

Abstract
In myotonic dystrophy (DM1), both inactivation of muscleblind proteins and increased levels of CUG-BP1 are reported. These events have been shown to contribute independently to aberrant splicing of a subset RNAs. We demonstrate that steady-state levels of the splice regulator, hnRNP H, are elevated in DM1 myoblasts and that increased hnRNP H levels in normal myoblasts results in the inhibition of insulin receptor (IR) exon 11 splicing in a manner similar to that observed in DM1. In normal myoblasts, overexpression of either hnRNP H or CUG-BP1 results in the formation of an RNA-dependent suppressor complex consisting of both hnRNP H and CUG-BP1, which is required to maximally inhibit IR exon 11 inclusion. Elevated levels of MBNL1 show RNA-independent interaction with hnRNP H and dampen the inhibitory activity of increased hnRNP H levels on IR splicing in normal myoblasts. In DM1 myoblasts, overexpression of MBNL1 in conjunction with si-RNA mediated depletion of hnRNP H contributes to partial rescue of the IR splicing defect. These data demonstrate that coordinated physical and functional interactions between hnRNP H, CUG-BP1 and MBNL1 dictate IR splicing in normal and DM1 myoblasts.
Keywords: CUG-BP1, hnRNP H, muscleblind, myotonic dystrophy 1, splicing
Introduction
Myotonic dystrophy 1 (DM1) is a multisystem disorder characterized by skeletal muscle disease, cardiac conduction disorders, endocrine dysfunction, which includes insulin resistance and psychiatric disease (Harper, 1989). The genetic defect in DM1 is a CTG repeat expansion, which is located in the 3′untranslated region of a protein kinase, DMPK, and found immediately 5′ of a homeo-domain encoding gene, SIX5 (Brook et al, 1992; Fu et al, 1992; Mahadevan et al, 1992; Boucher et al, 1995). Repeat expansion results both in the aggregation of the mutant DMPK RNA into abnormal nuclear foci and in decreased DMPK and SIX5 levels (Fu et al, 1993; Taneja et al, 1995; Klesert et al, 1997; Thornton et al 1997). Functional inactivation of Dmpk and Six5 in mice demonstrates that decreased levels of either gene results in a unique subset of DM1 features (Reddy and Paul, 2006). These data support the hypothesis that decreased DMPK and SIX5 levels contribute to DM1 pathology. Importantly, several lines of evidence demonstrate that expression of expanded CUG repeats per se plays an important role in the development of DM1 (Mankodi et al, 2000; Liquori et al, 2001; Seznec et al, 2001). Although, the mechanism of CUG RNA toxicity has yet to be completely elucidated, altered splice patterns in a subset of physiologically important RNAs appears to be an important pathological consequence of expanded CUG repeat expression in DM1 cells (Philips et al, 1998; Savkur et al, 2001; Buj-Bello et al, 2002; Charlet-B et al, 2002; Mankodi et al, 2002; Kimura et al, 2005).
The toxicity of CUG repeats has been hypothesized to result both from the abnormal sequestration of physiologically important proteins by the expanded CUG repeats and by downstream signaling events, which may alter the levels or function of one or more proteins involved in RNA processing. To date, the muscleblind (MBNL) family of proteins consisting of MBNL1, MBNL2 and MBNL3 have been shown to be sequestered by the expanded CUG repeats and are therefore functionally inactivated in DM1 cells (Miller et al, 2000; Fardaei et al, 2002). MBNL1 and MBNL2 are expressed at relatively high levels in muscle; however, MBNL3 expression is restricted primarily to the placenta (Fardaei et al, 2002). Other data demonstrate that level of the CUG-binding protein (CUG-BP1), the prototype of the CELF family of splicing regulators, is elevated in DM1 myoblasts as a consequence of CUG repeat expression (Timchenko et al, 1996, 2001; Ladd et al, 2001). Importantly, inactivation of MBNL1 or overexpression CUG-BP1 has been shown to result in identical splicing defects in a subset of RNAs (Kanadia et al, 2003; Ho et al, 2004, 2005; Dansithong et al, 2005). However, the numbers of splice regulators that serve to set the aberrant splice patterns in DM1 cells and their possible interactions in normal and DM1 cells are currently unknown.
In a previous study, Kim et al (2005) have demonstrated that the splice regulator, hnRNP H, binds in conjunction with a docking protein to both the CUG repeats and a splicing branch point in the DMPK RNA in vitro. In this study, we show that the steady-state level of hnRNP H is elevated in DM1 myoblasts. Increased hnRNP H levels are not causally related to the sequestration and loss of function of the muscleblind proteins in DM1 myoblasts. We demonstrate that elevated hnRNP H levels in normal myoblasts results in altered insulin receptor (IR) exon 11 splicing in a manner that duplicates the defects observed in DM1 myoblasts. These results therefore show that elevated hnRNP H levels contribute to aberrant IR splicing in DM1 myoblasts. Suppression of IR exon 11 splicing by elevated hnRNP H levels requires endogenous CUG-BP1 levels and conversely elevated levels of CUG-BP1 requires endogenous hnRNP H levels for maximum suppression of IR exon 11 inclusion. As both hnRNP H and CUG-BP1 demonstrate RNA-dependent interaction both in vivo and in vitro, these data demonstrate that elevated levels of either hnRNP H or CUG-BP1 results in the formation of an RNA-dependent suppressor complex that is required to maximally inhibit IR exon 11 inclusion in normal myoblasts. We further show that elevated levels of MBNL1 partially rescues the aberrant IR splice pattern resulting from the overexpression of hnRNP H in normal myoblasts. As our results demonstrate that MBNL1 binds to hnRNP H in an RNA-independent manner in normal myoblasts and effectively recruits hnRNP H to intranuclear CUG foci in DM1 myoblasts, these data are consistent with the hypothesis that MBNL1 physically binds to and dampens the inhibitory effect of elevated hnRNP H on IR splicing in normal myoblasts. Consistent with results obtained in normal myoblasts overexpression of MBNL1 in conjunction with siRNA-mediated inactivation of hnRNP H results in a partial rescue of the IR splicing defect in DM1 cells. Taken together, these results demonstrate that finely coordinated physical and functional interactions between hnRNP H, CUG-BP1 and MBNL1 serve to regulate aberrant IR splicing in DM1 myoblasts.
Results
Steady-state hnRNP H levels are elevated in DM1 myoblasts
To test the possible role of hnRNP H in DM1 muscle disease, we measured the levels of hnRNP H in both normal and DM1 myoblasts. We observe that steady-state levels of hnRNP H are ∼2.3- and ∼2.1-fold elevated in two DM1 myoblast lines when compared to the two normal myoblast lines (Figure 1A and E). A similar increase in CUG-BP1 levels (∼2.4- and ∼1.9-fold higher) was observed in the two DM1 myoblast lines studied when compared to normal myoblast lines (Figure 1A and E). The levels of other RNA regulatory proteins including hnRNP A1, SF2/ASF and polyadenylate binding protein nuclear 1 (PABPN1) were measured in both normal and DM1 myoblasts. The steady-state levels of hnRNP A1, SF2/ASF and PABPN1 were however not altered in DM1 myoblasts when compared to normal myoblasts (Supplementary Figure S1). As myoblast lines can be contaminated by fibroblasts, in other experiments, we transduced normal ((CTG)17) and DM1 ((CTG)167 and (CTG)667) fibroblasts with adenoviral vectors expressing both MyoD and green fluorescent protein (GFP) in order to confer a skeletal muscle cell fate on these cells. Fluorescent microscopic observation of fibroblasts 24 h after transduction demonstrated that ∼95% (±5%) of the transduced cells expressed GFP (data not shown). This second approach therefore ensures the homogeneity of the cells that are being examined. Endogenous hnRNP H and CUG-BP1 levels were elevated in DM1 fibroblasts when compared to normal fibroblasts transduced with MyoD expressing viruses. Specifically, both hnRNP H and CUG-BP1 levels increased in a CTG tract length dependent fashion showing ∼1.5- and ∼2.1-fold (hnRNP H) and ∼1.7- and ∼2.5-fold (CUG-BP1) increase in MyoD transduced DM1 fibroblasts encoding CTG167 and CTG667, respectively, when compared to control fibroblasts encoding CTG17 repeats (Figure 1B and E).
Figure 1.
