length protein is correctly folded. This is consistent with the native gel analysis, which indicates that, within the detection limits of the technique, full-length apoMb is not bound to the chaperone at equilibrium. The NMR resonance intensities for refolded full-length apoMb increase by an average 36(±15)% when DnaK-β is present in the refolding buffer (Figure 6). Since we found no evidence of chaperone-bound apoMb at equilibrium, we attribute this increase in intensity to the correct folding of an enhanced population offull-length protein, leading to higher overall refolding yields. DnaK-β must act transiently during the refolding process, given that native gel analysis proves that DnaK-β is not bound to the full-length protein at equilibrium. The presence of a rescuable misfolded population of apoMb in the absence of chaperone is consistent with the fact that chaperone-free apoMb is known to exhibit some degree of self-association upon refolding.26
NMR line broadening and polypeptide dynamics
1H,15N HSQC spectra of most N-terminal polypeptides contain peaks with a wide range of intensities, some broadened beyond detection. NMR line broadening is caused by either transverse relaxation or chemical exchange in the micro- to millisecond timescale. In order to learn more about the origin for the unusual line-shape distribution, we collected T2 relaxation data and assessed the role of micro- to millisecond chemical exchange by relating NMR parameters from SE-HSQC and SE-CPMG-HSQC experiments. We focused on a representative polypeptide, i.e. (1–119)apoMb, refolded at pH 6.0 in the absence of chaperone, given its sharpest resonancesamong all the chaperone-free polypeptides. We observed a wide range of R2 values for different (1–119)apoMb residues (Figure 7(a)).
Figure 7.

(a) T2 relaxation data for refolded (1–119)apoMb at pH 6.0 and 4 °C. Error bars denote T2 curve fitting errors. (b) Ratio of intensities for CPMG-HSQC21 and reference SE-HSQC27 resonances, ICPMG/IREF for refolded (1–119)apoMb at pH 6.0.
The CPMG pulse trains in SE-CPMG-HSQC reduce de-phasing of spin coherence due to exchange processes on the upper micro- to millisecond timescale. This minimizes exchange broadening. The distribution of the intensity ratios for SE-HSQC21 relative to SE-CMPG-HSQC27 (ICPMG/IREF) for (1–119)apoMb is shown in Figure 7(b). A similar analysis has also been carried out for (1–77)apoMb. While very few residues have ICPMG/IREF ratios significantly greater than 1, indicating the presence of chemical exchange, the majority of the amino acid residues have a ICPMG/IREF ratio approximately equal to 1, indicating that the contribution of conformational exchange in the upper micro- to millisecond timescale is small. Thus, the observed line broadening arises mainly from slow local and (or) global tumbling and not from conformational exchange. As a consequence, the wide-ranging R2 values support the presence of nano- to picosecond motions.
The SE-CPMG-HSQC NMR spectra for the N-terminal fragments show that the broadened or missing resonances in the absence of chaperone often correspond to the broader or missing peaks upon complex formation with chaperone. This phenomenon deserves further investigation.
Characterization of incomplete apoMb chains by NMR diffusion experiments
The translational diffusion coefficients D for unfolded and refolded (1–77), (1–119) and full-length apoMb in the absence of DnaK-β are reported in Figure 8. Specific numerical values and relevant equations are provided in Materials and Methods. At pH 2.5, D decreases as chain length increases, as expected for a constant molecular shape. The scaling law of polymer theory shows that the radius of gyration (Rg), which is proportional to the hydrodynamic radius (RH), is related to the number of monomers (N) according to Rg=aNv, where a is a constant and v=0.6 for a “good” solvent behaving as a Gaussian chain.28,29 The hydrodynamic radii for the unfolded species, determined from experimentally measured D values, were fit to RH=bNv, where N is number of residues and b is a constant. The v=0.65 best fit value is consistent with predictions for an expanded homopolymer in a good solvent. Therefore, all polypeptides behave as expanded chains at pH 2.5. Global fits using experimental data for a large number of different unfolded proteins resulted in a reported v value of 0.57 ± 0.02.30 Although a relatively small number of data points for a single protein chain were used in the current fit, the v value is in reasonable agreement with the theoretical value for expanded chains, and experimental value for unfolded proteins. This result highlights a general property of the unfolded state ensemble, and is compatible with the finding by Lietzow et al.,31 who identified the presence of transient long-range tertiary contacts in acid-unfolded apoMb. The combined evidence from our data, prior light-scattering experiments32 and Lietzow et al.31 concur in defining full-length acid unfolded apoMb as a predominantly expanded Gaussian chain in dynamic equilibrium with a smaller population of compact conformations. Our results also show that the expanded chain nature is common to both full-length protein and incomplete N-terminal chains.
