

Abstract
Epstein–Barr virus (EBV)-encoded small RNAs (EBERs) are nonpolyadenylated, untranslated RNAs, exist most abundantly in latently EBV-infected cells, and are expected to show secondary structures with many short stem–loops. Retinoic acid-inducible gene I (RIG-I) is a cytosolic protein that detects viral double-stranded RNA (dsRNA) inside the cell and initiates signaling pathways leading to the induction of protective cellular genes, including type I interferons (IFNs). We investigated whether EBERs were recognized by RIG-I as dsRNA. Transfection of RIG-I plasmid induced IFNs and IFN-stimulated genes (ISGs) in EBV-positive Burkitt's lymphoma (BL) cells, but not in their EBV-negative counterparts or EBER-knockout EBV-infected BL cells. Transfection of EBER plasmid or in vitro-synthesized EBERs induced expression of type I IFNs and ISGs in RIG-I-expressing, EBV-negative BL cells, but not in RIG-I-minus counterparts. EBERs activated RIG-I's substrates, NF-κB and IFN regulatory factor 3, which were necessary for type I IFN activation. It was also shown that EBERs co-precipitated with RIG-I. These results indicate that EBERs are recognized by RIG-I and activate signaling to induce type I IFN in EBV-infected cells.
Keywords: Burkitt's lymphoma, EBER, EBV, RIG-I, type I IFN
Introduction
Epstein–Barr virus (EBV) is widespread in the human population, is the causative agent of infectious mononucleosis, and is associated with various malignancies, including Burkitt's lymphoma (BL), nasopharyngeal carcinoma, gastric carcinoma, Hodgkin's lymphoma, and lymphomas in immunosuppressed patients (Rickinson and Kieff, 2001). It efficiently infects human resting B-lymphocytes in vitro and transforms them into indefinitely proliferating lymphoblastoid cell lines (LCLs). LCLs express 12 EBV gene products, including six EBV nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP), three latent membrane proteins (LMP1, LMP2A, and LMP2B), BamH1-A rightward transcripts (BARTs), and two EBV-encoded small RNAs (EBER1 and EBER2) (Kieff and Rickinson, 2001).
EBERs are the most abundant viral transcripts in cells with latent EBV infection (Rymo, 1979). EBERs are nonpolyadenylated, untranslated RNAs either 167 or 172 nucleotides long (Rosa et al, 1981; Glickman et al, 1988), and are structurally highly conserved (Arrand et al, 1989). Several reports have described growth-stimulatory activities of EBERs (Komano et al, 1999; Kitagawa et al, 2000; Ruf et al, 2000; Yamamoto et al, 2000; Iwakiri et al, 2003; Yang et al, 2004; Iwakiri et al, 2005). They are known to make complexes with several cellular proteins, such as RNA-activated protein kinase PKR (Clarke et al, 1991), ribosomal protein L22 (Toczyski and Steitz, 1991; Toczyski et al, 1994), and La (Lerner et al, 1981). Therefore, EBERs may exert various biological effects through their direct interaction with the cellular proteins. Among them, the significance of the interaction between EBERs and PKR, a key mediator of the antiviral effect of type I interferon (IFN-α and IFN-β), has been most extensively studied (Sharp et al, 1993; Yamamoto et al, 2000; Nanbo et al, 2002). We have shown that in BL cells, EBERs confer resistance to IFN-α-induced apoptosis by binding PKR and inhibiting its phosphorylation (Nanbo et al, 2002). Moreover, we have shown that EBERs induce the expression of various cellular cytokines—interleukin 10 (IL-10) in B-lymphocytes (Kitagawa et al, 2000), IL-9 in T-lymphocytes (Yang et al, 2004), and insulin-like growth factor 1 (IGF-1) in epithelial cells (Iwakiri et al, 2003, 2005)—each of which acts as an autocrine growth factor. However, the mechanisms by which EBERs induce these cytokines remain to be elucidated.
Retinoic acid-inducible gene I (RIG-I) is a cytosolic protein that detects viral double-stranded RNA (dsRNA) inside the cell and initiates signaling pathways leading to the induction of protective cellular genes, including type I IFNs and inflammatory cytokines. RIG-I contains an N-terminal caspase recruitment domain (CARD) and a C-terminal DExD/H-box RNA helicase domain (Yoneyama et al, 2004). The helicase domain is responsible for dsRNA recognition, and the CARD domain activates downstream signals, resulting in the activation of transcription factors, NF-κB and IFN regulatory factor 3 (IRF-3). RIG-I functions independently of toll-like receptor 3 (TLR3), which is involved in the recognition of viral dsRNA on the cell surface and induction of type I IFN responses. In latently EBV-infected cells, EBERs are expressed abundantly (Rymo, 1979) and are expected to form a stem–loop structure by intramolecular base-pairing, giving rise to dsRNA-like molecules (Rosa et al, 1981; Glickman et al, 1988). Thus, we hypothesized that EBERs were recognized by RIG-I as dsRNA and activated RIG-I signaling in EBV-infected cells. Hence, we analyzed the interaction of EBERs and RIG-I using BL-derived Mutu, Akata, and Daudi cell lines (Klein et al, 1968; Gregory et al, 1990; Takada et al, 1991). They were originally EBV-positive, and we isolated EBV-negative cell clones from these three BL cell lines (Shimizu et al, 1994; Nanbo et al, 2002). We here report that in BL cells, EBERs are recognized by RIG-I and activate signaling to induce type I IFN and IFN-stimulated genes (ISGs).
Results
RIG-I induces type I IFN in EBV-infected cells, but not in EBV-uninfected counterparts
To examine the RIG-I signaling pathway in the EBV-infected cells, we first used BL-derived Mutu cells (Gregory et al, 1990). They originated from an EBV-positive BL cell line and from which EBV-negative subclones have been isolated (Nanbo et al, 2002). EBV-positive Mutu cells (Mutu(+) cells) and EBV-negative Mutu cells (Mutu(−) cells) were transfected with a plasmid carrying green fluorescence protein (GFP)-tagged RIG-I or a control plasmid carrying only the GFP gene. The efficiency of transfection and expression of RIG-I in both Mutu(+) and Mutu(−) cells were checked at 24 h post-transfection by fluorescence and immunoblot assay (Figure 1A). Expression of RIG-I resulted in the activation of IFN-α, IFN-β, and ISG56 in Mutu(+) cells (Figure 1B), but not in Mutu(−) cells (Figure 1C). EBV-positive Mutu cells retained BL-type EBV expression (termed type I latency) that was characterized by expression of a restricted set of EBV latent genes, including EBNA1, BARTs, and EBERs (Nanbo et al, 2002). Among these latently expressed genes, we envisioned that EBERs would be responsible for RIG-I activation, because it had been demonstrated that EBERs functioned as dsRNA-like molecules (Glickman et al, 1988; Clarke et al, 1991) and that RIG-I specifically recognized dsRNA (Yoneyama et al, 2004).