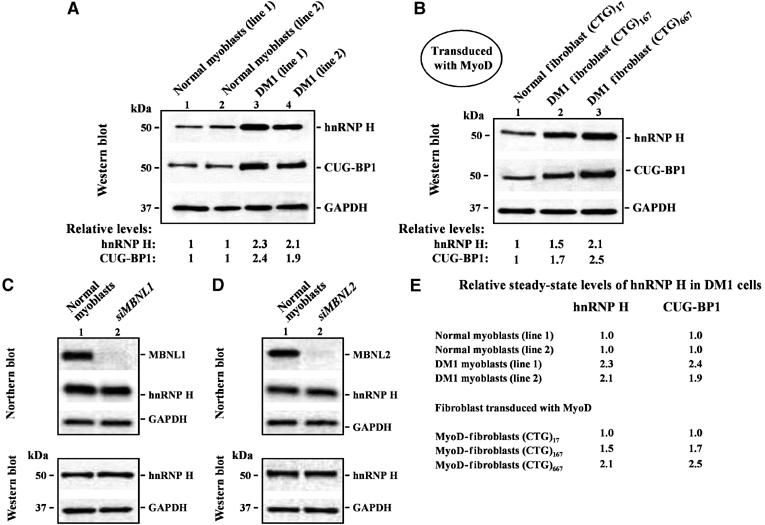
Steady-state hnRNP H levels are elevated in DM1 myoblasts by mechanisms unlinked to MBNL1 and MBNL2 loss. (A) Endogenous hnRNP H levels were measured in 10 μg of total protein from two normal and two DM1 myoblasts lines by Western blot analyses. The levels of hnRNP H in the DM1 myoblasts lines tested are ∼2.3- and ∼2.1-fold higher than that observed in normal myoblasts. CUG-BP1 levels in the DM1 myoblasts lines are ∼2.4- and ∼1.9-fold higher than that observed in the normal myoblast lines (n=3). (B) Fibroblasts containing 17, 167 and 667 CTG repeats were transduced with adenoviral vectors expressing both MyoD and GFP. Fluorescence microscopy study of fibroblasts 24 h after transduction demonstrated that ∼95% (±5%) of the transduced cells expressed GFP (data not shown). The levels of hnRNP H in the transduced DM1 fibroblasts were ∼1.5 fold (CTG)167 and ∼2.1 fold (CTG)667 elevated when compared with normal transduced fibroblasts (CTG)17 repeats (n=3). CUG-BP1 levels in the transduced DM1 fibroblasts were ∼1.7-fold (CTG)167 and ∼2.5-fold (CTG)667 higher than that observed in the normal transduced fibroblasts (CTG)17 repeats (n=3). (C, D) siRNA-mediated depletion of MBNL1 and MBNL2 in normal myoblasts does not result in increased steady state hnRNP H RNA or protein levels. Northern blot analyses demonstrate that MBNL1 or MBNL2 were ∼95 and ∼93% silenced, respectively. hnRNP H RNA and protein levels in normal myoblasts 5 days after transfection with siRNA directed against MBNL1 and MBNL2 were measured by Northern blot and Western blot analyses respectively. In both cases, the membranes were stripped and re-probed for GAPDH RNA and protein in parallel as an internal control. (E) Relative steady-state levels of hnRNP H and CUG-BP1 in DM1 cells when compared to normal controls is shown.
Elevated hnRNP H levels in DM1 myoblasts is unlinked to the functional inactivation of MBNL1 and MBNL2
The muscleblind proteins, MBNL1 and MBNL2, are functionally inactivated in DM1 cells due to their aberrant sequestration by the expanded CUG repeats located in the mutant DMPK RNA. To test if elevated hnRNP H levels result as a consequence of the functional inactivation of MBNL1 and MBNL2 in DM1 cells, we measured the steady-state levels of hnRNP H in normal myoblasts in which MBNL1 and MBNL2 mRNA levels were depleted by the cognate siRNAs. In these experiments, the steady-state RNA and protein levels of hnRNP H were not elevated as a consequence of reduced MBNL1 or MBNL2 levels (Figure 1C and D). Thus, the increased steady-state levels of hnRNP H in DM1 myoblasts do not result from the inactivation of MBNL1 and MBNL2. These data suggest that increased hnRNP H levels may be a consequence of signaling events occurring downstream of CTG repeat expansion in DM1 myoblasts.
Increased hnRNP H levels result in aberrant IR splicing in normal myoblasts
The 36 nt exon 11 of the alpha subunit of the IR RNA is alternatively spliced in normal myoblasts, with isoform A (IR-A), in which exon 11 is excluded or isoform B (IR-B), in which exon 11 is included being produced in approximately equal amounts (Figure 2A; Seino and Bell, 1989; Savkur et al, 2001; Dansithong et al, 2005). In contrast, DM1 cells demonstrate preferential exclusion of IR exon 11 (Savkur et al, 2001; Ho et al, 2004; Dansithong et al, 2005). To test the role of hnRNP H in the aberrant splice patterns observed in DM1, we studied the effect of altered hnRNP H dosage on IR exon 11 splicing in normal myoblasts and compared and contrasted these results with those observed for CUG-BP1 and the MBNL proteins.
Figure 2.
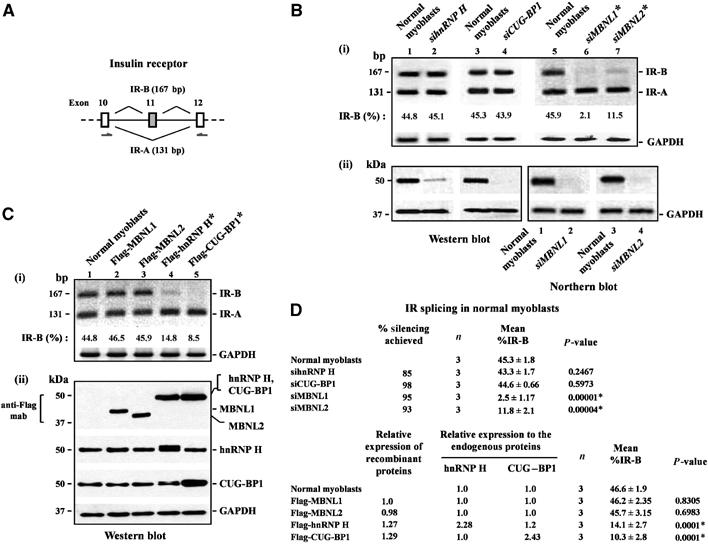
Overexpression of hnRNP H induces abnormal IR splicing in normal human myoblasts. (A) Schematic of the IR genomic sequence encoding exons 10, 11 and 12 is shown. Primers used to amplify the IR-B (167 nt; exon 11 is included) and the IR-A (131 nt; exon 11 is excluded) isoforms are indicated. (B) siRNA-mediated downregulation of hnRNP H does not alter IR splicing in normal myoblasts. Normal myoblasts were transfected with siRNAs directed against hnRNP H, CUG-BP1, MBNL1 and MBNL2, and total RNA was isolated 5 days after transfection and subjected to RT–PCR analysis using IR primers indicated. IR-B and IR-A levels were measured by densitometry analyses and % IR-B was calculated as described in Materials and methods. Levels of IR-B obtained in the experiment shown in (i) are indicated. (ii) siRNA-mediated silencing was studied by analyzing of 10 μg of protein by Western blot analyses for hnRNP H and CUG-BP1. 1.0 μg of mRNA was subjected to Northern blot analyses to measure the silencing achieved for MBNL1 and MBNL2. (C) Overexpression of Flag-hnRNP H results in decreased IR exon 11 splicing. Normal myoblasts were transfected with Flag tagged-MBNL1, MBNL2, hnRNP H and CUG-BP1, and total RNA was isolated 48 h later and subjected to RT–PCR analyses using IR primers as indicated. Levels of IR-B obtained in the experiment shown in (i) are indicated. (ii) Total protein (10 μg) was analyzed by Western blots to measure the relative expression of MBNL1, MBNL2, hnRNP H and CUG-BP1 using the anti-Flag, anti-hnRNP H and anti-CUG-BP1 antibodies, respectively. The blots were stripped and re-probed for GAPDH protein (Western blot) or GAPDH mRNA (Northern blot) expression as an internal control. For the RT–PCR analyses, GAPDH RNA was amplified as an internal control. (D) The results of three independent experiments of altered hnRNP H, CUG-BP1, MBNL1 and MBNL2 dosage on IR exon 11 splicing in normal myoblasts are tabulated. The asterisk (*) represents significant differences from the control (Student's two-tailed t-test; P<0.05).
To assess the role of endogenous hnRNP H on IR splicing, we decreased the levels hnRNP H, CUG-BP1, MBNL1 and MBNL2 using the cognate siRNAs in normal myoblasts. A reduction in the levels of hnRNP H and CUG-BP1 did not alter the equilibrium of IR exon 11 inclusion in normal myoblasts. In contrast, as previously reported, siRNA-mediated depletion of MBNL1 or MBNL2 resulted in the inhibition of IR exon 11 splicing, with MBNL1 depletion achieving almost complete repression of IR exon 11 inclusion and MBNL2 being slightly less efficient at inhibiting IR exon 11 splicing (Figure 2B and D). The relative levels of IR-B and IR-A were measured by RT–PCR analyses using the primers indicated in Figure 2A. These results demonstrate first, an absolute requirement of MBNL1 for IR exon 11 splicing and second, that endogenous hnRNP H and CUG-BP1 levels do not influence the equilibrium of IR exon 11 inclusion in normal myoblasts.