Figure 8.
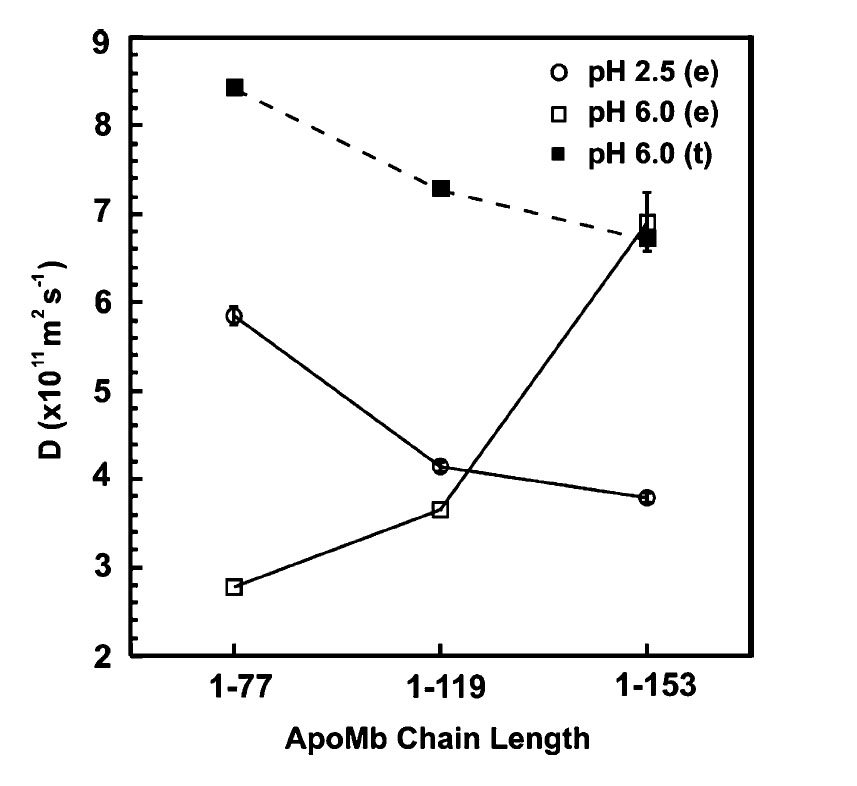
Diffusion coefficients D for (1–77)-, (1–119)- and full-length (1–153)apoMb at pH 2.5 (○) and pH 6.0 (□) determined experimentally (e) at 4 °C. Theoretical (t) D values calculated for a globular monomer at pH 6.0 (■).
In contrast with the pH 2.5 case, D increases with fragment length under refolding conditions (Figure 8). D values at pH 6.0 are lower than those at pH 2.5, indicating that hypothetical monomeric fragments would be more unfolded at pH 6.0 than at pH 2.5. Since this is not possible, we deduce that the fragments are self-associated. BPPSTE-HSQC33 data collection for (1–77)- and (1–119)apoMb could only be performed once, due to the long data collection times imposed by the poor NMR signal/noise displayed by these samples at pH 6.0.
The apparent molecular mass of full-length apoMb, obtained from the experimentally determined D value (using equations (1) and (2)), is 16,080(±2240) Da. This value is in good agreement, within experimental error, with the predicted mass of 15N-labeled apoMb, i.e. 17542 Da (at 98% 15N isotope enrichment). The apparent molecular masses of (1–77)- and (1–119)apoMb at pH 6.0 are 248 kDa and 109 kDa, corresponding to a 28-mer and an 8-mer, respectively. In summary, incomplete apoMb fragments behave as large self-associated species under refolding conditions, and the degree of self-association decreases as fragment length increases, in the absence of chaperone.