Figure 1.
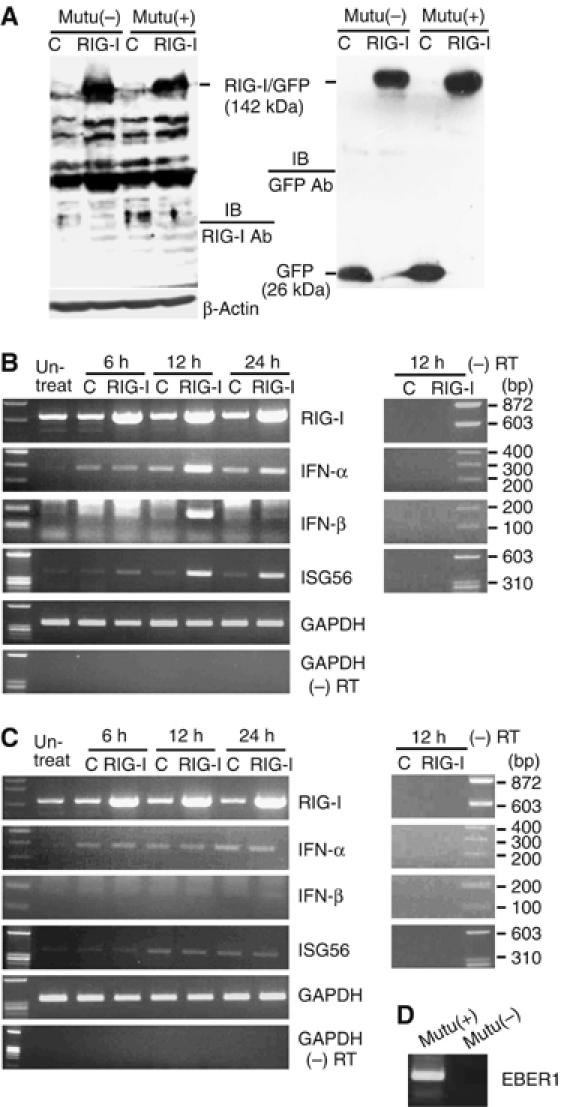
RIG-I induces type I IFN in EBV-infected cells. EBV-positive Mutu cells (Mutu(+)) and EBV-negative Mutu cells (Mutu(−)) (5 × 106 each) were transfected with 30 μg of GFP-tagged RIG-I plasmid or control GFP plasmid, and expression of IFNs and ISG56 was examined at the designated time by RT–PCR. (A) Immunoblot analysis for detection of RIG-I. The blots were probed with anti-RIG-I antibody (left panel) and anti-GFP antibody (right panel). Cell lysates were prepared after 24 h of transfection and 20 μg of protein sample was loaded per slot. (B) RT–PCR analysis of expression of IFN-α, IFN-β, and ISG56 in Mutu (+) cells. (C) RT–PCR analysis of expression of IFN-α, IFN-β, and ISG56 in Mutu (−) cells. (D) RT–PCR analysis of EBER1 expression in Mutu(+) and Mutu(−) cells.
RIG-I induces type I IFN in EBER-expressing cells
To test our hypothesis that EBERs were responsible for RIG-I activation, we used Akata cells infected with EBER-knockout EBV (EBER(−) EBV) or EBER-reintroduced EBV (EBER(+) EBV). The reasons for choosing Akata cells were that they were BL-derived cells with type I latency, and both EBER(−) EBV- and EBER(+) EBV-infected cell clones were available (Yajima et al, 2005). Transient expression of RIG-I resulted in the induction of IFN-β and ISG56 in EBER(+) EBV-infected Akata cells, but not in EBER(−) EBV-infected Akata cells (Figure 2A).
Figure 2.
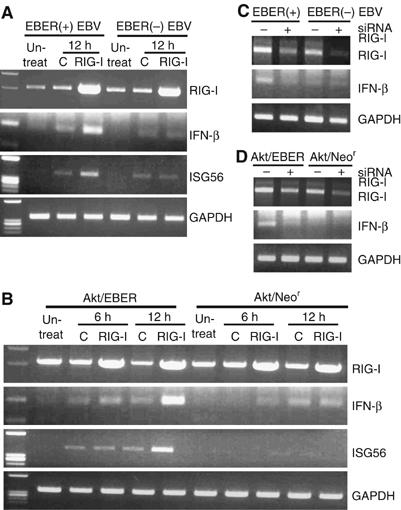
RIG-I induces type I IFN in EBER-expressing cells. BL-derived, EBER-positive and EBER-negative Akata cells (5 × 106 each) were transfected with 30 μg of GFP-tagged RIG-I plasmid or GFP plasmid, and expression of IFN-β and ISG56 was examined at the designated time by RT–PCR. (A) Expression of IFN-β and ISG56 in Akata cells infected with EBER-knockout EBV (EBER(−) EBV) or EBER-reintroduced EBV (EBER(+) EBV). After 12 h of transfection of the RIG-I plasmid or control plasmid, cells were subjected to RT–PCR analysis. (B) Expression of IFN-β and ISG56 in Akata cells stably transfected with EBER plasmid (Akt/EBER) or in those with control plasmid carrying the neomycin resistance gene (Akt/Neor). After 6 and 12 h of transfection of the RIG-I plasmid or GFP plasmid, cells were subjected to RT–PCR analysis. (C) EBER-positive EBV-infected and EBER-negative EBV-infected Akata cells were transfected with 100 nM of RIG-I siRNA or control siRNA. After 24 h, expression of RIG-I and IFN-β was checked by RT–PCR. (D) EBER-expressing Akata cells and control cells were also transfected with 100 nM of RIG-I siRNA/control siRNA and the expression of RIG-I and IFN-β was examined at 24 h post-transfection.
To further confirm that EBERs were responsible for RIG-I activation, we analyzed RIG-I function in EBV-negative Akata cells stably expressing EBERs and in control Akata cells carrying the neomycin resistance gene (Komano et al, 1999). Transient expression of RIG-I resulted in the expression of IFN-β and ISG56 in EBER-expressing Akata cells (Akt/EBER), but not in control Akata cells (Akt/Neor) (Figure 2B). Knockdown of RIG-I by siRNA resulted in downregulation of basal expression of IFN-β in EBER(+) EBV-infected Akata cells (Figure 2C) and also in EBER-expressing Akata cells (Figure 2D), but not in their negative counterparts. These results clearly demonstrated that EBERs activate RIG-I-mediated signaling cascades.