In a mirror image experiment hnRNP H, CUG-BP1, MBNL1 and MBNL2 were overexpressed in normal myoblasts. In these experiments, Flag-MBNL1, Flag-MBNL2, Flag-hnRNP H and Flag-CUG-BP1 were transfected into normal myoblasts and 48 h later the relative levels of IR-A and IR-B were measured by RT–PCR. Importantly, we observe that the percent of IR-B produced is significantly lower in normal myoblasts that express either ∼2.3-fold higher levels of hnRNP H (∼14.1%; P=0.0001) or ∼2.4-fold higher levels of CUG-BP1 (∼10.3%; P=0.0001) when compared to untransfected controls (∼46.6%) (Figure 2C and D). Inhibition of IR exon 11 inclusion achieved by hnRNP H was in the same range achieved by CUG-BP1, although overexpression of Flag-CUG-BP1 was slightly more efficient at inhibiting IR exon 11 splicing. Significantly, overexpression of the MBNL proteins was not sufficient to increase the efficiency of IR exon 11 splicing. Thus, these data demonstrate that the aberrant IR splice pattern in DM1 myoblasts can be recapitulated in normal myoblasts when either hnRNP H or CUG-BP1 levels are increased to approximate those observed in DM1 myoblasts.
Both hnRNP H and CUG-BP1 are required to maximally inhibit IR exon 11 splicing
As elevated levels of either hnRNP H or CUG-BP1 result in the reduction of IR-B levels in normal myoblasts, we tested if both proteins act in a coordinate fashion to suppress IR splicing in normal myoblasts. Thus, in a first set of experiments, we simultaneously down regulated CUG-BP1 levels by ∼97% using siRNAs and overexpressed hnRNP H in normal myoblasts. Significantly, the reduction in IR-B levels observed resulting from elevated hnRNP H levels (∼14.5%) was abolished in normal myoblasts that overexpress hnRNP H when endogenous CUG-BP1 levels are depleted (∼42.3%) (Figure 3A and C). In these experiments, a small increment (∼10–20%) in endogenous CUG-BP1 levels was observed when hnRNP H was overexpressed in normal myoblasts (Figure 3A(ii)).
Figure 3.
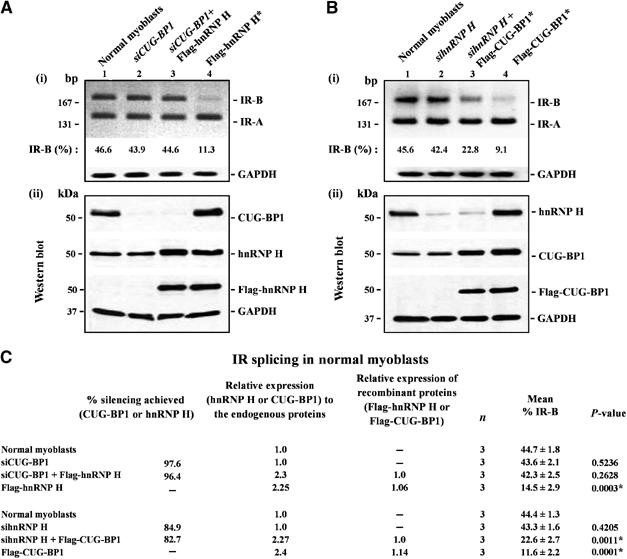
Abnormal IR splicing achieved by the overexpression of hnRNP H and CUG-BP1 in normal myoblasts requires endogenous levels of CUG-BP1 and hnRNP H. (A) Normal myoblasts were first transfected with siRNAs directed against CUG-BP1 and 3 days later the cells were re-transfected with vector or vector expressing Flag-hnRNP H. Following a 48-h incubation the cells were harvested for analyses. In parallel, normal myoblast cultures were transfected with Flag-hnRNP H and harvested 2 days post-transfection. In all cases, the harvested cells were split into two aliquots. From one aliquot, total RNA was isolated and subjected to RT–PCR analysis using IR primers. GAPDH RNA was amplified in parallel as an internal control. Levels of IR-B obtained in the experiment shown in (i) are indicated. (ii) Total protein (10 μg) from the second aliquot was analyzed by Western blots to measure the silencing achieved for CUG-BP1 using anti-CUG-BP1 mab staining. Relative levels of hnRNP H were measured using anti-hnRNP H or anti-Flag antibodies. (B) Normal myoblasts were first transfected with siRNAs directed against hnRNP H and 3 days later the cells were re-transfected with vector or vector expressing Flag-CUG-BP1. Following a 48-h incubation the cells were harvested for analyses. In parallel, normal myoblast cultures were transfected with Flag-CUG-BP1 and harvested 2 days post-transfection. Total RNA was isolated from harvested cells and subjected to RT–PCR analysis using IR and GAPDH primers (internal control). Levels of IR-B obtained in the experiment are shown in (i). (ii) Silencing achieved for hnRNP H and the relative levels of CUG-BP1 measured by Western blots using anti-hnRNP H, anti-CUG-BP1 and anti-Flag antibodies are shown. The blots were re-probed for GAPDH protein using anti-GAPDH polyclonal antibodies as a loading control. (C) Results from three independent experiments are tabulated. The asterisk (*) represents significant differences from the control (Student's two-tailed t-test; P<0.05).
In a second set of experiments, IR splicing was studied in normal myoblasts in which CUG-BP1 was overexpressed in conjunction with siRNA-mediated silencing of hnRNP H (silencing achieved for hnRNP H was ∼83%). In these experiments, we observed that the suppression of exon 11 inclusion was not as effective when CUG-BP1 was overexpressed in conjunction with the downregulation of endogenous hnRNP H levels (∼22.6%) when compared to the overexpression of CUG-BP1 alone (∼11.6%) (Figure 3B and C). Overexpression of CUG-BP1 did not result in altered steady-state hnRNP H levels in normal myoblasts (Figure 3B(ii)). These data demonstrate that both hnRNP H and CUG-BP1 are required to maximally suppress IR exon 11 splicing in normal myoblasts.
hnRNP H and CUG-BP1 form an RNA-dependent suppressor complex in normal myoblasts
To test if hnRNP H and CUG-BP1 interact in vivo, normal myoblasts were transduced with recombinant adenoviral vectors designed to express either Flag-CUG-BP1 or GFP. To detect both RNA-dependent and -independent interactions, extracts from normal myoblasts cultures overexpressing Flag-CUG-BP1 and GFP were first incubated with the anti-Flag antibody beads. Subsequently, the immunoprecipitates were divided into two aliquots, which were treated either with or without RNAse A as described in Materials and methods. After extensive washing, the bound proteins were eluted by competition with the Flag peptide, and the eluted proteins were immunoblotted and stained with Flag and hnRNP H antibodies, respectively. Immunoprecipitation experiments were carried out in triplicate and representative panels are shown in Figure 4. Expression of Flag-CUG-BP1 was confirmed by immunostaining with the anti-Flag monoclonal antibody (mab) (Figure 4A). In parallel, the eluted proteins were immunostained with anti-hnRNP H antibodies (Figure 4B). Endogenous hnRNP H co-immunoprecipitated with Flag-CUG-BP1 in RNase-free immunoprecipitates. Interaction between CUG-BP1 and hnRNP H was however lost upon RNAse A treatment. To verify the specificity of the RNA-dependent interaction observed between CUG-BP1 and hnRNP H, we studied the CUG-BP1 immunoprecipitates by Western blot analyses using antibodies directed against hnRNP A1, SF2/ASF and PABPN1. hnRNP A1, SF2/ASF and PABPN1 did not co-immunoprecipitate with Flag-CUG-BP1 either under non-RNAse or RNAse treatment conditions (Supplementary Figure S4). Thus, these data are consistent with the formation of an RNA-dependent suppressor complex consisting of both hnRNP H and CUG-BP1 in normal myoblasts that overexpress CUG-BP1.
Figure 4.
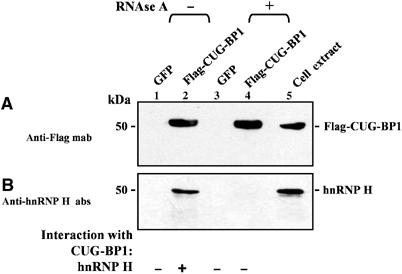
hnRNP H interacts with CUG-BP1 in RNA-dependent manner in vivo. Normal myoblasts were transduced with recombinant adenoviruses expressing Flag-CUG-BP1 or GFP. At 48 h post infection, total cell extracts were prepared and incubated with anti-Flag beads to immunoprecipitate proteins under non-RNAse and RNAse treatment conditions as described in Materials and methods. The eluted proteins from each immunoprecipitation were analyzed by Western blot staining with anti-Flag mab (A), anti-hnRNP H polyclonal antibodies (B). Staining with anti-Flag mab demonstrates the precipitation of Flag-CUG-BP1 (A). (B) Staining with anti-hnRNP H polyclonal antibodies demonstrates that hnRNP H co-immunoprecipitates with Flag-CUG-BP1 when the immunoprecipitates are not treated with RNAse. Co-immunoprecipitation of hnRNP H with Flag-CUG-BP1 does not occur when immunoprecipitates are treated with RNAse.