The combined transverse relaxation and diffusion data indicate that incomplete apoMb chains behave as relatively large self-associated species with spectroscopically detectable flexible regions experiencing motions in the pico- to nanosecond timescale regime. Residues in the core tumble very slowly and are broadened beyond detection. The observed resonances correspond to the most dynamic portions of the polypeptide chain, and the narrow ∼1 ppm 1H chemical shift dispersion indicates that these highly dynamic residues lack a well-defined tertiary structure. This is consistent with a model envisioning the self-associated chaperone-free fragments as made of a bulky relatively rigid core with highly flexible globally unfolded regions. Further relaxation and diffusion experiments in the presence of chaperone are underway and will be reported elsewhere.
Insights into the interaction of DnaK-β with N-terminal polypeptides of increasing length
The poor 1H-NMR chemical shift dispersion shows that there is no defined tertiary structure for the chaperone-bound apoMb N-terminal polypeptides of incomplete chain length, up to 119 residues. This rules out models II and III (Figure 1), since they imply the presence of well-defined tertiary interactions. Our data are consistent with model I, which envisions DnaK as a molecular timer, acting to postpone folding until late stages of chain elongation. Some of the missing resonances in DnaK-β-bound (1–119)apoMb might, in principle, be involved in motionally restricted spectroscopically undetectable tertiary structure. While this possibility cannot be ruled out, it appears unlikely that stable native-like tertiary structure modules (comprising B–G and B–E helices) might form, since more resonances than the missing ones would be affected. The general aspects for the chaperone–polypeptide interaction supported by our studies are illustrated in Figure 9. We propose that the substrate-binding domain of the chaperone interacts with incomplete chains unable to autonomously fold into a stable monomeric tertiary structure. The extent of interaction with the chaperone correlates with the tendency to misfold and self-associate in the absence of chaperone. When interaction with a particular chaperone is not effective at completely preventing aggregation (as for (1–60)apoMb), other chaperones are likely needed. As the polypeptide chain elongates to a sufficient degree to fold independently and achieve a stable tertiary structure, DnaK-β exerts its action only transiently, and it disassociates from the full-length folded protein at equilibrium.
Figure 9.

Proposed model illustrating the thermodynamic and kinetic aspects of the interaction between DnaK-β chaperone and elongating polypeptide chains, up to full-length protein.
The results reported here complement our knowledge on another well-studied Escherichia coli chaperone, SecB. This chaperone is involved in translocation and it is able to capture its substrates by kinetic partitioning, before they fold.34,35 In so doing, SecB keeps its substrates in a translocation-competent unfolded conformation.36 The model presented here shares some common features with SecB, in that it proposes that the nascent chain DnaK substrates are kept in a globally unfolded conformation by the chaperone until the polypeptide chain acquires the ability to fold.
The regulatory role of the ATPase domain of DnaK on binding affinity37,38 and folding is not addressed here and is beyond the scope of this study. However, it is expected that the qualitative trends as a function of polypeptide chain elongation identified here will serve as a representative bench-mark to further understand the role of full length DnaK within its regulatory cycle. Future studies utilizing full-length DnaK are needed to address this issue experimentally.
Conclusions
This study highlights DnaK's chain length-dependent “holder” function,1 i.e. its ability to prevent aggregation by interacting with solvent-accessible non-polar substrate regions. The resulting conformational ensemble is globally unfolded, and it acquires tertiary folding competence only upon generation of a full-length protein chain. It is important to note at this point that, while a globally unfolded chaperone-bound conformation best matches the available data and the mechanistic models proposed in Figure 1, the presence of a molten globular chaperone-bound conformation cannot be completely ruled out. For instance, the molten globular state of apomyoglobin at pH 4.0 has a backbone amide proton chemical shift dispersion similar to that of its unfolded state.22 The kinetic relationships between protein folding and DnaK's functional cycle require further studies utilizing the whole DnaK chaperone system, including co-chaperones and Mg-ATP. However, the demonstrated ability by DnaK-β to promote folding of full-length apoMb (by preventing the misfolding of a folding-incompetent subpopulation; Figure 5, lower panels) shows that this chaperone substrate-binding domain is indeed able to exert a “folding enhancer” action on full-length protein chains. Here we regard the term folding enhancer as denoting the ability to promote the folding of a polypeptide chain that has the intrinsic ability to become native but is unable to do so without the transient assistance by the chaperone. Future investigations are needed to explore whether Hsp70 acts by lowering the activation barriers for productive folding (via substrate binding), thereby serving as an enzyme, or whether its role results from simple kinetic partitioning. This work provides an initial glimpse at the chain length-dependent folding of a single-domain soluble protein in the presence of the co-translationally active chaperone DnaK. It also opens new avenues to more detailed structural investigations at atomic resolution, possibly taking into account the effect of other cellular factors (e.g. co-chaperones, nucleotides) influencing the evolution of polypeptide conformation as the chain elongates.