RIG-I-expressing stable clone recognizes EBERs
EBV-negative Daudi cells were transfected with the plasmid carrying GFP-tagged RIG-I or control plasmid carrying the GFP gene, and cell clones stably expressing RIG-I/GFP or GFP were isolated. The reasons for choosing EBV-negative Daudi cells to make stable clones were that they did not express TLR3 (data not shown), they were also BL-derived (Klein et al, 1968), and EBV-negative derivatives were available (Nanbo et al, 2002). The expression of RIG-I in the stable clone was analyzed by immunoblot assay using an anti-GFP antibody (Figure 3A). To express EBERs, we used EKS10 plasmid containing 10 tandem repeats of the SacI/EcoRI subfragment from the EcoRI K fragment of Akata EBV DNA (Komano et al, 1999), and transfected it into the RIG-I-expressing cell clone and control cell clone. Transient expression of EBERs resulted in the induction of IFN-β only in RIG-I-expressing clones. The induction of IFN-β peaked as early as 6 h after transfection, suggesting that EBERs readily interacted with RIG-I after being expressed from the transgene (Figure 3B).
Figure 3.
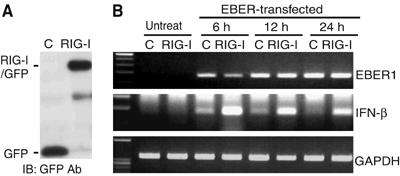
EBERs induce type I IFN in RIG-I-expressing cell clones. EBV-negative Daudi cells were transfected with the plasmid carrying GFP-tagged RIG-I or control plasmid carrying the GFP gene only, and cell clones stably expressing RIG-I/GFP or GFP were isolated. They (5 × 106 each) were transfected with 30 μg of EBER plasmid, and expression of type I IFN was examined at the designated time by RT–PCR. (A) Immunoblot analysis for detection of RIG-I. The blots were probed with the anti-GFP antibody. Protein samples amounting to 20 μg was loaded per slot. (B) RT–PCR analysis for detection of IFN-β.
Both EBER1 and EBER2 activate RIG-I to induce type I IFN
EBERs consist of EBER1 and EBER2. They have different primary nucleotide sequences and secondary structures (Rosa et al, 1981; Glickman et al, 1988). Thus, it is possible that EBER1 and EBER2 behave differently when they activate RIG-I. To resolve this issue, we synthesized both EBER1 and EBER2 by in vitro transcription and purified them as described previously (Sumpter et al, 2005). Analysis of purified in vitro-synthesized EBER1 and EBER2 in 5% denaturing PAGE (7 M urea) revealed a single band (Figure 4A, left panel), which was abolished after digestion with RNase A, indicating the purity of RNA and the absence of DNA contamination (Figure 4A, middle panel). It was also confirmed that in vitro-synthesized EBER1 and EBER2 were specifically amplified by EBER1- and EBER2-specific primers, respectively (Figure 4A, right panel).
Figure 4.
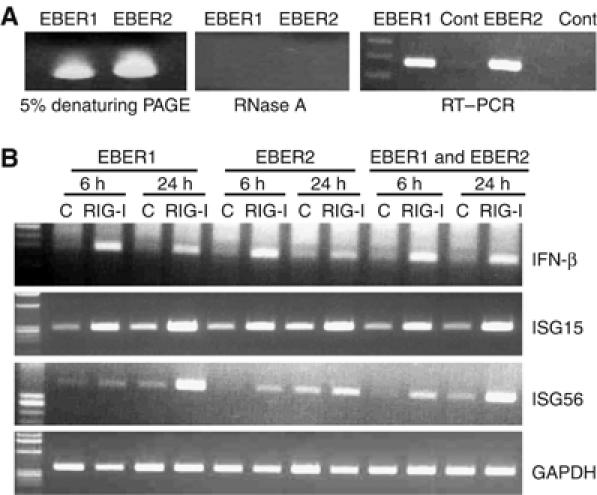
Both EBER1 and EBER2 activate RIG-I to induce type I IFN. (A) Analysis of in vitro-synthesized EBER1 and EBER2 in 5% denaturing PAGE before (left panel) and after (middle panel) RNase A treatment. RT–PCR analysis of EBER1 cDNA with EBER1 and EBER2 primers and also EBER2 cDNA with EBER2 and EBER1 primers (right panel). (B) RT–PCR analysis of expression of IFN-β, ISG56, and ISG15 after transfection of EBER1 and EBER2. RIG-I- or GFP- (5 × 106 each) expressing stable clones were transfected with 30 μg of EBER1 or EBER2, separately or in combination (1:1). Expression of IFN-β and ISGs was examined after 6 and 24 h of transfection.
Next, we transfected in vitro-synthesized EBER1 and EBER2 separately, as well as in combination (EBERs), into EBV-negative Daudi cell clones stably expressing RIG-I/GFP or GFP. Transfection of either EBER1 or EBER2 alone (30 μg each) or together (EBER1 and EBER2; 15 μg each) could induce expression of IFN-β, ISG56, and ISG15 in RIG-I-expressing cell clones, but not in control cell clones (Figure 4B). EBER1 and EBER2 had identical effects on IFN-β and ISG15 induction, whereas induction of ISG56 appeared to be greater for EBER1 than EBER2.
EBERs activate NF-κB and IRF-3
NF-κB and IRF-3 are downstream signals that are activated by RIG-I and are necessary for RIG-I-mediated type I IFN activation (Yoneyama et al, 2004; Li et al, 2005; Sumpter et al, 2005).
To determine the involvement of NF-κB in the induction of type I IFN by EBERs, we transfected in vitro-synthesized EBERs or polyI:C (as a positive control) into RIG-I-expressing EBV-negative Daudi cells and control cells. EBERs, as well as polyI:C, activated NF-κB, as evidenced by enhanced phosphorylation of NF-κB in RIG-I-expressing cells (Figure 5A, left panel), but not in control cells (Figure 5A, right panel). NF-κB reporter assay also expressed similar results (Figure 5B). To further confirm that EBERs activated NF-κB to induce type I IFN, RIG-I-expressing, EBV-negative Daudi cells were first transfected with an IκB-α plasmid, a specific inhibitor for NF-κB. At 36 h post-transfection, the cells were further transfected with in vitro-synthesized EBERs, and expression of IFN-β was examined at 8 h after the transfection of EBERs. The results demonstrated that transfection of IκB-α plasmid decreased IFN-β induction by EBERs in a dose-dependent manner (Figure 5C). The efficiency of the IκB-α plasmid to block NF-κB activation was confirmed by reporter assay, in which IκB-α plasmid blocked luciferase expression from the NF-κB promoter (Figure 5D).