Overexpression of MBNL1 partially rescues the IR splicing defect resulting from elevated levels of hnRNP H in normal myoblasts
We next tested if elevated levels of MBNL1 can rescue the IR splicing defect resulting from the overexpression of hnRNP H in normal myoblasts. Thus MBNL1 was overexpressed in conjunction with hnRNP H in normal myoblasts and the relative levels of IR-B and IR-A was measured by RT–PCR analyses. The percent IR-B produced was significantly higher in normal myoblasts which overexpress both Flag-MBNL1 and Flag-hnRNP H (∼21.3%), when compared with myoblasts that overexpress Flag-hnRNP H alone (∼11.9%) (Figure 5A and B). Thus, overexpression of MBNL1 can partially rescue the inhibitory effect resulting from elevated levels of hnRNP H on IR exon 11 inclusion in normal myoblasts. As overexpression of MBNL1 does not increase the amount of IR-B produced in normal myoblasts (Figure 5A and B), these data suggest that MBNL1 may partially repress the inhibitory activity of elevated hnRNP H levels on IR exon 11 inclusion by physical interaction.
Figure 5.
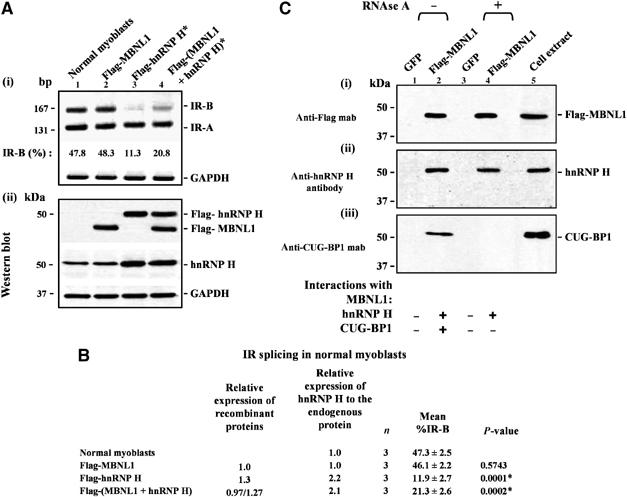
Overexpression of MBNL1 partially rescues the IR splicing defect resulting from elevated levels of hnRNP H in normal myoblasts and MBNL1 interacts with hnRNP H in an RNA-independent manner in vivo. (A) Normal myoblasts were transfected with Flag-MBNL1, Flag-hnRNP H or Flag-MBNL1 and Flag-hnRNP H in combination. Total RNA was isolated 48 h post-transfection and subjected to RT–PCR analyses using IR primers. GAPDH RNA was amplified in parallel as an internal control. Levels of IR-B obtained in the experiment shown in (i) are indicated. (ii) Total protein (10 μg) was analyzed by Western blots to measure the levels of expressed MBNL1 and hnRNP H using anti-Flag and anti-hnRNP H antibodies, respectively. The blots were re-probed for GAPDH as a loading control. (B) The results of three independent experiments are tabulated. The asterisk (*) represents significant differences from the control (Student's two-tailed t-test; P<0.05). (C) Normal myoblasts were transduced with recombinant adenoviruses expressing Flag-MBNL1 or GFP. At 48 h postinfection, total cell extracts were prepared and incubated with anti-Flag beads to immunoprecipitate proteins under non-RNAse and RNAse treatment conditions. The eluted proteins from each immunoprecipitation were analyzed by Western blot staining with anti-Flag mab (i), anti-hnRNP H polyclonal antibodies (ii), and anti-CUG-BP1 mab (iii). Staining with anti-Flag mab demonstrates the precipitation of Flag-MBNL1 (i). (ii) Staining with anti-hnRNP H polyclonal antibodies demonstrates that endogenous hnRNP H co-immunoprecipitates with Flag-MBNL1 both under non-RNAse and RNAse treatment conditions. (iii) CUG-BP1 mab staining demonstrates that CUG-BP1 co-immunoprecipitates with Flag-MBNL1 only under non-RNAse treatment conditions.
MBNL1 and hnRNP H demonstrate RNA-independent interaction in vivo
To test if MBNL1 interacts with hnRNP H in vivo, normal myoblasts were transduced with adenoviral vectors expressing Flag-MBNL1 or GFP. Flag antibodies were used to immunoprecipitate MBNL1 and the interaction of endogenous hnRNP H with flag-MBNL1 was tested under both RNase A and RNAse A free treatment conditions as described in Materials and methods. Immunoprecipitation experiments were carried out in triplicate and the representative panels are shown in Figure 5C. Expression of Flag-MBNL1 was confirmed by immunostaining with the anti-Flag mab (Figure 5C(i)). RNA-independent interaction between Flag-MBNL1 and hnRNP H and RNA-dependent interaction between Flag-MBNL1 and CUG-BP1 was observed in these experiments (Figure 5C(ii) and (iii)). As MBNL1 interacts with hnRNP H in an RNA-independent manner these data support the hypothesis that MBNL1 physically interacts with hnRNP H and partially represses its inhibitory activity on IR exon 11 splicing.
Overexpression of MBNL1 and MBNL2 recruits hnRNP H to DM1 foci
Physical interaction between MBNL1 and hnRNP H predicts that overexpression of MBNL1 should recruit hnRNP H away from its normal locale in the cell. To test this idea, we overexpressed Flag-MBNL1 and Flag-MBNL2 in conjunction with GFP tagged hnRNP H (GFP-hnRNP H) in DM1 myoblasts. Previous experiments have demonstrated that GFP-MBNL1 and GFP-MBNL2 co-localize with the expanded CUG repeats in DM1 cells (Fardaei et al, 2002). More recently, it has been reported that hnRNP H co-localizes with foci in cortical neurons and DM1 myoblasts to a limited extent (Jiang et al, 2004). As elevated levels of MBNL1 bind hnRNP H in an RNA-independent manner, we hypothesized that increasing levels of MBNL1 should facilitate a corresponding increase in the recruitment of hnRNP H to DM1 foci.
Consistent with previous results, transfection of vectors expressing GFP-MBNL1 and GFP-MBNL2 into DM1 myoblasts demonstrated co-localization of GFP-MBNL1 and GFP-MBNL2 with the mutant DMPK RNA, which was detected by in situ hybridization of a (CAG)10-Cy3 probe (Figure 6A–L and Table I). Polyclonal antibodies directed against hnRNP H demonstrate that hnRNP H is freely distributed in the nucleus in DM1 myoblasts. Although a small percentage of the foci colocalized with hnRNP H, no significant localization of either the endogenous protein or the GFP-hnRNP H to DM1 foci was observed in DM1 myoblasts (Figure 6M–R and Table I). However, when GFP-hnRNP H was co-expressed with either Flag-MBNL1 or Flag MBNL2 striking co-localization of GFP-hnRNP H to the DM1 foci, which approached the values observed for GFP-MBNL1 and GFP-MBNL2, was observed (Figure 6S–X and Table I). Thus, increasing MBNL1 or MBNL2 dosage can serve to recruit hnRNP H to the CUG RNA foci in DM1 myoblasts.
Figure 6.