Materials and Methods
Protein expression and purification
Full-length apoMb and its N-terminal fragments were overexpressed in E. coli and purified as described.2,33 Isotopically labeled fragments were obtained by growing the E. coli cells in 15N-enriched M9 minimal medium. The DNA sequence encoding for substrate-binding domain of DnaK (DnaK-β; residues 393–505 of full length DnaK, numbered including the initial Met) was constructed from the wild-type dnaK gene by PCR amplification and subcloning into a pET16b vector (Novagen, Madison, WI). The construct includes an N-terminal His-tag (MGHHHHHHHHHHSSGHIEGRH). Identity of the subcloned DnaK-β was confirmed by DNA sequencing. DnaK-β was overexpressed in E. coli BL21(DE3) cells according to standard methods. The protein is produced as inclusion bodies. The purification was carried out using Ni-chelating resin under denaturing conditions, according to standard manufacturer's instructions (Qiagen, Valencia, CA). The purified protein was refolded by drop-wise dilution into excess 10 mM sodium acetate buffer (pH 6.0), followed by concentration and overnight dialysis against the same buffer.
Native gels
Non-denaturing PAGE gels were run on a continuous McLellan buffer system at pH 5.8.39 Gels were run with a vertical electrophoresis apparatus (Bio-Rad, Hercules, CA) at a constant voltage of 200 V for 35 min at 4 °C. Ferguson plot analysis was performed on native gels of different acrylamide percentages, employing pararosanilin (Sigma-Aldrich Corp., St. Louis, MO) as tracking dye. Ferguson plots were constructed to map protein mobility (relative to tracking dye) versus gel percentage. The plot slopes are known to correlate with the apparent molecular mass of the proteins.19,20 Pyruvate kinase (237 kDa), GAPDH (144 kDa), creatine phosphokinase (81 kDa), proteinase K (29 kDa) and horse heart Mb (17 kDa) were used as standards to construct the calibration curve.
SDS-PAGE analysis of native gel bands
Gel bands from different horizontal regions of a native gel similar to the one shown in Figure 3 were excised. The sides of the band were excluded, since they may contain spurious protein smearing. The gel was stained with Coomassie dye using SimplyBlue Safestain (Invitrogen Corp., Carlsbad, CA). The gel pieces were chopped into small pieces and incubated overnight in 10% (w/v) SDS-containing gel loading buffer, and then loaded onto a three-layered 16.5%(w/v) Tris–tricine SDS-PAGE gel. The gel was run at 100 V for 100 min, and silver-stained using the SilverSNAP Stain kit (Pierce, Rockford, IL).
Mass spectrometry analysis
In-gel tryptic digest for protein band identification was performed at the mass spectrometry facility in University of Wisconsin Biotechnology Center. Gel bands were excised as explained for SDS-PAGE analysis. Peptide fingerprint analysis was performed on a Bruker BIFLEX III MALDI time-of-flight (MALDI-TOF) mass spectrometer (Bruker Daltonics, Billerica, MA). The expected masses of the proteolytic fragments were estimated using the PeptideMass tool of ExPASy.40
While several of the predicted peaks were detected experimentally, a subset of the fragments could not be observed. This is not surprising, and it is likely due to either inherent instrumental limitations or post-proteolysis peptide degradation. In addition, we observed a few fragments that did not match the predictions, likely the result of incomplete proteolysis or further degradation of the expected fragments. Yet, the results enabled unequivocal protein identification in the gel samples.