Figure 5.
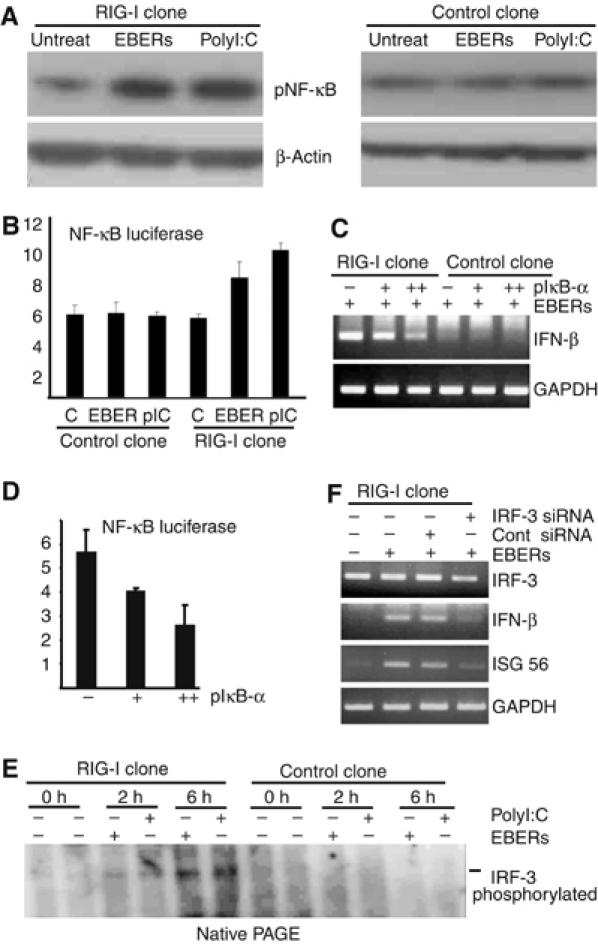
EBERs activate NF-κB and IRF-3. (A) Phosphorylation of NF-κB in RIG-I- or GFP-expressing stable clone was analyzed after they (5 × 106 cells each) were transfected with 30 μg of in vitro-synthesized EBERs (1:1) or polyI:C (positive control). After 6 h of transfection, whole-cell lysates (20 μg of protein for each sample) were subjected to immunoblot analysis for detection of phosphorylated NF-κB. (B) NF-κB reporter assay. NF-κB plasmid was transfected into both RIG-I-expressing Daudi cell clone and control clone and after 36 h of transfection, EBERs or polyI:C were transfected. After 12 h of EBER or polyI:C transfection, luciferase activity was measured in cell lysates (Figure 5B). Results are shown as the means±standard errors from three independent experiments. (C) Effect of IκB-α, a specific inhibitor of NF-κB, on EBER-induced phosphorylation of NF-κB. RIG-I-expressing stable clones (5 × 106 cells) were first transfected with 10 μg (+) or 20 μg (++) of IκB-α plasmid. After 36 h, they were further transfected with 30 μg of in vitro-synthesized EBERs (1:1), and expression of IFN-β was examined by RT–PCR after 8 h of transfection. (D) Inhibition of NF-κB activation by IκB-α plasmid. RIG-I-expressing stable clones (5 × 106 cells) were transfected with 10 μg of pNF-κB/luc (firefly luciferase) and 100 ng of pCMV/luc (Renilla luciferase as an internal control), along with 10 μg (+) or 20 μg (++) of IκB-α plasmid. After 36 h, luciferase activity was quantified in cell lysates using a Dual-Luciferase reporter assay system (Promega). Results are shown as the means±standard errors from three independent experiments. (E) Phosphorylation of IRF-3 in EBV-negative Daudi cells stably expressing RIG-I. RIG-I- or GFP- (5 × 106 cells each) expressing stable clones were transfected with 30 μg of in vitro-synthesized EBERs (15 μg each) or polyI:C (positive control). After 2 and 6 h of transfection, whole-cell lysates (30 μg of protein for each sample) were separated with native PAGE gel (8%) to analyze the phosphorylation of IRF-3 by EBERs or polyI:C. The blot was probed with anti-phosho-IRF-3 antibody. This antibody reacted with phosphorylated IRF-3, but not with unphosphorylated IRF-3. (F) RIG-I-expressing stable clone was treated with 100 nM of IRF-3 siRNA or control siRNA. After 24 h, cells were transfected with 30 μg of in vitro-synthesized EBERs, and 6 h later, expression of IRF-3, IFN-β, and ISG56 was examined by RT–PCR.
To examine whether IRF-3 was activated by EBERs in RIG-I-expressing cells, RIG-I-expressing, EBV-negative Daudi cells were transfected with in vitro-synthesized EBERs or polyI:C (as a positive control). Immunoblot analysis with native PAGE indicated that EBERs, as well as polyI:C, phosphorylated IRF-3, as evidenced by detection of the phosphorylated IRF-3 in RIG-I-expressing cells, but not in control cells (Figure 5E). Knockdown of IRF-3 by siRNA resulted in downregulation of IFN-β and ISG56 induction by EBERs in RIG-I-expressing cells (Figure 5F).
These results suggested that the induction of type I IFN by EBERs could be mediated by activation of the RIG-I signaling pathway through NF-κB and IRF-3, in agreement with other dsRNA-mediated signaling reported earlier (Yoneyama et al, 2004; Li et al, 2005; Sumpter et al, 2005).
EBER is recognized by full-length RIG-I
RIG-I consists of an RNA-binding helicase domain and a signal-transducing CARD domain. RIG-I recognizes dsRNA through its helicase domain (Yoneyama et al, 2004). To analyze the functional interaction of EBERs with the helicase domain and CARD domain of RIG-I, we established two EBV-negative Daudi cell clones stably expressing the deletion mutants of RIG-I, as described previously (Yoneyama et al, 2004). The first cell clone expressed an N-terminal CARD domain of RIG-I (N-RIG) tagged with GFP, and the second cell clone expressed a C-terminal helicase domain (C-RIG) tagged with GFP (Figure 6A). The expressions of N-RIG and C-RIG in stable clones were checked by immunoblot assay using anti-RIG-I antibody and also with anti-GFP antibody (Figure 6B). Next, we transfected in vitro-synthesized EBERs (15 μg each of EBER1 and EBER2) into N-RIG-expressing and C-RIG-expressing cell clones. The results indicated that deletion of the helicase domain, as well as the CARD domain, abrogated RIG-I function to induce IFN-β and ISGs (Figure 6C). Transfection of polyI:C also revealed the same result (Figure 6C). Thus, we concluded that both CARD domain and helicase domain of RIG-I were necessary to activate IFN signaling that is triggered by EBERs.