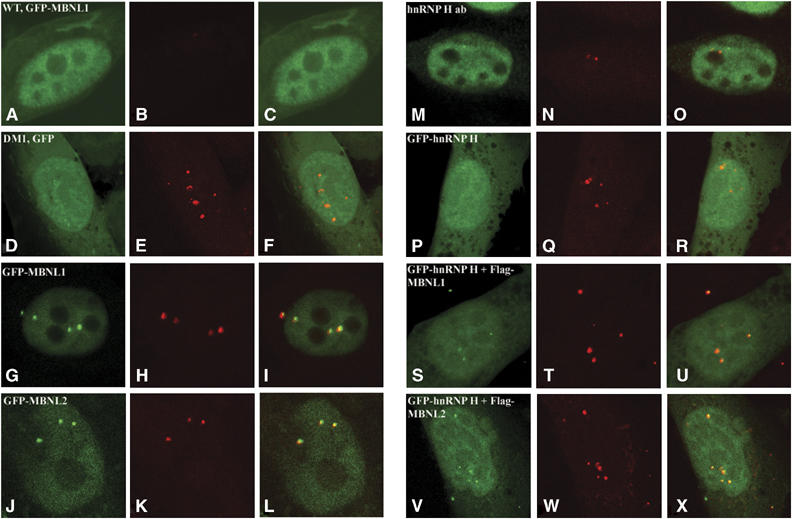
Overexpression of MBNL1 and MBNL2 increases recruitment of hnRNP H to DM1 foci. Normal myoblasts expressing GFP-MBNL1 (A–C) and DM1 myoblasts expressing GFP (D–F), GFP-MBNL1 (G–I), GFP-MBNL2 (J–L), GFP-hnRNP H (P–R), or co-expressing GFP-hnRNP H and Flag-MBNL1 (S–U) or GFP-hnRNP H and Flag-MBNL2 (V–X) are shown. Distribution of endogenous hnRNP H (M–O) was studied using anti-hnRNP H polyclonal antibodies conjugated with FITC (green signal). GFP tagged proteins and endogenous hnRNP H are visualized as a green signal (A, D, G, J, M, P, S and V). The mutant DMPK transcripts encoding the expanded CUG tract was detected by hybridization with a (CAG)10-Cy3 probe (red signal: B, E, H, K, N, Q, T and W). Transcripts containing expanded repeat were not observed in the normal myoblasts (B). Merged images (C, F, I, L, O, R, U and X) where super-imposition of green and red signals are observed as yellow signals demonstrate that GFP-MBNL1 (I), GFP-MBNL2 (L) and GFP-hnRNP H co-expressed with Flag-MBNL1 (U) or Flag-MBNL2 (X) co-localize with the mutant DMPK RNA in DM1 myoblasts. However, GFP alone in DM1 myoblasts (F), endogenous hnRNP H (O) and GFP-hnRNP H (R) did not co-localize significantly with the mutant DMPK RNA. The percent of foci that co-localize with each protein are tabulated in Table I. The asterisk (*) represents significant differences from GFP (Student's two-tailed t-test; P<0.05).
Table 1.
Co-localization of proteins with DM1 foci
| Proteins | Cell no. | Foci no. | No. of foci that colocalize with proteins | % of foci that colocalize with proteins | P-value |
|---|---|---|---|---|---|
| GFP | 13 | 40 | 1 | 1.5 | |
| GFP-MBNL1 | 23 | 61 | 50 | 81.5 | 0.0001* |
| GFP-MBNL2 | 19 | 55 | 40 | 72.4 | 0.0006* |
| hnRNP H | 34 | 58 | 10 | 15.7 | 0.1915 |
| GFP-hnRNP H | 19 | 54 | 12 | 19.8 | 0.1201 |
| GFP-hnRNP H+Flag-MBNL1 | 16 | 45 | 36 | 79.8 | 0.0008* |
| GFP-hnRNP H+Flag-MBNL2 | 15 | 43 | 30 | 71.6 | 0.0032* |
| Significantly different from GFP (Student's t-test; P<0.05). | |||||
siRNA-mediated downregulation of hnRNP H and CUG-BP1 does not rescue aberrant IR splicing in DM1 myoblasts
In the normal human myoblast lines studied, IR-B makes up ∼46% of the IR RNA isoforms (Figure 2B–D). In contrast, in DM1 myoblasts, IR exon 11 is almost completely excluded and the percent of IR-B produced drops to 5.7% (Figure 7A and C; Dansithong et al, 2005). As DM1 myoblasts have elevated levels of hnRNP H and increased levels of hnRNP H result in the reduction of IR-B levels in normal myoblasts, we tested if siRNA-mediated silencing of hnRNP H can rescue the IR splicing defect in DM1 myoblasts. However, no significant increase in IR-B levels was observed in DM1 myoblasts when hnRNP H levels were downregulated to ∼74.3% (Figure 7A and C). These results are similar to that observed when CUG-BP1 is silenced in DM1 myoblasts (Figure 7A and C; Dansithong et al, 2005). IR exon 11 inclusion was also not achieved by a combinatorial approach where both hnRNP H and CUG-BP1 were depleted simultaneously in DM1 myoblasts (Figure 7A and C). Transfection of a scrambled RNA did not alter IR splicing in DM1 myoblasts (data not shown). Thus, siRNA-mediated silencing of either hnRNP H or CUG-BP1 or hnRNP H and CUG-BP1 was not sufficient to rescue the IR splicing defect in DM1 myoblasts.
Figure 7.
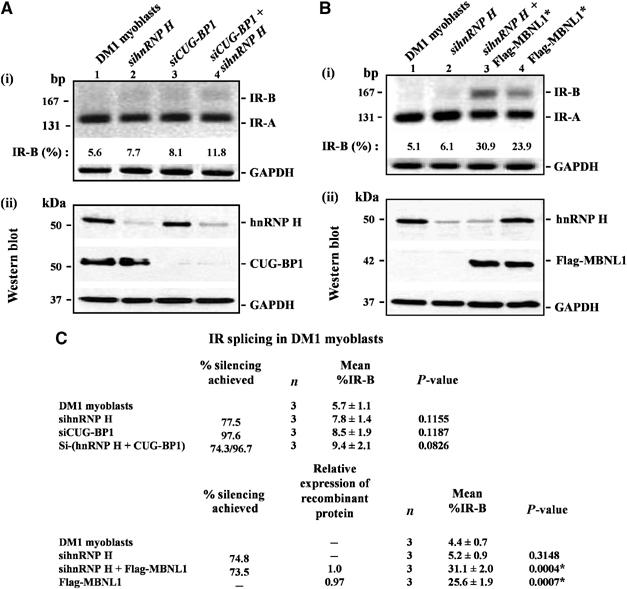
Overexpression of MBNL1 in conjunction with decreased hnRNP H levels partially rescues aberrant IR splicing in DM1 myoblasts. (A) DM1 myoblasts were transfected with siRNAs directed against hnRNP H or CUG-BP1 or hnRNPH and CUG-BP1 and total RNA isolated 5 days post-transfection was subjected to RT–PCR analysis using IR primers. Levels of IR-B obtained in the experiment shown in (i) are indicated. (ii) Total protein (10 μg) was analyzed by Western blot to measure the silencing achieved for hnRNP H and CUG-BP1 using anti-hnRNP H and anti-CUG-BP1 antibodies, respectively. (B) DM1 myoblasts were transfected with siRNAs directed against hnRNP H. After 3 days, the culture was transduced with recombinant adenoviruses expressing Flag-MBNL1. At 48 h post-transduction, total RNA was isolated and subjected to RT–PCR analysis and the percentage of IR-B was measured. Levels of IR-B obtained in the experiment shown in (i) are indicated. (ii) Total protein (10 μg) was analyzed by Western blot to measure the silencing achieved for hnRNP H and the levels of Flag-MBNL1 expressed using anti-hnRNP H and anti-Flag antibodies, respectively. The blots were re-probed for GAPDH protein as a loading control. In the RT–PCR analyses, GAPDH RNA was amplified as an internal control. (C) The results from three independent experiments are tabulated. The asterisk (*) represents significant differences from the control (Student's two-tailed t-test; P<0.05).
Overexpression of MBNL1 in conjunction with decreased hnRNP H levels allow partial restoration of normal IR splicing in DM1 myoblasts
In DM1 cells, MBNL1 is functionally inactivated as this protein is sequestered by the mutant DMPK RNA. To test if a combinatorial approach in which hnRNP H levels are downregulated simultaneously with the overexpression of MBNL1 results in restoration of normal IR splice patterns, we studied IR exon 11 inclusion in DM1 myoblasts overexpressing either MBNL1 or overexpressing MBNL1 in conjunction with siRNA-mediated down regulation of hnRNP H. We observe that overexpression of MBNL1 was primarily responsible for the rescue achieved (∼25.6%; P=0.0007 for a two way comparison between DM1 myoblasts and DM1 myoblasts overexpressing MBNL1). However, a significant incremental improvement in IR-B levels was achieved when hnRNP H was downregulated in conjunction with the overexpression of MBNL1 (∼31.1%; P=0.027 for a two-way comparison between DM1 myoblasts overexpressing MBNL1 and DM1 myoblasts overexpressing MBNL1 in conjunction with siRNA-mediated downregulation of hnRNPH) (Figure 7B and C). Thus, overexpression of MBNL1 in combination with down regulation of hnRNP H allows a partial rescue of IR splicing in DM1 myoblasts.