NMR sample preparation
NMR samples were prepared starting from an identical stock solution of 15N-labeled unfolded polypeptides in HCl at pH 2.5. All NMR samples were in 20 mM sodium acetate, 5 mM KCl and 5% (v/v) 2H2O adjusted to the final pH, unless otherwise stated. The final concentration of the apoMb fragments was 100 μM.
pH 2.5 samples
Aliquots of the polypeptide stock solutions were diluted to their final concentrations at pH 2.5.
pH 6.0 samples
Polypeptides were diluted into sodium acetate buffer at pH 6.0. The solution pH was adjusted to 6.0 with 0.1 M KOH.
pH 6.0 DnaK-β containing samples
Polypeptides were diluted in sodium acetate buffer (pH 6.0) which included DnaK-β. The final pH value was adjusted as described above. Final DnaK-β concentration was 100 μM. NMR samples were equilibrated at 4 °C overnight prior to data acquisition. The concentration of polypeptide stock solutions were determined spectrophotometrically using the experimentally determined extinction coefficients ε280=890 cmK1 MK1, ε280= 9305 cmK1 MK1, ε280=14,134 cmK1 MK1 for (1–60)- and (1–77)apoMb, (1–119)apoMb, full-length (1–153)apoMb, respectively. The DnaK-β concentration was determined by a Coomassie reagent protein assay (Pierce, Rockford, IL), based on a calibration obtained according to amino acid analysis (University of Iowa Molecular Analysis Facility).
NMR spectroscopy
All experiments were performed on a Varian INOVA-600 MHz NMR spectrometer at 4 °C. Varian triple resonance probes, i.e. either 1H{13C, 15N} triple axis gradient or 1H{31P, X} single axis gradient, were used for diffusion, SE CPMG-HSQC21 and SE-HSQC27 experiments. 15N transverse relaxation experiments were performed using a Varian triple resonance 1H{13C, 15N} triple axis gradient cold probe. The NMRPipe41 and NMRView42 software packages were used for 2D NMR data processing. Spectral widths were set to 8000 Hz and 2370 Hz in the 1H and 15N dimensions, respectively, unless otherwise specified. Relaxation delay between scans was set to 1.7 s for CPMG-HSQC and SE-HSQC experiments and to 1.5 s for diffusion and T2 relaxation experiments.
CPMG-HSQC experiments
1H,15N HSQC spectra were acquired using the SE CPMG-HSQC pulse sequence. Data were acquired either as 256(t1) × 4096(t2) or as 192(t1) × 4096(t2) total points, apodized with an unshifted Gaussian window function and zero filled twice in both dimensions.
NMR transverse relaxation measurements
The 15N transverse relaxation rate (R2) was acquired using the pulse sequence described by Farrow et al.43 Data were recorded as 256(t1) × 2048(t2) total points at each T2 relaxation delay of 10, 30, 50, 70, 90, 110, 130 and 150 ms. It was then apodized with an unshifted Gaussian window function and zero filled to 512(t1) × 4096(t2) total points. Spectral widths were set to 8000 Hz and 1900 Hz in 1H and 15N dimensions, respectively. The maximum peak height was used in fitting relaxation data to a single-exponential two parameter fit using NMRView software.42 Reported T2 error values reflect the deviation from the best fit curve. Resonances adequately fitting to a single exponential whose R2 values were larger than one standard deviation were discarded and not included in the analysis.
Diffusion experiments
Diffusion coefficients were determined by 1H detected BPPSTE-HSQC.33 15N chemical shift evolution delays were not incremented, to yield a 1D version of the pulse sequence. Gradient amplitudes were calibrated according to published methods,44 using the known H2HO diffusion coefficient in 2H2O at 5 °C.45 Diffusion delays were set to 253 ms for (1–77)- and (1–119)apoMb at pH 6.0, and to 153 ms for full-length apoMb at pH 6.0, respectively. Diffusion-encoding gradients (g0) ranged from 0 to 60 G cmK1.