Figure 6.
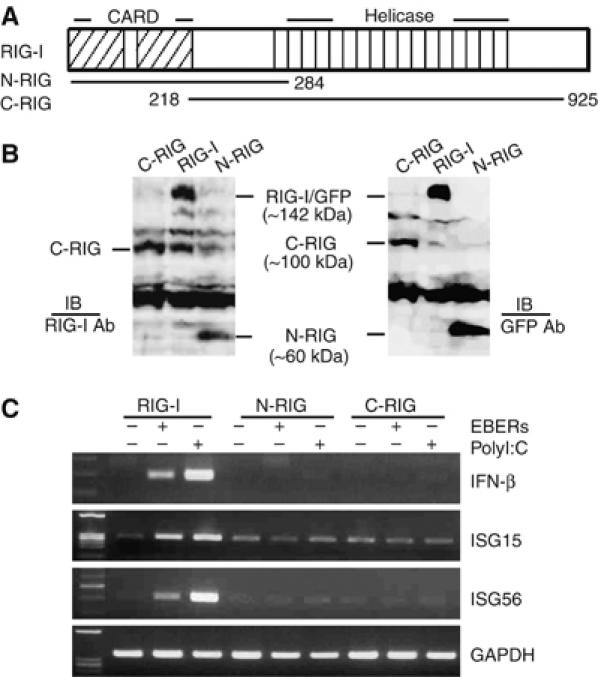
EBER is recognized by full-length RIG-I. (A) Schematic diagram of mutant plasmids containing the CARD domain (1–284 aa) or the helicase domain (218–925 aa). (B) Immunoblot analysis for detection of mutant RIG-I expressions in EBV-negative Daudi cell clones stably transfected with the deletion plasmids containing GFP-tagged CARD domain (N-RIG) or GFP-tagged helicase domain (C-RIG). Expression of N-RIG and C-RIG was detected by anti-RIG-I antibody (left panel) and also after reprobing with anti-GFP antibody (right panel). (C) Expression of type I IFN and ISGs in EBV-negative Daudi cells stably transfected with the deletion plasmids of RIG-I. The cells (5 × 106 cells each) were transfected with 30 μg of EBERs or polyI:C, and after 6 h, subjected to RT–PCR for detection of IFN-β, ISG15, and ISG56.
EBERs co-precipitate with RIG-I
Next, we examined whether EBERs could co-precipitate with RIG-I. As anti-RIG-I antibody, which was used in this study, was not available for immunoprecipitation, we used GFP-tagged full-length RIG-I to examine the binding of EBERs with RIG-I. The full-length RIG-I plasmid (RIG-I/GFP) or control plasmid (containing only the GFP gene) was transfected into Mutu cells infected with EBER-positive EBV (Mutu(+) cells). The same set of plasmids was also transfected into EBV-negative Mutu cells (Mutu(−)) and also EBV-negative Mutu cells stably expressing EBERs (Mutu/EBER cells). After 48 h of transfection, cells were treated with UV irradiation and then with RNases to remove unbound RNAs, and subjected to immunoprecipitation with anti-GFP antibody. RNA was isolated from immunoprecipitants, and the amounts of EBER1 and EBER2 were measured by RT–PCR. The result revealed that EBERs co-precipitated along with RIG-I when the cells were transfected with RIG-I/GFP and subjected to immunoprecipitation (Figure 7). By contrast, little EBERs were found in the precipitates when the cells were transfected with the control plasmid. Thus, we concluded that EBERs bound RIG-I or were at least part of the same complex.
Figure 7.
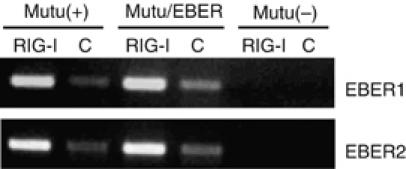
Binding assay for detection of association of EBERs with RIG-I. The full-length RIG-I plasmid (RIG-I/GFP) or control plasmid (containing the GFP gene only) was transfected into EBV-negative Mutu cells (Mutu(−) cells), EBER-positive EBV-infected Mutu cells ((Mutu(+) cells), and EBER-transfected Mutu cells (Mutu/EBER cells). After 48 h, cells were treated with UV irradiation, digested with RNases, and subjected to immunoprecipitation with the anti-GFP antibody. RNA was isolated from immunoprecipitants, and EBER1 and EBER2 were measured by RT–PCR.
RIG-I recognizes EBERs in the early phase of EBV infection
The results described above indicated that RIG-I-expressing cells could be activated by EBERs to induce type I IFN and ISGs. We speculated, therefore, that in primary EBV infection, newly synthesized EBERs could be recognized by RIG-I and activate the induction of type I IFN. To test this hypothesis, we used EBV-negative Daudi cell clones stably expressing RIG-I/GFP or GFP. They were infected with EBER(+) EBV or EBER(−) EBV (Yajima et al, 2005), and induction of IFN-β was examined at the designated time of EBV infection. The results indicated that IFN-β was induced by EBER(+) EBV infection in RIG-I-expressing cells, but not in control cells (Figure 8). On the other hand, IFN-β was not induced by EBER(−) EBV infection in either RIG-I-expressing cells or control cells (Figure 8), although both EBER(+) EBV and EBER(−) EBV infected the cells with similar infectivities as revealed by the expression of EBNA2. These results, considered together, indicated that EBERs were recognized by RIG-I in the early phase of EBV infection resulting in the induction of type I IFN in BL cells.
Figure 8.
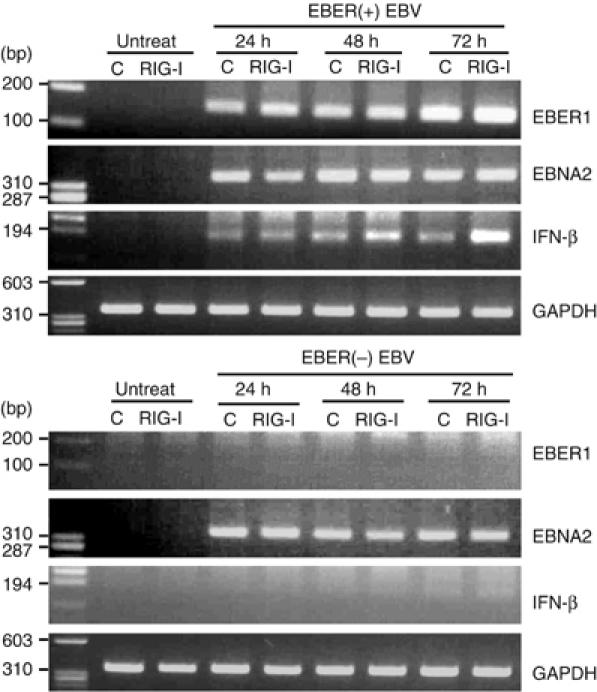
RIG-I recognizes EBERs in the early phase of EBV infection. EBV-negative Daudi cell clones stably expressing RIG-I/GFP or GFP were infected with EBER(+) EBV or EBER(−) EBV, and expression of EBER1, EBNA2, and IFN-β was examined at the designated time of EBV infection by RT–PCR.