MBNL1, hnRNP H and CUG-BP1 bind directly to the human IR RNA in vitro
To test if MBNL1, hnRNP H and CUG-BP1 bind to the IR RNA, we expressed a human IR mini gene encoding IR exons 10, 11 and 12 (Figure 8A; Kosaki et al, 1998) in MBNL1 depleted normal myoblasts. Similarly, the IR minigene was co expressed with Flag-hnRNP H or Flag-CUG-BP1 in normal myoblasts. Examination of the splice pattern of the IR minigene using mini gene specific primers demonstrated that exon 11 splicing was repressed when MBNL1 was depleted or when hnRNP H and CUG-BP1 were overexpressed in normal myoblasts, in a manner similar to that observed for the endogenous IR gene (data not shown). We therefore used a UV crosslinking analysis to assess the binding of MBNL1, hnRNP H and CUG-BP1 to the human IR minigene RNA. GST (control) and His tagged-MBNL1, hnRNP H, and CUG-BP1 were expressed in bacteria and the recombinant proteins were purified as described in Supplementary Materials and methods. 32P-labeled human IR-B minigene RNA was transcribed in vitro using [α-32P]UTP and used in a UV crosslinking assay with the purified recombinant proteins (GST, MBNL1, hnRNP H, CUG-BP1) as described in Materials and methods. Results from these experiments demonstrate that MBNL1, hnRNP H and CUG-BP1 bind directly to IR-B minigene RNA in vitro (Figure 8B).
Figure 8.
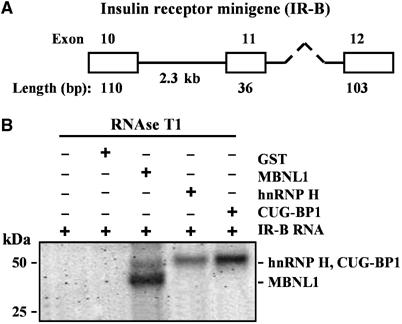
MBNL1, hnRNP H and CUG-BP1 bind directly to human IR-B RNA in vitro. (A) Depicts the schematic of the human IR-B minigene (Kosaki et al, 1998). (B) UV crosslinking experiments were carried out using uniformly 32P-labeled IR-B RNA and 700 ng purified GST or His-MBNL1, His-hnRNP H and His-CUG-BP1. After incubation, reaction mixtures were UV irradiated and digested with RNase T1. The samples were analyzed by SDS–PAGE. The position of each protein-RNA complex is shown by an arrow.
Discussion
In this study, we demonstrate that the levels of the splice regulator hnRNP H are elevated in DM1 myoblasts. Overexpression of hnRNP H in normal myoblasts represses IR exon 11 inclusion in a manner that is similar to that observed in DM1 cells. These data therefore demonstrate that elevated hnRNP H levels contribute to aberrant RNA splicing, a characteristic feature of DM1. We demonstrate that elevated levels of either hnRNP H or CUG-BP1 results in the formation of an RNA-dependent suppressor complex comprising of both hnRNP H and CUG-BP1, which is required for maximal repression of IR exon 11 inclusion in normal myoblasts. As elevated levels of MBNL1 cannot increase IR exon 11 splicing in normal myoblasts but does serve to partially rescue the repression of IR exon 11 splicing resulting from elevated hnRNP H levels, our data support the hypothesis that overexpression of MBNL1 can in part dampen the inhibitory activity of hnRNP H. In support of this hypothesis, immunoprecipitation and co-localization studies demonstrate RNA-independent interaction between MBNL1 and hnRNP H in both normal and DM1 myoblasts. Consistent with the absolute requirement of MBNL1 for IR exon 11 inclusion, we show that siRNA-mediated downregulation of either hnRNP H or CUG-BP1 does not rescue IR splicing in DM1 myoblasts. However, a partial rescue of the IR splicing defect is achieved by the coordinate upregulation of MBNL1 in conjunction with siRNA-mediated downregulation of hnRNP H. Thus, these data demonstrate that physical and functional interactions between the splice regulators, hnRNP H, CUG-BP1 and MBNL1 serve to set the equilibrium of splice site selection in both normal and DM1 myoblasts.
A prominent deleterious effect resulting from the expression of expanded CUG repeats is the aberrant splicing of a subset of physiologically important target RNAs. Abnormal splicing is not a global phenomenon in DM1 cells, rather several lines of evidence demonstrate that aberrant splice site selection in a subset of alternatively spliced exons may result in the development of a key pathophysiological feature of DM1 (Philips et al, 1998; Savkur et al, 2001; Buj-Bello et al, 2002; Charlet-B et al, 2002; Mankodi et al, 2002). Expression of expanded CUG repeats has therefore been hypothesized to dysregulate one or more splice regulators, which may act either independently or coordinately to disturb the equilibrium of splice site selection in DM1 cells. Several studies demonstrate that MBNL1, MBNL2 and CUG-BP1 function as splicing regulators whose altered dosage results in aberrant splice patterns both in normal and DM1 cells (Kanadia et al, 2003; Ho et al, 2004, 2005; Dansithong et al, 2005). However, the number of splice regulators that are dysfunctional in DM1 cells and the functional interplay between such factors that dictates aberrant splice site selection in DM1 is currently unclear.
In this study, we demonstrate that the levels of the alternative splice regulator, hnRNP H, are elevated in DM1 myoblasts (Figure 1). We show that the aberrant IR splice pattern in DM1 myoblasts is recapitulated when the levels of either hnRNP H or CUG-BP1 in normal myoblasts are elevated to approximate levels observed in DM1 myoblasts (Figure 2). Therefore, the levels of two splice regulators, CUG-BP1 and hnRNP H, are elevated in DM1 and either protein can serve to deregulate IR splicing. Although overexpression of both proteins can play an inhibitory role, CUG-BP1 is slightly more effective than hnRNP H in inhibiting IR exon 11 inclusion (Figure 2).
Elevated levels of hnRNP H in DM1 cells are not a consequence of the functional inactivation of MBNL1 and MBNL2, rather the increase in hnRNP H levels appear to result from signaling events occurring as a consequence of CTG repeat expansion (Figure 1). Thus, two sets of events contribute to aberrant splicing in DM1: First, splice regulators, such as MBNL1 and MBNL2, are inactivated as a consequence of their abnormal sequestration by the CUG repeats. Second, a set of splice regulators, such as hnRNP H and CUG-BP1, appear to be deregulated as a consequence of signaling events occurring downstream of the CTG repeat expansion. To test if these two sets of proteins alter IR splice site choice by independent mechanisms or alternatively act in a coordinate fashion, we carried out a series of experiments. Thus, IR splicing was studied under defined conditions in which expression levels of one or more of these splice regulators was altered in both normal and DM1 myoblasts. The results from these experiments allow the following important insights into the mechanism by which IR splice site selection is regulated both in normal and DM1 myoblasts.
Experiments conducted in normal myoblasts demonstrate that IR exon inclusion does not occur in the absence of the MBNL1 (Figure 2). MBNL2 depletion was a little less effective at inhibiting IR exon 11 inclusion when compared to MBNL1 depletion (Figure 2). Thus, these data demonstrate an absolute requirement for MBNL1 in IR exon 11 splicing. However, as overexpression of MBNL1 cannot increase the amount of IR-B produced in normal myoblasts (Figure 2), MBNL1 may require a limiting factor in order to facilitate splicing or alternatively, the ability of MBNL1 to recruit or stabilize spliceosome factors may be suboptimal even under saturating conditions. The equilibrium between IR exon 11 inclusion and exclusion is unaffected by endogenous levels of CUG-BP1 and hnRNP H in normal myoblasts (Figure 2). Therefore, the decision to splice IR exon 11 rests on the levels of MBNL1 and MBNL2 in normal myoblasts and is unaffected by endogenous levels of hnRNP H and CUG-BP1 (Figure 9A).
Figure 9.
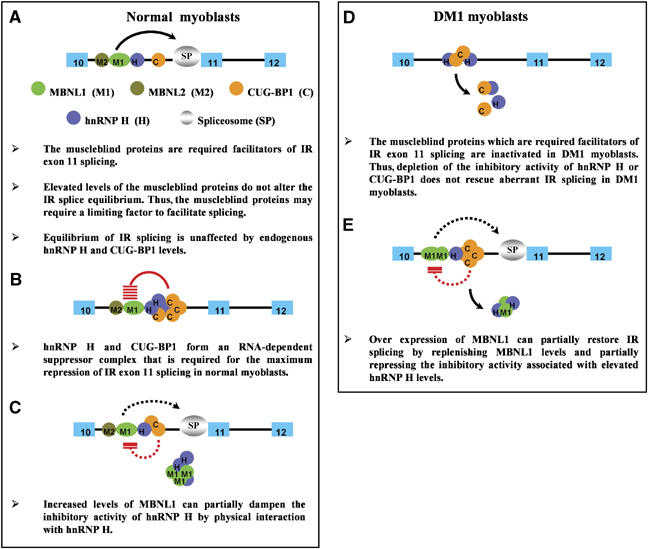
Coordinate regulations of IR splicing in normal and DM1 cells. A model for the regulation of IR exon 11 splicing in normal and DM1 myoblasts by MBNL1, MBNL2, hnRNP H and CUG-BP1 is shown in panels A–E.