Data processing was performed by the VNMR software (Varian) on a Sun workstation. No window functions were used except for (1–77)apoMb at pH 6.0, where, due to a poor signal-to-noise ratio, a 5.86 Hz line broadening coupled to exponential multiplication was applied. Diffusion coefficients were measured from the slope of ln(I/I0) versus plots, where I denotes the peak intensity at a particular g0 value, and I0 denotes the signal intensity for g0=0. Observed intensities were determined by peak area integration. Baseline correction was applied prior to integration.
Molecular mass estimation from diffusion data
The hydrodynamic radius RH of a molecule is related to its translation diffusion coefficient D according to:
| 1 |
where kB is the Boltzmann constant, T is the temperature, η is the solution viscosity and F is the Perrin shape factor. For a sphere, F=1. The apparent molecular mass M can be determined from RH via the relation:46
| 2 |
where NA is Avogadro's number, V̄2 is the specific volume of the target molecule, δ-1 is the mass of bound hydration water per gram of molecule and V̄2 is the specific volume of water. For globular proteins, typical values of V̄1 and δ-1 are 0.73 cm3 g-1 and 0.3 g H2O (g protein)-1, respectively.46
The translational diffusion coefficients D for refolded (1–77)-, (1–119)- and full-length (1–153)apoMb, in the absence of DnaK-β at pH 6.0, are 2.78 × 10-11 m2 s-1, 3.65 × 10-11 m2 s-1 and 6.91 ± 0.32 × 10-11 m2 sK1, respectively.
The error in D measurements was calculated as ±1 standard deviation from the mean of three independent measurements. Average intensities and their associated errors were plotted as a function of . Least-squares linear fits were then performed by the Excel software package (Microsoft). Errors in the “best fit” slopes were calculated as one half of the difference between the slopes of the two limiting “worst-fit” lines covering the error bars of all points.
Apparent molecular masses M calculated from equation (2) may overestimate the actual aggregation state in case molecular shapes deviate significantly from spherical. For instance, modeling a self-associated species as an ellipsoid, rather than a sphere, yields a lower apparent aggregation state, since ellipsoids have a Perrin shape factor F greater than 1,47 and a lower D value, i.e. they translationally diffuse more slowly, than corresponding spherical objects of identical molecular mass.46
Acknowledgements
This research was supported by National Institutes of Health grant GM068535-01A1, by the Research Corporation (Research Innovation Award to S.C.) and by the American Heart Association (postdoctoral fellowship to N.K.). We are grateful to Elizabeth A. Craig and Linda K. Nicholson for useful comments about this work. We thank Carolina Vega, Meng-Tse Lee, Scott Veglahn, Ye-Jin Eun, Lisa M. Jungbauer and Grzegorz Sabat for technical assistance.
Footnotes
- HSQC
- heteronuclear single quantum coherence
- CPMG
- Carr–Purcell–Meiboom–Gill
- SE
- sensitivity enhanced
- apoMb
- apomyoglobin
Edited by K. Kuwajima
References
- 1.Agashe V, Hartl F. Roles of molecular chaperones in cytoplasmic protein folding. Semin. Cell Dev. Biol. 2000;11:15–25. doi: 10.1006/scdb.1999.0347. [DOI] [PubMed] [Google Scholar]
- 2.Chow C, Chow C, Rhagunathan V, Huppert T, Kimball E, Cavagnero S. The chain length dependence of apomyoglobin folding: structural evolution from misfolded sheets to native helices. Biochemistry. 2003;42:7090–7099. doi: 10.1021/bi0273056. [DOI] [PubMed] [Google Scholar]
- 3.Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- 4.Teter S, Houry W, Ang D, Tradler T, Rockabrand D, Fischer G, et al. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 1999;197:755–765. doi: 10.1016/s0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 5.Mayer M, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slepenkov S, Witt S. The unfolding story of the Escherichia coli Hsp70 DnaK: is DnaK a holdase or an unfoldase? Mol. Microbiol. 2002;45:1197–1206. doi: 10.1046/j.1365-2958.2002.03093.x. [DOI] [PubMed] [Google Scholar]