Discussion
It is well established that RIG-I is activated by dsRNA that is synthesized as a replicative intermediate during replication of RNA viruses, including Newcastle disease virus, vesicular stomatitis virus (Yoneyama et al, 2004), and hepatitis C virus (Sumpter et al, 2005). Here, we demonstrated that EBV-encoded, non-coding RNAs, EBERs, were recognized by RIG-I and activated its downstream signaling, leading to the induction of type I IFN and other IFN-inducible genes in EBV-infected B-lymphocytes. This is the first report showing that DNA virus-encoded RNAs are recognized by RIG-I.
It is known that in viral infection, IFN-β is first expressed and then the induced IFN-β activates the expression of IFN-α (Levy et al, 2003). This will be the reason why IFN-β is expressed earlier than IFN-α in our studies.
The primary sequence similarity between EBER1 and EBER2 is only 54%, but both EBER1 and EBER2 show striking similarity in their secondary structures (Rosa et al, 1981; Glickman et al, 1988). Activation of RIG-I by both EBER1 and EBER2 highlights the importance of double stranded-like structures of EBERs. EBERs have extensive structural similarity to adenoviruses VA1 and VA2 and cell U6 small RNAs, which are small nonpolyadenylated RNAs like EBERs (Rosa et al, 1981). It would be interesting to test whether VAs and U6 RNAs are recognized by RIG-I and induce type I IFN signaling.
Production of type I IFN by EBERs induces apoptosis of EBV-infected cells. To evade IFN attack, EBV has developed several protective viral genes, including BHRF1, LMP1, and EBERs (Henderson et al, 1991, 1993; Nanbo et al, 2002, 2005). BHRF1 protein, a viral homolog of Bcl-2, protects EBV-infected cells from apoptotic cell death. LMP1 has been shown to induce Bcl-2, thus protecting EBV-infected cells from apoptosis. Moreover, we have shown that EBERs counteract PKR activation, which is a key mediator of the antiviral effect of type I IFN. PKR is induced by IFN and its autophosphorylation is stimulated by viral dsRNA, thereby activating two cellular substrates, eIF-2α and IκB-α, leading to the apoptosis of virus-infected cells (Moran et al, 2005). We have shown that EBERs bind PKR, inhibit its phosphorylation, and confer resistance to IFN-α-induced apoptosis in EBV-infected cells. These findings, together with the present findings, indicate that EBV evades antiviral activities of IFN through activation of protective viral genes, and suggest that EBV latent infection is maintained under the balance of actions of these viral and cellular gene products.
Another aspect of the functions of RIG-I that we have not dealt here is the induction of various inflammatory cytokines. It is well known that activation of RIG-I results in production of type I IFN as well as inflammatory cytokines such as IL-6, IL-8, and IL-12p40 (Foy et al, 2005; Kato et al, 2005). Previously, we have reported that EBERs induce transcription of various cytokines, including IL-10 (Kitagawa et al, 2000), IL-9 (Yang et al, 2004), and IGF-1 (Iwakiri et al, 2003, 2005), and that the produced cytokines act as autocrine growth factors. However, the mechanism of cytokine induction by EBERs is still not known. It is very likely, and is worth testing, that EBERs induce transcription of these cytokines through activation of RIG-I.
Further studies will be needed to elucidate the overall picture of the interaction of EBERs and RIG-I and to uncover strategies by which EBV evades and/or utilizes this surveillance mechanism to establish stable infection not only in BL cells but also in other EBV-associated malignancies.
Materials and methods
Cells and culture
EBV-negative and EBV-positive Mutu cells (Gregory et al, 1990) and EBV-negative Daudi cells (Nanbo et al, 2002) were maintained in RPMI 1640 medium containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA). Akata cells infected with EBER-knockout EBV or EBER-reintroduced EBV (Yajima et al, 2005) and Akata cells stably transfected with the EBER plasmid or control plasmid with the neomycin resistance gene (Komano et al, 1999) were maintained in the same medium with the addition of 500 μg/ml of G418 (Sigma, St Louis, MO).
Plasmids, transfection, and establishment of stable clones
Human RIG-I cDNA was amplified from LCL and cloned into pSG5/EGFPN1 vector, thereby making a fused RIG-I expression construct (pSG5/EGFPN1/RIG-I) with the GFP gene tagged at the C-terminus. RIG-I deletion constructs containing only the CARD domain (N-RIG) or helicase domain (C-RIG) with GFP gene at the C-terminus were also made as described earlier (Yoneyama et al, 2004). EKS10 plasmid, containing 10 tandem repeats of EBER1 and EBER2, was made as described previously (Komano et al, 1999). pNF-κB/luc (encoding firefly luciferase) and pCMV/luc (encoding Renilla luciferase as an internal control) were obtained commercially (BD Biosciences, San Jose, CA), and pIκB-α, a specific inhibitor of NF-κB, was a kind gift of Bill Sugden (Mitchell and Sugden, 1995). Endotoxin-free plasmid DNA was prepared by using an endo-free Mega Prep kit (Invitrogen). For transient or stable expression of plasmids, we performed electroporation. Stable transfectants expressing full-length RIG-I (RIG-I), helicase-deleted RIG-I (N-RIG), CARD-deleted RIG-I (C-RIG), and GFP clones were selected by culturing with 1000 μg/ml of G418 (Sigma).
Virus production and infection
For EBER-positive EBV and EBER-negative EBV production, we followed the protocol described earlier (Yajima et al, 2005).
For EBV infection, EBV-negative Daudi cells stably expressing RIG-I (RIG-I/GFP) or GFP (106 cells each) were suspended in 1 ml virus soup and incubated for 90 min at 37°C with continuous gentle mixing. Then, the cells were washed with serum-free medium, resuspended in 2 ml of growth medium, and incubated for 24, 48, and 72 h in 12-well cell culture plates.