However, when either hnRNP H or CUG-BP1 levels are elevated in normal myoblasts, we demonstrate the formation of a suppressor complex that effectively abrogates the facilitatory activity of MBNL1 and MBNL2 (Figure 2). Both hnRNP H and CUG-BP1 are required to maximally repress IR splicing, as the data demonstrate that maximal repression achieved by the overexpression of either hnRNP H or CUG-BP1 requires endogenous levels of CUG-BP1 and hnRNP H, respectively. Specifically, if hnRNP H is silenced, overexpression of CUG-BP1 is approximately half as effective at repressing IR exon 11 splicing. Similarly, siRNA-mediated downregulation of CUG-BP1 in conjunction with elevated hnRNP H levels prevent hnRNP H from inducing a repressive effect on IR exon splicing (Figure 3). In other experiments, we demonstrate that overexpression of CUG-BP1 in normal myoblasts results in an RNA-dependent interaction between hnRNP H and CUG-BP1 both in vivo and in vitro (Figures 4 and 8). Taken together, these data demonstrate that overexpression of either hnRNP H or CUG-BP1 results in the formation of an RNA-dependent suppressor complex that requires both CUG-BP1 and hnRNP H to achieve maximum repression of IR exon 11 splicing (Figure 9B). The mechanism of action of the suppressor complex is currently unclear, but as with other splice regulators hnRNP H and CUG-BP1 may recruit each other to form a stable contiguous complex on the IR RNA and thus prevent spliceosome assembly. Alternatively, CUG-BP1 and hnRNP H may bind at independent sites on the RNA and occlude the binding of unique components of the spliceosome. A third possibility is that binding of hnRNP H and CUG-BP1 to silencer elements in introns may physically loop out IR exon11 and cause IR exon 11 skipping. The exact mechanism of repression mediated by CUG-BP1 and hnRNP H however requires the mapping of the binding sites for hnRNP H and CUG-BP1 on the IR RNA.
Significantly, we observe that overexpression of MBNL1 can partially rescue the inhibitory effect of elevated levels of hnRNP H on IR exon 11 splicing (Figure 5). As elevated levels of MBNL1 cannot alter IR splicing in normal myoblasts in which hnRNP H levels are not elevated (Figure 2), we hypothesized that overexpression of MBNL1 binds to hnRNP H and thus abolishes its repressive effect on IR splicing. To test this hypothesis, we carried out two experiments: First, we overexpressed MBNL1 in normal myoblasts and tested if MBNL1 interacts with hnRNP H. Consistent with our model, we observe that MBNL1 is able to bind to hnRNP H in an RNA-independent manner in vivo (Figure 5). In a second experiment, we determined if overexpression of MBNL1 was able to bind and recruit hnRNP H away from its normal cellular locale. Specifically, when GFP-hnRNP H is expressed in conjunction with elevated levels of Flag-MBNL1 or Flag-MBNL2 in DM1 myoblasts, we observe that the recruitment of hnRNP H to CUG foci is strikingly increased (Figure 6 and Table I). Thus, these data are consistent with the hypothesis that overexpression MBNL1 can bind and partially inhibit the repressive activity of hnRNP H on IR exon 11 splicing (Figure 9C).
The data from normal myoblasts make the following predictions about the mechanism of action of these proteins in mediating abnormal IR splicing in DM1 myoblasts. In DM1 cells, where MBNL1 and MBNL2 are sequestered by the expanded CUG tracts, downregulation of hnRNP H or CUG-BP1 levels should not rescue the aberrant IR splice pattern, as the MBNL proteins, which are the required facilitators for IR exon 11 splicing, are functionally inactivated. Consistent with this prediction, we demonstrate that siRNA-mediated downregulation of either hnRNP H or CUG-BP1 does not rescue the IR splicing defect in DM1 myoblasts (Figures 7 and 9D). The role of the muscleblind proteins in DM1 cells cannot be accurately tested by rescue experiments, as the expanded CUG repeats may sequester a fraction of the overexpressed muscleblind proteins. None the less data from normal myoblasts predict that overexpression of MBNL1 should partially but not wholly restore IR splicing as overexpression of MBNL1 would first, serve to replenish MBNL1 levels and second, partially squelch the repressive activity of the suppressor complex resulting from elevated levels of hnRNP H. Consistent with this prediction, we observe that overexpression of MBNL1 in DM1 myoblasts results in a partial rescue of IR splicing, which is incrementally improved by a further siRNA mediated silencing of hnRNP H (Figure 7). Thus, taken together these data demonstrate that IR splicing in normal and DM1 myoblasts reflects finely orchestrated interactions between the required facilitator MBNL1 and a suppressor complex made up of hnRNP H and CUG-BP1 (Figure 9E). Importantly, these results show that although deregulation of the MBNL proteins, hnRNP H and CUG-BP1 have independent etiologies in DM1 cells, it is the coordinate activity between the these sets of proteins that dictates aberrant IR splice patterns in DM1 myoblasts.
Materials and methods
DNA constructs
cDNA sequences for MBNL1 (Y13829), MBNL2 (AF061261), CUG-BP1 (NM-006560) and hnRNP H (L22009) were derived from the NCBI DNA database and used to design primers to PCR-amplify the coding sequences of these genes from a HeLa cell cDNA library. All cDNAs were subsequently sequenced and cloned into the appropriate vectors.
siRNAs
All siRNA oligonucleotides were synthesized by Dharmacon (USA), deprotected and the complementary strands were annealed. The sequences of the siRNAs used in this study were: MBNL1: 5′-CACUGGAAGUAUGUAGAGAdTdT-3′; MBNL2: equimolar amounts of two siRNAs, 5′-CACCGUAACCGUUUGUAUGdTdT-3′ and 5′-GAGGAACAUGCUCACGCUCdTdT-3′; CUG-BP1: 5′-GUUACGACAAUCCUGUUUCdTdT-3′ and hnRNP H: 5′-AAUCAGAAGAUGAAGUCAAAUdTdT-3′.
Cell culture and transfection
Normal and DM1 myoblasts were a generous gift of Dr Charles Thornton and have been previously described in Dansithong et al (2005). Myoblast lines were cultured and maintained in SKGM media (Cambrex, MD) containing 10% fetal bovine serum. Overexpression of proteins and siRNA-mediated depletion of proteins were carried out using the methods previously described by Dansithong et al (2005). Briefly, for the overexpression of proteins, myoblasts (2 × 106) were seeded on 100 mm dishes and incubated overnight. The cells were transiently transfected with 30 μg of the DNA constructs and the cells were harvested for analyses 48 h post-transfection. For siRNA-mediated depletion of proteins, myoblasts (1 × 106) were plated on 100 mm dishes overnight and siRNAs at a concentration of 100 nM were transfected using the oligofectamin kit (Invitrogen), according to the manufacturer's protocol. Prior standardization experiments showed that maximum silencing was achieved 5 days post-transfection.
In situ fluorescence hybridization analyses
FISH analyses were carried out primarily as described by Dansithong et al (2005). Briefly, myoblasts were plated on chamber slides and transfected with plasmids. At 48 h post-transfection, cells were fixed in 4% paraformaldehyde in PBS for 20 min at room temperature and stored in 70% ethanol at 4°C. For FISH studies, a Cy3 conjugated (CAG)10 oligonucleotide probe (Operon), was used to detect CUG repeat expansions as described by Taneja et al (1995). Endogenous hnRNP H was detected using hnRNP H polyclonal antibodies (Santa Cruz) at a dilution of 1:200. The secondary antibodies conjugated with FITC (Santa Cruz Inc., USA) were used at a dilution 1:100. The confocal microscopy facility in the Doheny Eye Institute at the University of Southern California was used for the FISH analyses.
RNA extraction and measurement of IR exon 11 splicing
Total RNA was isolated from DM1 and normal myoblasts using the RNAeasy mini kit (Qiagen), according to the manufacturer's protocol. cDNA was synthesized from 5 μg of total RNA using the cDNA synthesis kit (Amersham Bioscience Inc., USA). PCR was carried out using 150 ng of the synthesized cDNA. IR primers (5′-CCAAAGACAGACTCTCAGAT-3′ and 5′-AACATCGCCAAGGGACCTGC-3′) used to measure IR exon 11 splicing hybridized to the 5′ end of exon 10 and 3′ end of exon 12. PCR conditions used for the amplification of IR-A and IR-B are primarily as described by Savkur et al (2001). To ensure that PCR amplification was occurring in the linear range prior standardization experiments were conducted as described in Supplementary Methods (Supplementary Figure S3). The resulting PCR products (IR-B and IR-A) were cloned and verified by sequencing. The band intensities of the products were measured by densitometry analyses using Gene Tool (Syngene Inc., USA). % IR-B was calculated as (IR-B/IR-A+IR-B) × 100.
Northern blot
Total cellular RNA was isolated using the RNAeasy mini kit according to the manufacturer's protocol. The blots were hybridized with radioactive MBNL1, MBNL2 and hnRNP H probes using standard techniques. The probes correspond to bases 512–877 of MBNL1 (Y13829), 783–1262 of MBNL2 (AF061261), and 73–670 of hnRNP H (L22009), respectively. The blots were subsequently re-hybridized with a GAPDH probe as an internal control. The relative band intensities were measured by densitometry analyses using Gene Tool.