- 7.Deuerling E, Bukau B. Chaperone-assisted folding of newly synthesized proteins in the cytosol. Crit. Rev. Biochem. Mol. Biol. 2004;39:261–277. doi: 10.1080/10409230490892496. [DOI] [PubMed] [Google Scholar]
- 8.Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellecchia M, Montgomery D, Stevens S, Vander Kooi C, Feng H, Gierasch L, Zuiderweg E. Structural insights into substrate binding by the molecular chaperone DnaK. Nature Struct. Biol. 2000;7:298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- 10.Stevens S, Cai S, Pellecchia M, Zuiderweg E. The solution structure of the bacterial HSP70 chaperone protein domain DnaK(393–507) in complex with the peptide NRLLLTG. Protein Sci. 2003;12:2588–2596. doi: 10.1110/ps.03269103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder H, Langer T, Hartl F, Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palleros D, Shi L, Reid K, Fink A. Hsp70–protein complexes–complex stability and conformation of bound substrate protein. J. Biol. Chem. 1994;269:13107–13114. [PubMed] [Google Scholar]
- 13.Horwich A. Cell biology: sight at the end of the tunnel. Nature. 2004;431:520–522. doi: 10.1038/431520a. [DOI] [PubMed] [Google Scholar]
- 14.Ferbitz L, Maier T, Patzelt H, Bukau B, Deuerling E, Ban N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature. 2004;431:590–596. doi: 10.1038/nature02899. [DOI] [PubMed] [Google Scholar]
- 15.Kramer G, Patzelt H, Rauch T, Kurz T, Vorderwulbecke S, Bukau B, Deuerling E. Trigger factor peptidyl-prolyl cis/trans isomerase activity is not essential for the folding of cytosolic proteins in Escherichia coli. J. Biol. Chem. 2004;279:14165–14170. doi: 10.1074/jbc.M313635200. [DOI] [PubMed] [Google Scholar]
- 16.Zhu XT, Zhao X, Burkholder W, Gragerov A, Otaga C, Gottesman M, Hendrickson W. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erbse A, Mayer M, Bukau B. Mechanism of substrate recognition by Hsp70 chaperones. Biochem. Soc. Trans. 2004;32:617–621. doi: 10.1042/BST0320617. [DOI] [PubMed] [Google Scholar]
- 18.Popp S, Packschies L, Radzwill N, Vogel KP, Steinhoff HJ, Reinstein J. Structural dynamics of the DnaK–peptide complex. J. Mol. Biol. 2005;347:1039–1052. doi: 10.1016/j.jmb.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 19.Hedrick J, Smith A. Size and charge isomer separation and estimation of molecular weights of proteins by disc gel electrophoresis. Arch. Biochem. Biophys. 1968;126:155–164. doi: 10.1016/0003-9861(68)90569-9. [DOI] [PubMed] [Google Scholar]
- 20.Chrambach A, Rodbard D. Polyacrylamide gel electrophoresis. Science. 1971;172:440–451. doi: 10.1126/science.172.3982.440. [DOI] [PubMed] [Google Scholar]
- 21.Mulder F, Spronk C, Slijper M, Kaptein R, Boelens R. HSQC improved experiments for the observation of exchange broadened signals. J. Biomol. NMR. 1996;8:223–228. doi: 10.1007/BF00211169. [DOI] [PubMed] [Google Scholar]
- 22.Eliezer D, Yao J, Dyson H, Wright P. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nature Struct. Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 23.Cavagnero S, Kurt N.Murphy R, Tsai A.Folding and misfolding as a function of polypeptide chain elongation: conformational trends and implications for intracellular events Misbehaving Proteins: Protein (Mis)Folding, Aggregation and Stability 2005Springer; New York: In the press [Google Scholar]
- 24.Rudiger S, Freund S, Veprintsev D, Fersht A. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc. Natl Acad. Sci. USA. 2002;99:11085–11090. doi: 10.1073/pnas.132393699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eliezer D, Wright P. Is apomyoglobin a molten Globule? Structural characterization by NMR. J. Mol. Biol. 1996;263:531–538. doi: 10.1006/jmbi.1996.0596. [DOI] [PubMed] [Google Scholar]