In vitro synthesis of EBERs and RNA transfection
EBER1 and EBER2 were amplified from LCL (Akata) and cloned into pGEMT easy vector (Promega, Madison, WI). From pGEMT/EBER1 and pGEMT/EBER2, T7 promoter-tagged (at the 5′ end) EBER1 and EBER2 were synthesized by PCR (primer pairs: EBER1, 5′-TGT AAT ACG ACT CAC TAT AGG GAC CTA CGC TGC CCT AGA GGT TTT GCT; 5′-AAA ACA TGC GGA CCA GC; EBER2, 5′-TGT AAT ACG ACT CAC TAT AGG GAC AGC CGT TGC CCT AGT GGT TTC GGA; 5′-AAA ACA GCG GAC AAG CCG AAT ACC). One microgram of purified DNA (PCR product) was used as a template per reaction, and in vitro transcription was carried out for 6 h following the manufacturer's protocol (Epicenter, Madison, WI). Purified EBER1 and EBER2 (3 μg each) before and after RNase A treatment were checked in 5% denaturing polyacrylamide gel and by RT–PCR. For RNA transfection, EBER1, EBER2, EBER1 and EBER2 together (1:1), or polyI:C (30 μg each) was transfected by electroporation.
RNA extraction and RT–PCR analysis
Reverse transcription was carried out with oligo-dT primer using total cellular RNA. For EBERs, gene-specific 3′ end primers were used. One-tenth of cDNA was subjected to PCR using primers specific for IFN-α (5′-ATC CAG CAG ATC TTC AAT CT; 5′-AAG AAA AAG ATC TCA TGA TT; 35 cycles), IFN-β (5′-GAT TCA TCG AGC ACT GGC TGG; 5′-CTT CAG GTA ATG CAG AAT CC; 30 cycles), TLR3 (5′-TCA CTT GCT CAT TCT CCC TT; 5′-GAC CTC TCC ATT CCT GGC; 35 cycles), ISG15 (5′-GGT GGA CAA ATG CGA CGA AC; 5′-ATG CTG GTG GAG GCC CTT AG; 28 cycles), ISG56 (5′-TAG CCA ACA TGT CCT CAC AGA C; 5′-TCT TCT ACC ACT GGT TTC ATG C; 30 cycles), EBER1 (5′-AGG ACC TAC GCT GCC CTA GA; 5′-AAA ACA TGC GGA CCA GC; 18 cycles), EBER2 (5′-AGG GAC AGC CGT TGC CCT AGT GGT TTC GGA; 5′-AAA ACA GCG GAC AAG CCG AAT ACC; 18 cycles), RIG-I (5′-GCA TAT TGA CTG GAC GTG GCA; 5′-CAG TCA TGG CTG CAG TTC TGT C; 30 cycles), EBNA2 (5′-GCT GCT ACG CAT TAG AGA CC; 5′-CAG CAC TGG CGT GTG ACG TGG TGT AAA GTT; 35 cycles), IRF-3 (5′-CACAGCAGGAGGATTTCGG; 5′-CCTG GGTATCAGAAGTAC; 26 cycles), and GAPDH (5′-GCC TCC TGC ACC ACC AAC TG; 5′-CGA CGC CTG CTT CAC CAC CTT CT; 20 cycles). Control reactions were performed in parallel in the absence of reverse transcriptase.
Immunoblot analysis
Cell lysates containing equal quantities of protein (20–30 μg) were analyzed in 8% SDS–PAGE gel. The membranes were treated with the primary antibodies for RIG-I (Imaizumi et al, 2004), GFP (BD Biosciences), phospho-NF-κB p65 (Ser-536), and β-actin (Cell Signaling, Beverly, MA) at 4oC overnight, followed by reaction with HRP-conjugated anti-rabbit (1:2000) or anti-mouse (1:5000) IgG antibodies (Amersham Biosciences, Buckinghamshire, UK).
For IRF-3 phosphorylation analysis, whole-cell lysates were prepared after 2 or 6 h of EBERs or polyI:C transfection into RIG-I-expressing or control cell clones. Cell lysate preparation and immunoblotting with an anti-phospho-IRF-3 antibody (IBL, Takasaki, Japan) were performed, as described previously (Mori et al, 2004), using 8% Tris–HCl native PAGE ready gel (Invitrogen). Protein bands were visualized using ECL Plus Western blotting detection reagents (Amersham Biosciences, UK).
Immunoprecipitation and RT–PCR
Co-precipitation of EBERs with RIG-I was analyzed by UV crosslinking and immunoprecipitation of RIG-I and EBER complexes. GFP-tagged RIG-I or GFP was transiently expressed in EBER(+) EBV-infected Mutu cells, EBER-expressing EBV-negative Mutu cells, and EBV-negative Mutu cells (5 × 106 cells each). After 48 h, cells were washed with pre-chilled PBS and UV irradiated. Cell lysates were treated with RNase T1 and RNase A to remove unbound RNAs and immunoprecipitated with anti-GFP antibody. Total RNA was isolated from the immunoprecipitants, and one-third of the RNA sample was subjected to 30 cycles of RT–PCR with EBER1- and EBER2-specific primers. The compositions of buffers and the methodology for cell lysate preparation, RNA extraction, and RT–PCR analysis were described previously (Nanbo et al, 2002).
RNA interference
For siRNA experiments, dsRNA duplexes composed of 25-nucleotide sense and antisense oligonucleotides were synthesized from Invitrogen. RNA oligonucleotides used for targeting RIG-I and IRF-3 were as follows: RIG-I sense 5′-UUAGGAUUCUCAUUGCUGGGAUCCC-3′, antisense 5′-GGGAUCCCAGCAAUGAGAAUCCUAA-3′ and IRF-3 sense 5′-UAAACGCAACCCUUCUUUGCGGUUG-3′ and antisense 5′-CAACCGCAAAGAAGGGUUGCGUUUA-3′. Lipofectamine RNAiMax (Invitrogen) was used for reverse transfection of cells following the manufacturer's protocol (Invitrogen).
Acknowledgments
We thank Y Ando for technical assistance. This work was supported by grants-in-aid from the Ministry of Education, Science, Sports, Culture and Technology, Japan.