Immunoprecipitations
Flag-MBNL1 and Flag-CUG-BP1 were cloned into the adenovirus expression vector (pAdTrack-CMV, generously provided by Coralie Poizet and Larry Kedes, USC) and normal myoblasts were infected with recombinant adenoviruses. At 48 h after infection, the cells were harvested and whole-cell extracts were prepared using a lysis buffer (25 mM Tris–HCl, pH 7.6, 200 mM NaCl, 0.5% NP-40, 2.0 mM EDTA, 2.0 mM MgCl2, and protease inhibitor (Sigma Inc., USA)). The extracts were split into two aliquots and the both aliquots were incubated with anti-Flag beads (Sigma) at 4°C for 2 h with mild agitation. After several washes with a buffer (10 mM Tris–HCl, pH 7.6, 10 mM HEPES, 200 mM NaCl, 2.0 mM MgCl2, 0.5 mM EDTA, 0.05% NP-40, 10% glycerol, 1.0 mM DTT and 0.1 mM PMSF), each immunoprecipitate was treated either with or without RNAse A (1.0 mg/ml) and both immunoprecipitates were incubated at 37°C for 20 min. Immunoprecipitates were then washed extensively and the bound proteins were eluted with an elution buffer (2% glycerol in PBS with 200 ng/μl of the Flag peptide). The eluted proteins were analyzed by SDS–PAGE and Western blot analyses.
Western blot analyses
Whole-cell extracts were prepared from harvested cells and equal amounts of protein (10 μg) were resolved by SDS–PAGE and transferred onto Hybond P membranes (Amersham). After blocking with 5% skim milk in 0.1% Tween-20 in PBS, the membranes were incubated with the desired antibodies for 2 h at room temp or overnight at 4°C. After incubation, the membranes were washed with 0.1% Tween-20 in PBS and then incubated with the corresponding secondary antibodies conjugated with HRP. Signals were detected by using the ECL plus detection reagents (Amersham), according to the manufacturer's protocol. To ensure that the signals were not saturated prior standardization experiments were conducted as described in Supplementary Methods (Supplementary Figure S2). The primary antibody dilutions were 1:8000 for Flag M2 mab (4.9 mg/ml), 1:3000 for goat anti-hnRNP H (N-16) polyclonal antibodies (200 μg/ml) and 1:4000 for CUG-BP1 (3B1) mab (200 μg/ml). The secondary antibody dilutions were 1:8000 for goat anti-mouse IgG (1 mg/ml), and 1:5000 for donkey anti-goat IgG (400 μg/ml). To control for protein quality and loading the membranes were re-probed with goat anti-GAPDH (V-18) polyclonal antibodies (200 μg/ml) at a dilution of 1:3000. The relative band intensities were measured by densitometry analyses using Gene Tool (Syngene). All antibodies other than anti-Flag M2 mab and goat anti-mouse IgG peroxidase conjugate (Sigma) were purchased from Santa Cruz Inc., USA.
In vitro RNA binding assay
Methods for purification of recombinant His tagged-MBNL1, hnRNP H and CUG-BP1 for the in vitro RNA binding assay are described in the Supplementary Methods. 32P-labeled IR-B RNA was transcribed in vitro using [α-32P]UTP (MP Biomedicals, LLC, USA). UV crosslinking assays were carried out primarily as described by Ladd et al (2001) with some modifications. Purified recombinant proteins (700 ng) (His tagged-MBNL1, hnRNP H, and CUG-BP1 or GST) and labeled RNA were incubated at 25°C for 15 min with 1 μg of tRNA (Ambion Inc., USA), and 1 μg of bovine serum albumin in the binding buffer (20 mM HEPES (pH 7.9), 2 mM magnesium acetate, 2 mM ATP, 65 mM potassium glutamate, 0.16 mM EDTA, 0.4 mM DTT and 10% glycerol). The reaction mixtures were UV irradiated at 400 mJ (twice). Samples were subsequently digested with 0.5 μg of RNase T1 (Ambion) at 37°C for 20 min. An equal volume of protein loading buffer was added and samples were denatured at 80°C and run on a SDS–PAGE gel. The gels were dried and autoradiographed.
Supplementary Material
Supplementary Materials and methods
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Acknowledgments
This work was supported by grants from NIH and the MDA to S Reddy.
References
- Boucher CA, King SK, Carey N, Krahe R, Winchester CL, Rahman S, Creavin T, Meghji P, Bailey MES, Chartier FL, Brown SD, Siciliano MJ, Johnson KJ (1995) A novel homeodomain-encoding gene is associated with a large CpG island interrupted by the myotonic dystrophy unstable (CTG)n repeat. Hum Mol Genet 4: 1919–1925 [DOI] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburtani H, Hunter K, Stanton VP, Thirion J-P, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, Housman DE (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68: 799–808 [DOI] [PubMed] [Google Scholar]
- Buj-Bello A, Furling D, Tronchere H, Laporte J, Lerouge T, Butler-Browne GS, Mandel JL (2002) Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet 11: 2297–2307 [DOI] [PubMed] [Google Scholar]
- Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA (2002) Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 1: 45–53 [DOI] [PubMed] [Google Scholar]
- Dansithong W, Paul S, Comai L, Reddy S (2005) MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem 280: 5773–5780 [DOI] [PubMed] [Google Scholar]
- Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD (2002) Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet 11: 805–814 [DOI] [PubMed] [Google Scholar]
- Fu Y-H, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizutti A, Ashizawa T, Perryman MB, Scarlato G, Fenwick RGJ, Caskey CT (1993) Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science 260: 235–238 [DOI] [PubMed] [Google Scholar]
- Fu Y-H, Pizutti A, Fenwick RG, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, Wieringa B, Korneluk R, Perryman MB, Epstein HF, Caskey CT (1992) An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255: 1256–1258 [DOI] [PubMed] [Google Scholar]
- Harper PS (1989) Myotonic Dystrophy. 2nd edn. Philadelphia: Saunders [Google Scholar]
- Ho TH, Bundman D, Armstrong DL, Cooper TA (2005) Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet 14: 1539–1547 [DOI] [PubMed] [Google Scholar]
- Ho TH, Charlet-B N, Poulos MG, Singh G, Swanson MS, Cooper TA (2004) Muscleblind proteins regulate alternative splicing. EMBO J 23: 3103–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 13: 3079–3088 [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS (2003) A muscleblind knockout model for myotonic dystrophy. Science 302: 1978–1980 [DOI] [PubMed] [Google Scholar]
- Kim DH, Langlois MA, Lee KB, Riggs AD, Puymirat J, Rossi JJ (2005) HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucl Acids Res 33: 3866–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S (2005) Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet 14: 2189–2200 [DOI] [PubMed] [Google Scholar]
- Klesert TR, Otten AD, Bird TD, Tapscott SJ (1997) Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat Genet 16: 402–406 [DOI] [PubMed] [Google Scholar]
- Kosaki A, Nelson J, Webster NJ (1998) Identification of intron and exon sequences involved in alternative splicing of insulin receptor pre-mRNA. J Biol Chem 273: 10331–10337 [DOI] [PubMed] [Google Scholar]
- Ladd AN, Charlet BN, Cooper TA (2001) The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol 21: 1285–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293: 864–867 [DOI] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K, Leblond S, Earle-Macdonald J, De Jong PJ, Wieringa Bé, Korneluk RG (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255: 1253–1255 [DOI] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289: 1769–1773 [DOI] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA (2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 1: 35–44 [DOI] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19: 4439–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips AV, Timchenko LT, Cooper TA (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280: 737–741 [DOI] [PubMed] [Google Scholar]
- Reddy S, Paul S (2006) Cis effects of CTG expansion in myotonic dystrophy type 1. In Genetic Instabilities and Neurological Diseases. Wells R, Ashizawa T (eds). 2nd edn. Elsevier-Academic Press [Google Scholar]
- Savkur RS, Philips AV, Cooper TA (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29: 40–47 [DOI] [PubMed] [Google Scholar]
- Seino S, Bell GI (1989) Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun 159: 312–316 [DOI] [PubMed] [Google Scholar]
- Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, Fouquet C, Butler-Browne G, Delacourte A, Junien C, Gourdon G (2001) Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet 10: 2717–2726 [DOI] [PubMed] [Google Scholar]
- Taneja KL, McCurrach ME, Shalling M, Housman D, Singer R (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128: 995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CA, Wymer JP, Simmons Z, McClain C, Moxley RT III (1997) Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat Genet 16: 407–409 [DOI] [PubMed] [Google Scholar]
- Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS (1996) Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucl Acids Res 24: 4407–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT (2001) RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem 276: 7820–7826 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and methods
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4