- 26.Chow C, Kurt N, Murphy R, Cavagnero S.Structural characterization of apomyoglobin self-associated species in aqueous buffer and urea solution Biophys. J 2005. In the press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992;114:10663–10665. [Google Scholar]
- 28.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; Ithaca, NY: 1953. [Google Scholar]
- 29.De Gennes PG. Cornell University Press; Ithaca, NY: 1979. Scaling Concepts in Polymer Physics. [Google Scholar]
- 30.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 31.Lietzow M, Jamin M, Dyson H, Wright P. Mapping long-range contacts in a highly unfolded protein. J. Mol. Biol. 2002;322:655–662. doi: 10.1016/s0022-2836(02)00847-1. [DOI] [PubMed] [Google Scholar]
- 32.Gast K, Damaschun H, Misselwitz R, Muller-Frohne M, Zirwer D, Damaschun G. Compactness of protein molten globules: temperature-induced structural changes of the apomyoglobin folding intermediate. Eur. Biophys. J. 1994;23:297–305. doi: 10.1007/BF00213579. [DOI] [PubMed] [Google Scholar]
- 33.Rajagopalan S, Chow C, Raghunathan V, Fry CG, Cavagnero S. NMR spectroscopic filtration of polypeptides and proteins in complex mixtures. J. Biomol. NMR. 2004;29:505–516. doi: 10.1023/B:JNMR.0000034354.30702.de. [DOI] [PubMed] [Google Scholar]
- 34.Topping T, Randall L. Chaperone SecB from Escherichia coli mediates kinetic partitioning via a dynamic equilibrium with its ligands. J. Biol. Chem. 1997;272:19314–19318. doi: 10.1074/jbc.272.31.19314. [DOI] [PubMed] [Google Scholar]
- 35.Hardy S, Randall L. A kinetic partitioning model of selective binding of nonnative proteins by the bacterial chaperone SecB. Science. 1991;251:439–443. doi: 10.1126/science.1989077. [DOI] [PubMed] [Google Scholar]
- 36.Randall L, Hardy S. SecB, one small chaperone in the complex milieu of the cell. Cell. Mol. Life Sci. 2002;59:1617–1623. doi: 10.1007/PL00012488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayer MP, Schroder H, Rudiger S, Paal K, Laufen T, Bukau B. Multistep mechanism of substrate binding determines chaperone activity of HSP70. Nature Struct. Biol. 2000;7:586–593. doi: 10.1038/76819. [DOI] [PubMed] [Google Scholar]
- 38.Schmid D, Baici A, Gehring H, Philipp C. Kinetics of molecular chaperone action. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 39.McLellan T. Electrophoresis buffers for polyacrylamide gels at various pH. Anal. Biochem. 1982;126:94–99. doi: 10.1016/0003-2697(82)90113-0. [DOI] [PubMed] [Google Scholar]
- 40.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins M, Appel R, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker J, editor. The Proteomics Protocols Handbook. Humana Press; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Johnson B, Blevins R. NMRView–a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 43.Farrow N, Muhandiram R, Singer A, Pascal S, Kay S, Gish G, et al. Backbone dynamics of a free and a phosphopeptide-complexed SRC homology-2 domain studies by N-15 NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 44.Antalek B. Using pulsed gradient spin echo NMR for chemical mixture analysis: how to obtain optimum results. Conc. Magn. Reson. 2002;14:225–258. [Google Scholar]
- 45.Mills R. Self-diffusion in normal and heavy water in the range 1–45 degrees. J. Phys. Chem. 1973;77:685–688. [Google Scholar]
- 46.Cantor CR, Schimmel PR. Biophysical Chemistry, II. W. H. Freeman and Company; New York: 1980. [Google Scholar]
- 47.Scheraga HA. Protein Structure. Academic Press; New York: 1961. [Google Scholar]
- 48.Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, et al. The Protein Data Bank. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuriyan J, Wilz S, Karplus M, Petsko G. X-ray structure and refinement of carbon-monoxy (Fe II)-myoglobin at 1.5 Å resolution. J. Mol. Biol. 1986;192:133–154. doi: 10.1016/0022-2836(86)90470-5. [DOI] [PubMed] [Google Scholar]
- 50.Guex N, Peitsch M. SWISS-MODEL and the Swiss-PdbViewer: an enviroment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]