References
- Arrand JR, Young LS, Tugwood JD (1989) Two families of sequences in the small RNA-encoding region of Epstein–Barr virus (EBV) correlate with EBV types A and B. J Virol 63: 983–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PA, Schewemmle M, Schickinger J, Hilse K, Clemens MJ (1991) Binding of Epstein–Barr virus small RNA EBER-1 to the double stranded RNA-activated protein kinase DAI. Nucleic Acids Res 19: 243–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy E, Li K, Sumpter RJ, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale MJ (2005) Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci USA 102: 2986–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman JN, Howe JG, Steitz JA (1988) Structural analyses of EBER1 and EBER2 ribonucleoprotein particles present in Epstein–Barr virus-infected cells. J Virol 62: 902–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory CD, Rowe M, Rickinson AB (1990) Different Epstein–Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. J Gen Virol 71: 1481–1495 [DOI] [PubMed] [Google Scholar]
- Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, Kieff E, Rickinson A (1991) Induction of bcl-2 expression by Epstein–Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 65: 1107–1115 [DOI] [PubMed] [Google Scholar]
- Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A (1993) Epstein–Barr virus-encoded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci USA 90: 8479–8493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi T, Yagihashi N, Hatakeyama M, Yamashita K, Ishikawa A, Taima K, Yoshida H, Inoue I, Fujita T, Yagihashi S, Satoh K (2004) Expression of retinoic acid-inducible gene-I in vascular smooth muscle cells stimulated with IFN-γ. Life Sci 75: 1171–1180 [DOI] [PubMed] [Google Scholar]
- Iwakiri D, Eizuru Y, Tokunaga M, Takada K (2003) Autocrine growth of Epstein–Barr virus-positive gastric carcinoma cells mediated by an Epstein–Barr virus-encoded small RNA. Cancer Res 63: 7062–7067 [PubMed] [Google Scholar]
- Iwakiri D, Sheen TS, Chen JY, Huang DP, Takada K (2005) Epstein–Barr virus-encoded small RNA induces insulin-like growth factor 1 and supports growth of nasopharyngeal carcinoma-derived cell lines. Oncogene 24: 1767–1773 [DOI] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23: 19–28 [DOI] [PubMed] [Google Scholar]
- Kieff E, Rickinson AB (2001) Epstein–Barr virus and its replication. In Fields Virology, Fields BN, Knipe DM, Howley PM (eds) 4th edn, pp 2511–2573. Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- Kitagawa N, Goto M, Kurozumi K, Maruo S, Fukayama M, Naoe T, Yasukawa M, Hino K, Suzuki T, Todo S, Takada K (2000) Epstein–Barr virus-encoded poly(A)− RNA supports Burkitt's lymphoma growth through interleukin-10 induction. EMBO J 19: 6742–6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E, Klein G, Nadkarni JS, Nadkarni JJ, Wigzell H, Clifford P (1968) Surface IgM- specificity on a Burkitt lymphoma cell in vivo and in derived culture lines. Cancer Res 28: 1300–1310 [PubMed] [Google Scholar]
- Komano J, Maruo S, Kurozumi K, Oda T, Takada K (1999) Oncogenic role of Epstein–Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol 73: 9827–9831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Marie I, Praksh A (2003) Ringing the interferon alarm: differential regulation of gene expression at the interface between innate and adaptive immunity. Curr Opin Immunol 15: 52–58 [DOI] [PubMed] [Google Scholar]
- Lerner MR, Andrews NC, Miller G, Steitz JA (1981) Two small RNAs encoded by Epstein–Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc Natl Acad Sci USA 78: 805–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Chen Z, Kato N, Gale M Jr, Lemon SM (2005) Distinct poly(I–C) and virus-activated signaling pathways leading to interferon-β production in hepatocytes. J Biol Chem 280: 16739–16747 [DOI] [PubMed] [Google Scholar]
- Mitchell T, Sugden B (1995) Stimulation of NF-κB-mediated transcription by mutant derivatives of the latent membrane protein of Epstein–Barr Virus. J Virol 69: 2968–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JM, Buller RML, Corbett JA (2005) The cellular response to virus infection: the role of PKR and novel PKR-independent pathways. Cell Sci Rev 2: 108–131 [Google Scholar]
- Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T (2004) Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem 279: 9698–9702 [DOI] [PubMed] [Google Scholar]
- Nanbo A, Inoue K, Adachi-Takasawa K, Takada K (2002) Epstein–Barr virus RNA confers resistance to interferon-α-induced apoptosis in Burkitt's lymphoma. EMBO J 21: 954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A, Yoshiyama H, Takada K (2005) Epstein–Barr Virus-encoded poly(A)− RNA confers resistance to apoptosis mediated through Fas by blocking the PKR pathway in human epithelial intestine 407 cells. J Virol 79: 12280–12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson AB, Kieff E (2001) Epstein–Barr virus. In Fields Virology, 4th edn, Fields BN, Knipe DM, Howley PM (eds) pp 2575–2627. Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- Rosa MD, Gottlieb E, Lerner MR, Steitz JA (1981) Striking similarities are exhibited by two small Epstein–Barr virus-encoded ribonucleic acids and the adenovirus associated ribonucleic acids VAI and VAII. Mol Cell Biol 1: 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf IK, Rhyne PW, Yang C, Cleveland JL, Sample JT (2000) Epstein–Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J Virol 74: 10223–10228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymo L (1979) Identification of transcribed regions of Epstein Barr-virus DNA in Burkitt lymphoma-derived cells. J Virol 32: 8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Schwemmle M, Jeffrey I, Laing K, Mellor H, Proud CG, Hilse K, Clemens MJ (1993) Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein–Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res 21: 4483–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Tanabe-Tochikura A, Kuroiwa Y, Takada K (1994) Isolation of Epstein–Barr Virus (EBV)-negative cell clones from the EBV-positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J Virol 68: 6069–6073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter RJ, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M Jr (2005) Regulating intracellular antiviral defense and permissiveness to hepatitis C Virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79: 2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Horinouchi K, Ono Y, Aya T, Osato T, Takahashi M, Hayasaka S (1991) An Epstein–Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes 5: 147–156 [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Steitz JA (1991) EAP, a highly conserved cellular protein associated with Epstein–Barr virus small RNAs (EBER). EMBO J 10: 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski DP, Matera AG, Ward DC, Steitz JA (1994) The Epstein–Barr virus (EBV) small RNA EBER1 binds and relocalizes ribosomal protein L22 in EBV-infected human B lymphocytes. Proc Natl Acad Sci USA 91: 3463–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima M, Kanda T, Takada K (2005) Critical role of Epstein–Barr virus (EBV)-encoded RNA in efficient EBV-induced B-lymphocyte growth transformation. J Virol 79: 4298–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Takizawa T, Iwanaga Y, Shimizu N, Yamamoto N (2000) Malignant transformation of B lymphoma cell line BJAB by Epstein–Barr virus-encoded small RNAs. FEBS Lett 484: 153–158 [DOI] [PubMed] [Google Scholar]
- Yang L, Aozasa K, Oshimi K, Takada K (2004) Epstein–Barr virus (EBV)-encoded RNA promotes growth of EBV infected T cells through interleukin 9 induction. Cancer Res 64: 5332–5337 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]